
Rhinovirus and Cell Death


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Search Strategy
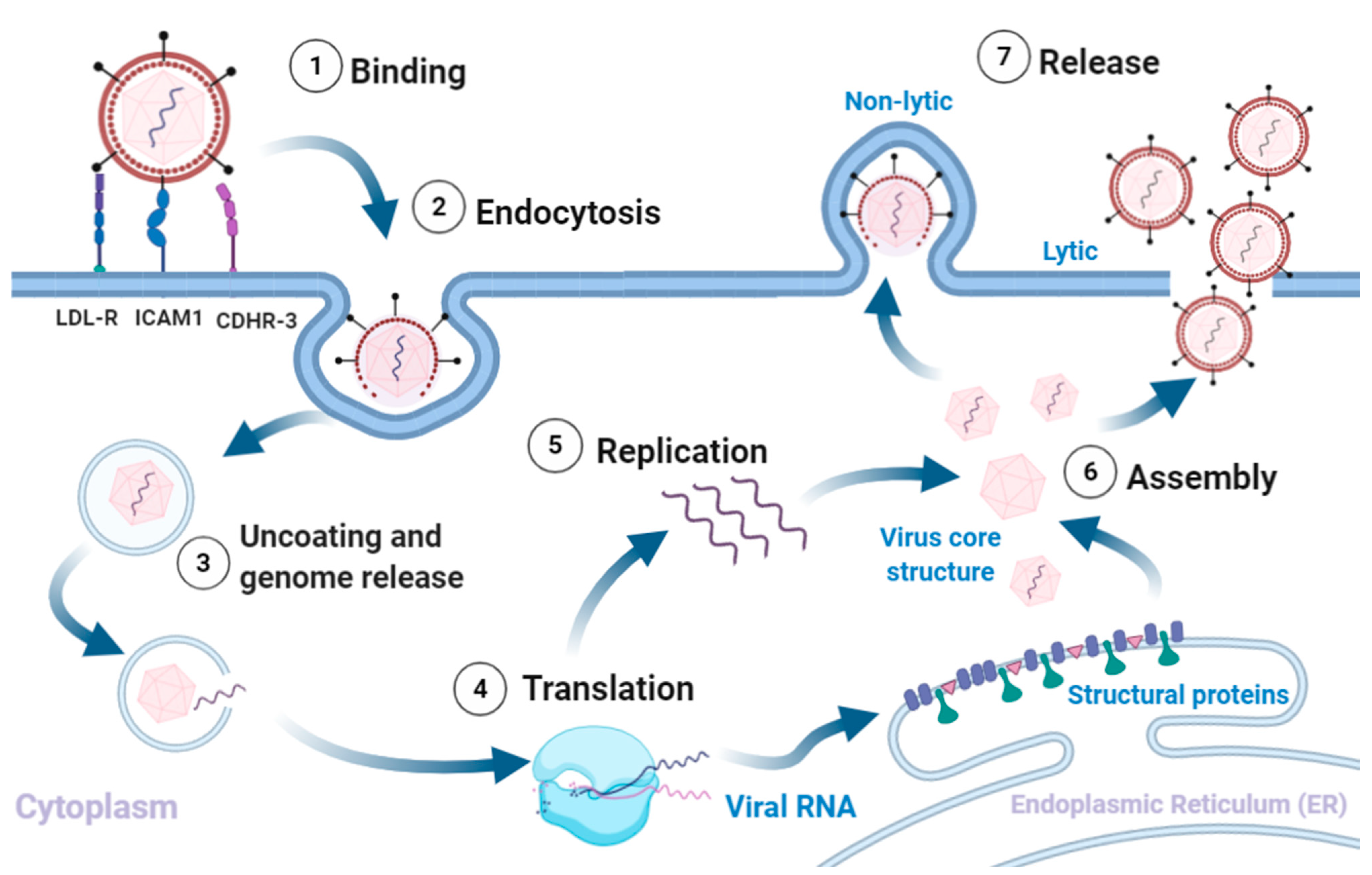
3. Virion Structure and Lifecycle
3.1. Virion Structure
3.2. Viral Replication, Assembly and Release
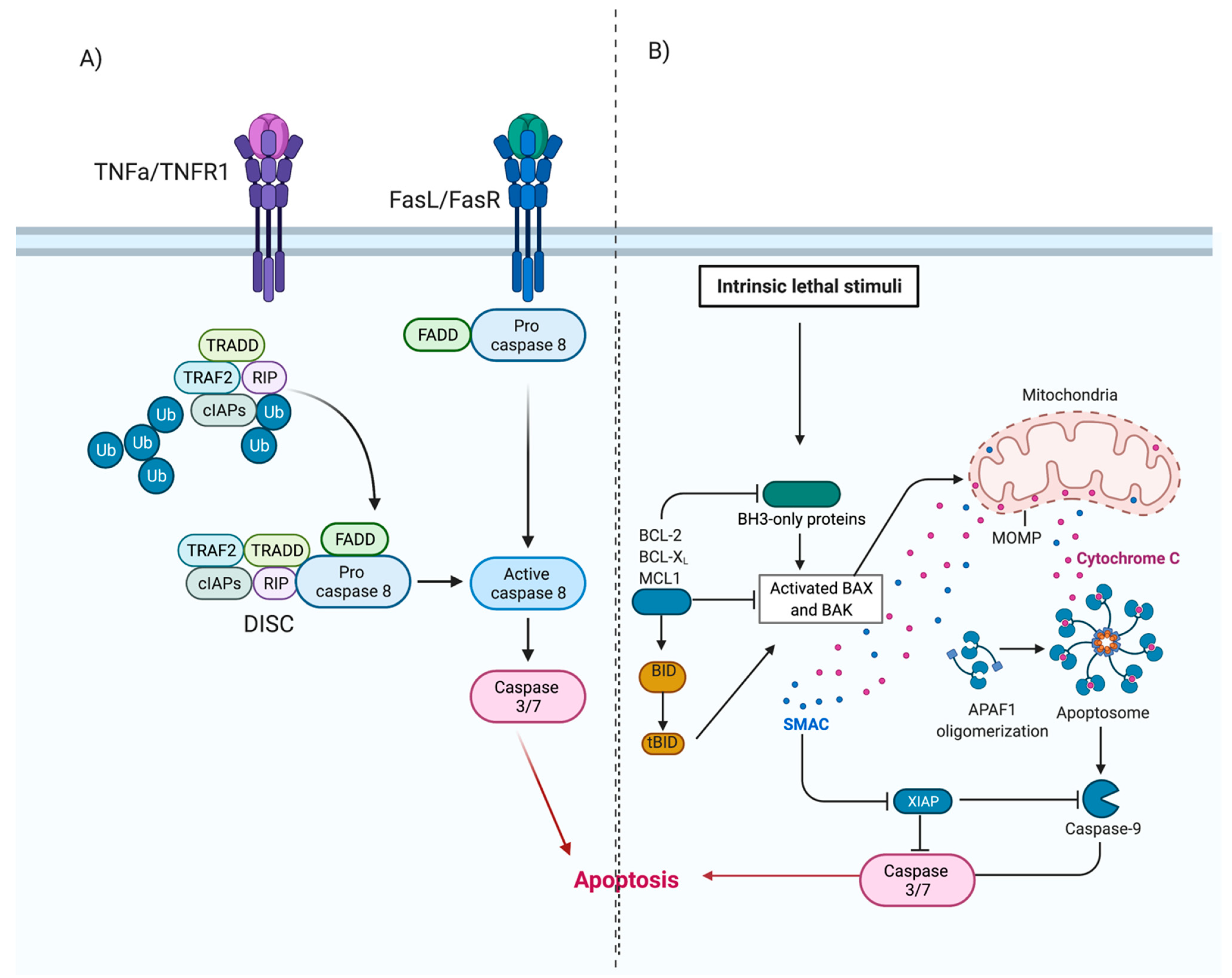
4. Cell Death Pathways
4.1. Apoptosis
Rhinovirus Can Modulate Apoptosis
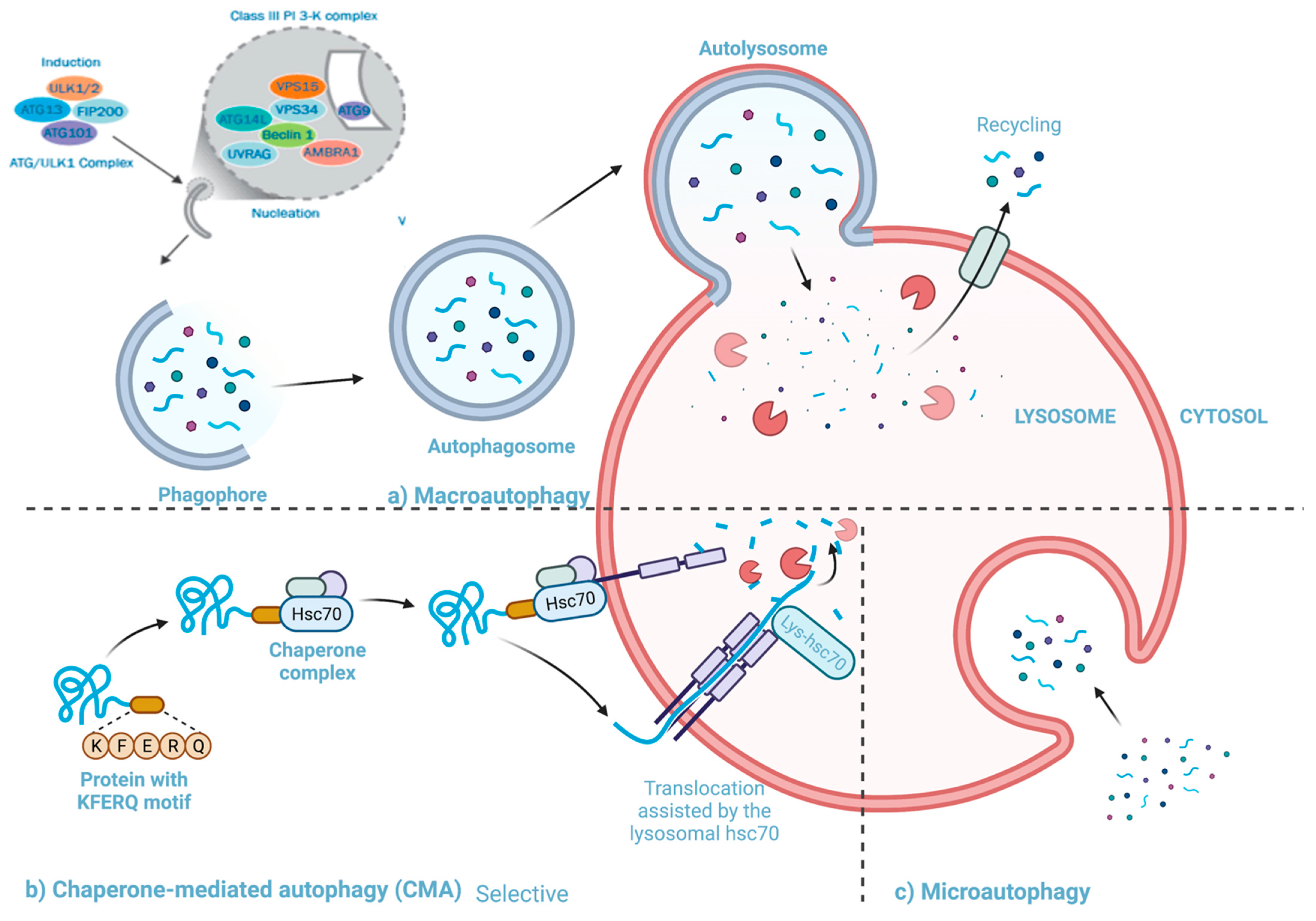
4.2. Autophagy
4.2.1. The Pathway and Mechanisms of Autophagy
4.2.2. The Interactions between Autophagy and Rhinovirus Infection
4.3. Necrosis
Modulation of Necrosis by Rhinovirus
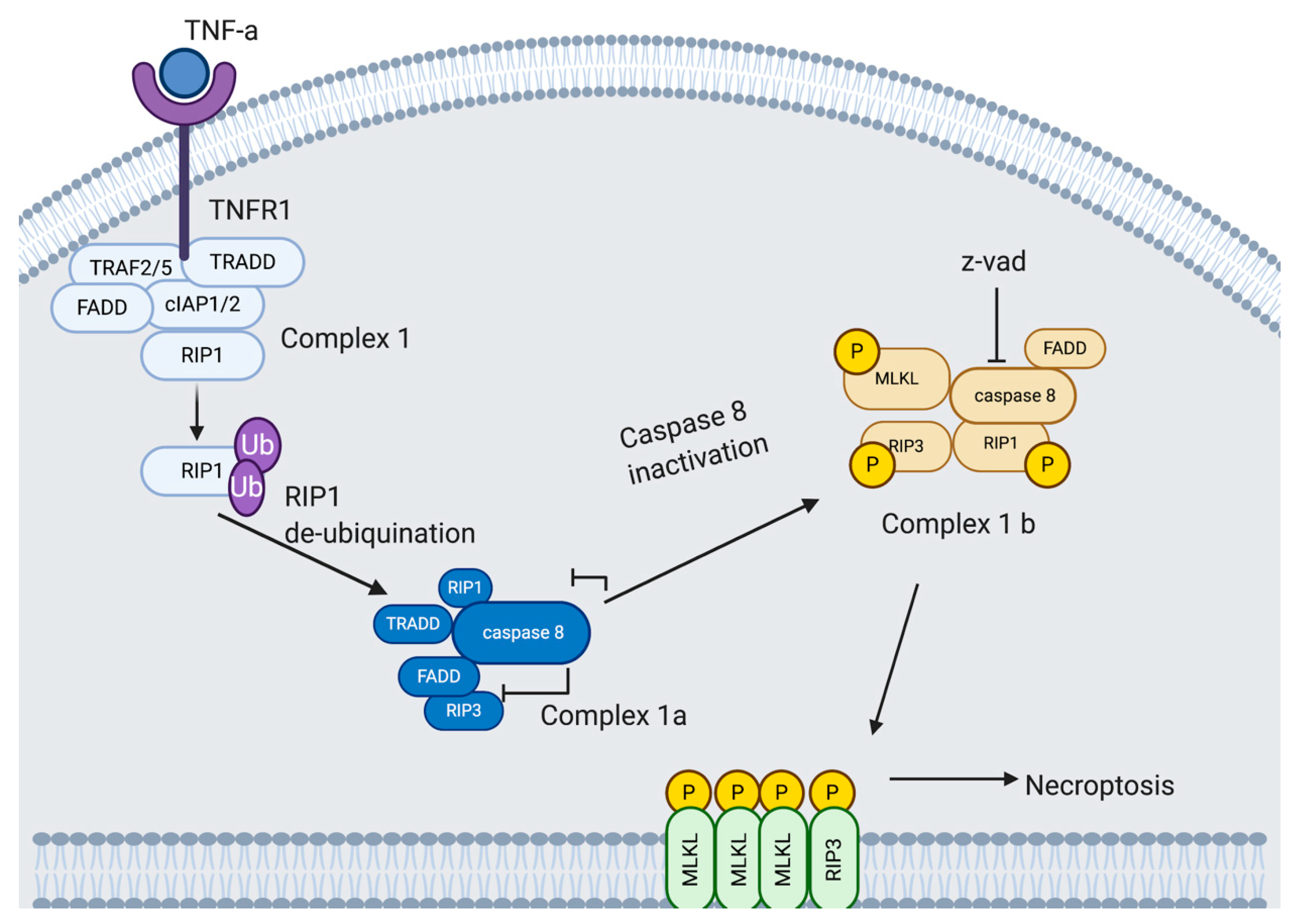
4.4. Necroptosis
4.4.1. Necroptosis Pathway
4.4.2. Potential Exploitation of Necroptosis by Rhinovirus
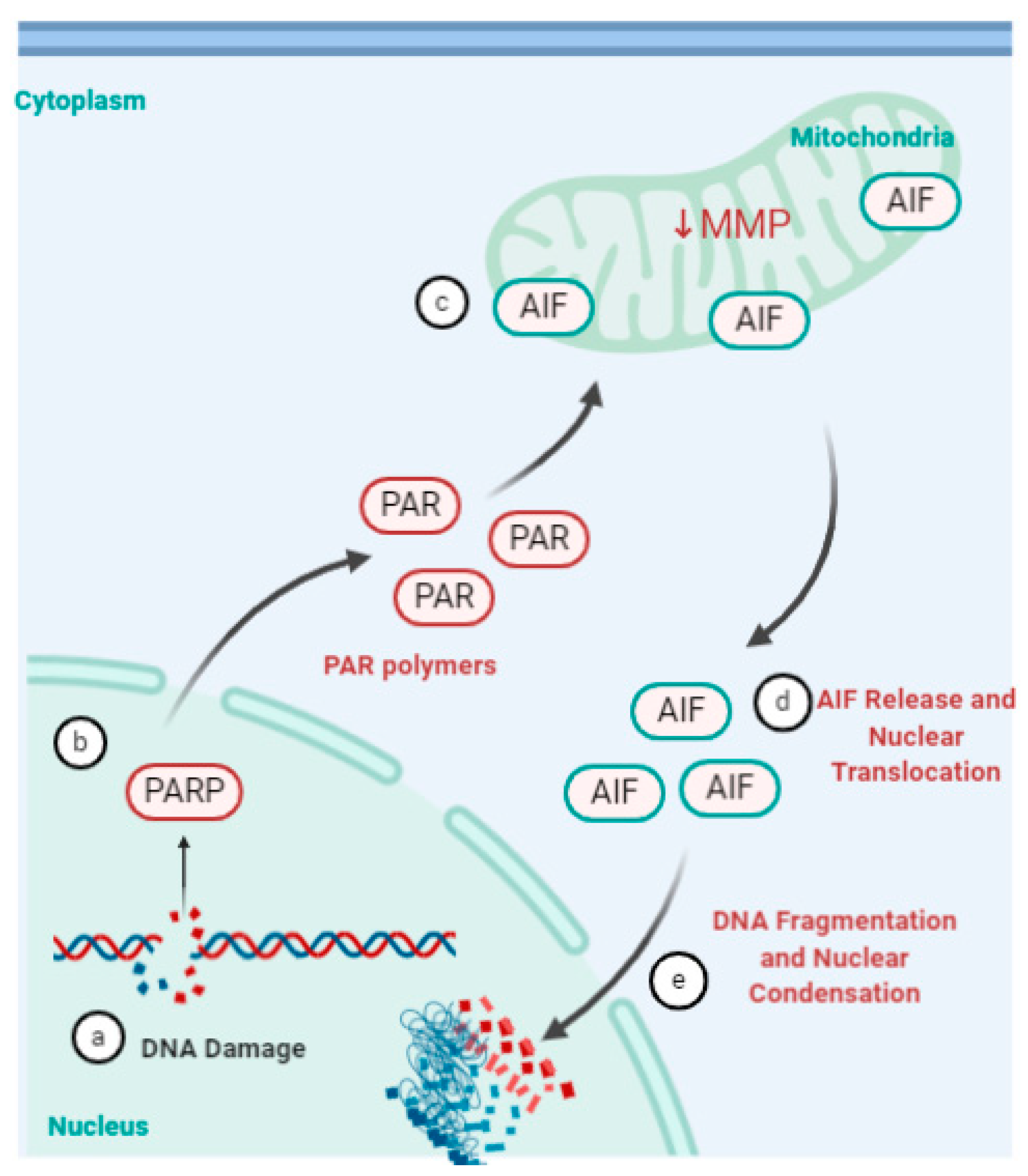
4.5. Parthanatos, Stress and Rhinovirus
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, T.; Yin, C.; Boyd, D.F.; Quarato, G.; Ingram, J.P.; Shubina, M.; Ragan, K.B.; Ishizuka, T.; Crawford, J.C.; Tummers, B.; et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 2020, 180, 1115–1129. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-Y.; Chiu, V.; Hsieh, P.-C.; Huang, C.-Y.; Huang, S.J.; Tzeng, I.S.; Tsai, F.-M.; Chen, M.-L.; Liu, C.-T.; Chen, Y.-R. Chrysophanol attenuates hepatitis B virus X protein-induced hepatic stellate cell fibrosis by regulating endoplasmic reticulum stress and ferroptosis. J. Pharmacol. Sci. 2020, 144, 172–182. [Google Scholar] [CrossRef]
- Suwanmanee, S.; Luplertlop, N. Immunopathogenesis of Dengue Virus-Induced Redundant Cell Death: Apoptosis and Pyroptosis. Viral Immunol. 2017, 30, 13–19. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, J.; Roh, J.; Park, C.S.; Seoh, J.Y.; Hwang, E.S. Reactive oxygen species-induced parthanatos of immunocytes by human cytomegalovirus-associated substance. Microbiol. Immunol. 2018, 62, 229–242. [Google Scholar] [CrossRef]
- Croft, S.N.; Walker, E.J.; Ghildyal, R. Picornaviruses and Apoptosis: Subversion of Cell Death. mBio 2017, 8, e01009-17. [Google Scholar] [CrossRef] [PubMed]
- Zlateva, K.T.; de Vries, J.J.C.; Coenjaerts, F.E.J.; van Loon, A.M.; Verheij, T.; Little, P.; Butler, C.C.; Goossens, H.; Ieven, M.; Claas, E.C.J. Prolonged shedding of rhinovirus and re-infection in adults with respiratory tract illness. Eur. Respir. J. 2014, 44, 169. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Self, W.H.; Wunderink, R.G.; Fakhran, S.; Balk, R.; Bramley, A.M.; Reed, C.; Grijalva, C.G.; Anderson, E.J.; Courtney, D.M.; et al. Community-Acquired Pneumonia Requiring Hospitalization among U.S. Adults. N. Engl. J. Med. 2015, 373, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Feddema, J.J.; Claassen, E. Prevalence of viral respiratory infections amongst asthmatics: Results of a meta-regression analysis. Respir. Med. 2020, 173, 106020. [Google Scholar] [CrossRef]
- Ortega, H.; Nickle, D.; Carter, L. Rhinovirus and asthma: Challenges and opportunities. Rev. Med. Virol. 2020, e2193. [Google Scholar] [CrossRef]
- Hershenson, M.B. Rhinovirus-Induced Exacerbations of Asthma and COPD. Scientifica 2013, 2013, 405876. [Google Scholar] [CrossRef]
- Iwane, M.K.; Prill, M.M.; Lu, X.; Miller, E.K.; Edwards, K.M.; Hall, C.B.; Griffin, M.R.; Staat, M.A.; Anderson, L.J.; Williams, J.V.; et al. Human rhinovirus species associated with hospitalizations for acute respiratory illness in young US children. J. Infect. Dis. 2011, 204, 1702–1710. [Google Scholar] [CrossRef]
- Palmenberg, A.C.; Gern, J.E. Classification and evolution of human rhinoviruses. Methods Mol. Biol. 2015, 1221, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.E.; Lamson, D.M.; St George, K.; Walsh, T.J. Human rhinoviruses. Clin. Microbiol. Rev. 2013, 26, 135–162. [Google Scholar] [CrossRef] [PubMed]
- Stobart, C.C.; Nosek, J.M.; Moore, M.L. Rhinovirus Biology, Antigenic Diversity, and Advancements in the Design of a Human Rhinovirus Vaccine. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef]
- Baggen, J.; Thibaut, H.J.; Strating, J.; van Kuppeveld, F.J.M. The life cycle of non-polio enteroviruses and how to target it. Nat. Rev. Microbiol. 2018, 16, 368–381. [Google Scholar] [CrossRef]
- Tuthill, T.J.; Groppelli, E.; Hogle, J.M.; Rowlands, D.J. Picornaviruses. Curr. Top. Microbiol. Immunol. 2010, 343, 43–89. [Google Scholar] [CrossRef]
- Uncapher, C.R.; DeWitt, C.M.; Colonno, R.J. The major and minor group receptor families contain all but one human rhinovirus serotype. Virology 1991, 180, 814–817. [Google Scholar] [CrossRef]
- Fuchs, R.; Blaas, D. Productive Entry Pathways of Human Rhinoviruses. Adv. Virol. 2012, 2012, 826301. [Google Scholar] [CrossRef]
- Hofer, F.; Gruenberger, M.; Kowalski, H.; Machat, H.; Huettinger, M.; Kuechler, E.; Blaas, D. Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc. Natl. Acad. Sci. USA 1994, 91, 1839–1842. [Google Scholar] [CrossRef]
- Bochkov, Y.A.; Gern, J.E. Rhinoviruses and Their Receptors: Implications for Allergic Disease. Curr. Allergy Asthma Rep. 2016, 16, 30. [Google Scholar] [CrossRef]
- Vlasak, M.; Roivainen, M.; Reithmayer, M.; Goesler, I.; Laine, P.; Snyers, L.; Hovi, T.; Blaas, D. The minor receptor group of human rhinovirus (HRV) includes HRV23 and HRV25, but the presence of a lysine in the VP1 HI loop is not sufficient for receptor binding. J. Virol. 2005, 79, 7389–7395. [Google Scholar] [CrossRef]
- Fuchs, R.; Blaas, D. Uncoating of human rhinoviruses. Rev. Med. Virol. 2010, 20, 281–297. [Google Scholar] [CrossRef]
- Prchla, E.; Kuechler, E.; Blaas, D.; Fuchs, R. Uncoating of human rhinovirus serotype 2 from late endosomes. J. Virol. 1994, 68, 3713–3723. [Google Scholar] [CrossRef]
- Garriga, D.; Pickl-Herk, A.; Luque, D.; Wruss, J.; Castón, J.R.; Blaas, D.; Verdaguer, N. Insights into Minor Group Rhinovirus Uncoating: The X-ray Structure of the HRV2 Empty Capsid. PLoS Pathog. 2012, 8, e1002473. [Google Scholar] [CrossRef]
- Panjwani, A.; Strauss, M.; Gold, S.; Wenham, H.; Jackson, T.; Chou, J.J.; Rowlands, D.J.; Stonehouse, N.J.; Hogle, J.M.; Tuthill, T.J. Capsid protein VP4 of human rhinovirus induces membrane permeability by the formation of a size-selective multimeric pore. PLoS Pathog. 2014, 10, e1004294. [Google Scholar] [CrossRef] [PubMed]
- Chase, A.J.; Semler, B.L. Viral subversion of host functions for picornavirus translation and RNA replication. Future Virol. 2012, 7, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Racaniello, V.R. Fields Virology—Picornaviridae: The Viruses and Their Replication, 4th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott/Raven: Philadelphia, PA, USA, 2001. [Google Scholar]
- Jiang, P.; Liu, Y.; Ma, H.-C.; Paul, A.V.; Wimmer, E. Picornavirus morphogenesis. Microbiol. Mol. Biol. Rev. 2014, 78, 418–437. [Google Scholar] [CrossRef]
- Kroemer, G.; El-Deiry, W.S.; Golstein, P.; Peter, M.E.; Vaux, D.; Vandenabeele, P.; Zhivotovsky, B.; Blagosklonny, M.V.; Malorni, W.; Knight, R.A.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005, 12, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Chicheportiche, Y.; Bourdon, P.R.; Xu, H.; Hsu, Y.M.; Scott, H.; Hession, C.; Garcia, I.; Browning, J.L. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J. Biol. Chem. 1997, 272, 32401–32410. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef]
- Peter, M.E.; Krammer, P.H. Mechanisms of CD95 (APO-1/Fas)-mediated apoptosis. Curr. Opin. Immunol. 1998, 10, 545–551. [Google Scholar] [CrossRef]
- Suliman, A.; Lam, A.; Datta, R.; Srivastava, R.K. Intracellular mechanisms of TRAIL: Apoptosis through mitochondrial-dependent and -independent pathways. Oncogene 2001, 20, 2122–2133. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Moscardo, F.; Blesa, D.; Mestre, C.; Siebert, R.; Balasas, T.; Benito, A.; Rosenwald, A.; Climent, J.; Martinez, J.I.; Schilhabel, M.; et al. Characterization of 8p21.3 chromosomal deletions in B-cell lymphoma: TRAIL-R1 and TRAIL-R2 as candidate dosage-dependent tumor suppressor genes. Blood 2005, 106, 3214–3222. [Google Scholar] [CrossRef]
- Deszcz, L.; Gaudernak, E.; Kuechler, E.; Seipelt, J. Apoptotic events induced by human rhinovirus infection. J. Gen. Virol. 2005, 86, 1379–1389. [Google Scholar] [CrossRef]
- Croft, S.N.; Walker, E.J.; Ghildyal, R. Human Rhinovirus 3C protease cleaves RIPK1, concurrent with caspase 8 activation. Sci. Rep. 2018, 8, 1569. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- van Loo, G.; van Gurp, M.; Depuydt, B.; Srinivasula, S.M.; Rodriguez, I.; Alnemri, E.S.; Gevaert, K.; Vandekerckhove, J.; Declercq, W.; Vandenabeele, P. The serine protease Omi/HtrA2 is released from mitochondria during apoptosis. Omi interacts with caspase-inhibitor XIAP and induces enhanced caspase activity. Cell Death Differ. 2002, 9, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef]
- Chinnaiyan, A.M. The apoptosome: Heart and soul of the cell death machine. Neoplasia 1999, 1, 5–15. [Google Scholar] [CrossRef]
- Hill, M.M.; Adrain, C.; Duriez, P.J.; Creagh, E.M.; Martin, S.J. Analysis of the composition, assembly kinetics and activity of native Apaf-1 apoptosomes. EMBO J. 2004, 23, 2134–2145. [Google Scholar] [CrossRef]
- Joza, N.; Susin, S.A.; Daugas, E.; Stanford, W.L.; Cho, S.K.; Li, C.Y.; Sasaki, T.; Elia, A.J.; Cheng, H.Y.; Ravagnan, L.; et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 2001, 410, 549–554. [Google Scholar] [CrossRef]
- Li, L.Y.; Luo, X.; Wang, X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001, 412, 95–99. [Google Scholar] [CrossRef]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Jiang, W.; Liu, Z.; Liu, S.; Liang, X. Virus Infection and Death Receptor-Mediated Apoptosis. Viruses 2017, 9, 316. [Google Scholar] [CrossRef]
- Lai, Y.; Wang, M.; Cheng, A.; Mao, S.; Ou, X.; Yang, Q.; Wu, Y.; Jia, R.; Liu, M.; Zhu, D.; et al. Regulation of Apoptosis by Enteroviruses. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Ghildyal, R.; Jordan, B.; Li, D.; Dagher, H.; Bardin, P.G.; Gern, J.E.; Jans, D.A. Rhinovirus 3C Protease Can Localize in the Nucleus and Alter Active and Passive Nucleocytoplasmic Transport. J. Virol. 2009, 83, 7349. [Google Scholar] [CrossRef]
- Gustin, K.E.; Sarnow, P. Inhibition of nuclear import and alteration of nuclear pore complex composition by rhinovirus. J. Virol. 2002, 76, 8787–8796. [Google Scholar] [CrossRef] [PubMed]
- Ferrando-May, E. Nucleocytoplasmic transport in apoptosis. Cell Death Differ. 2005, 12, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Lotzerich, M.; Roulin, P.S.; Boucke, K.; Witte, R.; Georgiev, O.; Greber, U.F. Rhinovirus 3C protease suppresses apoptosis and triggers caspase-independent cell death. Cell Death Dis. 2018, 9, 272. [Google Scholar] [CrossRef] [PubMed]
- Brisac, C.; Téoulé, F.; Autret, A.; Pelletier, I.; Colbère-Garapin, F.; Brenner, C.; Lemaire, C.; Blondel, B. Calcium Flux between the Endoplasmic Reticulum and Mitochondrion Contributes to Poliovirus-Induced Apoptosis. J. Virol. 2010, 84, 12226–12235. [Google Scholar] [CrossRef] [PubMed]
- Campanella, M.; de Jong, A.S.; Lanke, K.W.; Melchers, W.J.; Willems, P.H.; Pinton, P.; Rizzuto, R.; van Kuppeveld, F.J. The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J. Biol. Chem. 2004, 279, 18440–18450. [Google Scholar] [CrossRef] [PubMed]
- de Jong, A.S.; de Mattia, F.; Van Dommelen, M.M.; Lanke, K.; Melchers, W.J.; Willems, P.H.; van Kuppeveld, F.J. Functional analysis of picornavirus 2B proteins: Effects on calcium homeostasis and intracellular protein trafficking. J. Virol. 2008, 82, 3782–3790. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Yim, W.W.-Y.; Mizushima, N. Lysosome biology in autophagy. Cell Discov. 2020, 6, 6. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S. Choose Delicately and Reuse Adequately: The Newly Revealed Process of Autophagy. Biol. Pharm. Bull. 2015, 38, 1098–1103. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Biologicals, N. Autophagy-Signalling Pathway. Available online: www.novusbio.com/research-areas/autophagy/signaling-pathway (accessed on 28 January 2021).
- Ahmad, L.; Mostowy, S.; Sancho-Shimizu, V. Autophagy-Virus Interplay: From Cell Biology to Human Disease. Front. Cell Dev. Biol. 2018, 6. [Google Scholar] [CrossRef]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef]
- Klein, K.A.; Jackson, W.T. Human rhinovirus 2 induces the autophagic pathway and replicates more efficiently in autophagic cells. J. Virol. 2011, 85, 9651–9654. [Google Scholar] [CrossRef]
- Brabec-Zaruba, M.; Berka, U.; Blaas, D.; Fuchs, R. Induction of autophagy does not affect human rhinovirus type 2 production. J. Virol. 2007, 81, 10815–10817. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; van Dyk, L.F.; Jiang, D.; Dakhama, A.; Li, L.; White, S.R.; Gross, A.; Chu, H.W. Interleukin-1 receptor-associated kinase M (IRAK-M) promotes human rhinovirus infection in lung epithelial cells via the autophagic pathway. Virology 2013, 446, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Liu, P.; Li, J. Necroptosis: An emerging form of programmed cell death. Crit. Rev. Oncol. Hematol. 2012, 82, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Nobre, A.; Alarico, S.; Maranha, A.; Mendes, V.; Empadinhas, N. The molecular biology of mycobacterial trehalose in the quest for advanced tuberculosis therapies. Microbiology 2014, 160, 1547–1570. [Google Scholar] [CrossRef]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [PubMed]
- Golstein, P.; Kroemer, G. Cell death by necrosis: Towards a molecular definition. Trends Biochem. Sci. 2007, 32, 37–43. [Google Scholar] [CrossRef]
- Festjens, N.; Vanden Berghe, T.; Vandenabeele, P. Necrosis, a well-orchestrated form of cell demise: Signalling cascades, important mediators and concomitant immune response. Biochim. Biophys. Acta BBA Bioenerg. 2006, 1757, 1371–1387. [Google Scholar] [CrossRef]
- Montgomery, S.T.; Frey, D.L.; Mall, M.A.; Stick, S.M.; Kicic, A.; Arest, C.F. Rhinovirus Infection Is Associated With Airway Epithelial Cell Necrosis and Inflammation via Interleukin-1 in Young Children With Cystic Fibrosis. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, W.; Guan, Z.; Yu, W.; Fan, B.; Xu, N.; Liao, D.J. There are only four basic modes of cell death, although there are many ad-hoc variants adapted to different situations. Cell Biosci. 2018, 8, 6. [Google Scholar] [CrossRef]
- Zhao, H.; Jaffer, T.; Eguchi, S.; Wang, Z.; Linkermann, A.; Ma, D. Role of necroptosis in the pathogenesis of solid organ injury. Cell Death Dis. 2015, 6, e1975. [Google Scholar] [CrossRef] [PubMed]
- Hanson, B. Necroptosis: A new way of dying? Cancer Biol. Ther. 2016, 17, 899–910. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, T.; Lei, T.; Zhang, D.; Du, S.; Girani, L.; Qi, D.; Lin, C.; Tong, R.; Wang, Y. RIP1/RIP3-regulated necroptosis as a target for multifaceted disease therapy (Review). Int. J. Mol. Med. 2019, 44, 771–786. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, Y.; He, W.; Sun, L. Necrosome core machinery: MLKL. Cell. Mol. Life Sci. CMLS 2016, 73, 2153–2163. [Google Scholar] [CrossRef]
- Zhou, W.; Yuan, J. Necroptosis in health and diseases. Semin. Cell Dev. Biol. 2014, 35, 14–23. [Google Scholar] [CrossRef]
- Chen, J.; Kos, R.; Garssen, J.; Redegeld, F. Molecular Insights into the Mechanism of Necroptosis: The Necrosome as a Potential Therapeutic Target. Cells 2019, 8, 1486. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, K.; Shen, H.; Yao, X.; Sun, Q.; Chen, G. Necroptosis: A novel manner of cell death, associated with stroke (Review). Int. J. Mol. Med. 2018, 41, 624–630. [Google Scholar] [CrossRef]
- Diagnostics, C. Necroptosis Signaling Pathway. Available online: https://www.creative-diagnostics.com/necroptosis-signaling-pathway.htm (accessed on 15 January 2021).
- Kelly, J.T.; Busse, W.W. Host immune responses to rhinovirus: Mechanisms in asthma. J. Allergy Clin. Immunol. 2008, 122, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Gern, J.E.; Dick, E.C.; Lee, W.M.; Murray, S.; Meyer, K.; Handzel, Z.T.; Busse, W.W. Rhinovirus enters but does not replicate inside monocytes and airway macrophages. J. Immunol. 1996, 156, 621–627. [Google Scholar] [PubMed]
- Jackson, D.J.; Makrinioti, H.; Rana, B.M.; Shamji, B.W.; Trujillo-Torralbo, M.B.; Footitt, J.; Jerico, D.-R.; Telcian, A.G.; Nikonova, A.; Zhu, J.; et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am. J. Respir. Crit. Care Med. 2014, 190, 1373–1382. [Google Scholar] [CrossRef]
- Calvén, J.; Akbarshahi, H.; Menzel, M.; Ayata, C.K.; Idzko, M.; Bjermer, L.; Uller, L. Rhinoviral stimuli, epithelial factors and ATP signalling contribute to bronchial smooth muscle production of IL-33. J. Transl. Med. 2015, 13, 281. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nomura, M.; Ueno, A.; Saga, K.; Fukuzawa, M.; Kaneda, Y. Accumulation of cytosolic calcium induces necroptotic cell death in human neuroblastoma. Cancer Res. 2014, 74, 1056–1066. [Google Scholar] [CrossRef] [PubMed]
- Gazina, E.V.; Harrison, D.N.; Jefferies, M.; Tan, H.; Williams, D.; Anderson, D.A.; Petrou, S. Ion transport blockers inhibit human rhinovirus 2 release. Antivir. Res. 2005, 67, 98–106. [Google Scholar] [CrossRef]
- Wark, P.A.; Johnston, S.L.; Moric, I.; Simpson, J.L.; Hensley, M.J.; Gibson, P.G. Neutrophil degranulation and cell lysis is associated with clinical severity in virus-induced asthma. Eur. Respir. J. 2002, 19, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Cerps, S.C.; Menzel, M.; Mahmutovic Persson, I.; Bjermer, L.; Akbarshahi, H.; Uller, L. Interferon-β deficiency at asthma exacerbation promotes MLKL mediated necroptosis. Sci. Rep. 2018, 8, 4248. [Google Scholar] [CrossRef] [PubMed]
- Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Mitochondrial and nuclear cross talk in cell death: Parthanatos. In Annals of the New York Academy of Sciences; Wiley-Blackwell: Hoboken, NJ, USA, 2008; Volume 1147, pp. 233–241. [Google Scholar]
- Berger, N.A.; Sims, J.L.; Catino, D.M.; Berger, S.J. Poly(ADP-ribose) polymerase mediates the suicide response to massive DNA damage: Studies in normal and DNA-repair defective cells. Princess Takamatsu Symp. 1983, 13, 219–226. [Google Scholar]
- Crawford, K.; Bonfiglio, J.J.; Mikoč, A.; Matic, I.; Ahel, I. Specificity of reversible ADP-ribosylation and regulation of cellular processes. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 64–82. [Google Scholar] [CrossRef]
- Wark, P.; Pathinyake, P.; Hsu, A.; Parsons, K.; Wood, L. Effect of oxidative stress and rhinovirus infection on mitochondrial/endoplasmic reticular function in human primary bronchial epithelial cells. Eur. Respir. J. 2016, 48, PA3989. [Google Scholar] [CrossRef]
- Hudy, M.H.; Traves, S.L.; Wiehler, S.; Proud, D. Cigarette smoke modulates rhinovirus-induced airway epithelial cell chemokine production. Eur. Respir. J. 2010, 35, 1256. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.-Y.; Yang, Y.; Zhang, Y.-Y.; Xie, Z.; Zhao, X.-Y.; Sun, Y.; Kong, W.-J. The dual role of poly(ADP-ribose) polymerase-1 in modulating parthanatos and autophagy under oxidative stress in rat cochlear marginal cells of the stria vascularis. Redox Biol. 2018, 14, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Contoli, M. Rhinovirus vaccination: The case against. Eur. Respir. J. 2011, 37, 5. [Google Scholar] [CrossRef] [PubMed]
- Danthi, P. Viruses and the Diversity of Cell Death. Annu. Rev. Virol. 2016, 3, 533–553. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerr, S.-L.; Mathew, C.; Ghildyal, R. Rhinovirus and Cell Death. Viruses 2021, 13, 629. https://doi.org/10.3390/v13040629
Kerr S-L, Mathew C, Ghildyal R. Rhinovirus and Cell Death. Viruses. 2021; 13(4):629. https://doi.org/10.3390/v13040629
Chicago/Turabian StyleKerr, Shannic-Le, Cynthia Mathew, and Reena Ghildyal. 2021. "Rhinovirus and Cell Death" Viruses 13, no. 4: 629. https://doi.org/10.3390/v13040629
APA StyleKerr, S.-L., Mathew, C., & Ghildyal, R. (2021). Rhinovirus and Cell Death. Viruses, 13(4), 629. https://doi.org/10.3390/v13040629