
Population Disequilibrium as Promoter of Adaptive Explorations in Hepatitis C Virus

 , , , ,
, , , ,  ,
,
Abstract
1. Introduction: HCV Quasispecies in Cell Culture and In Vivo
2. Materials and Methods
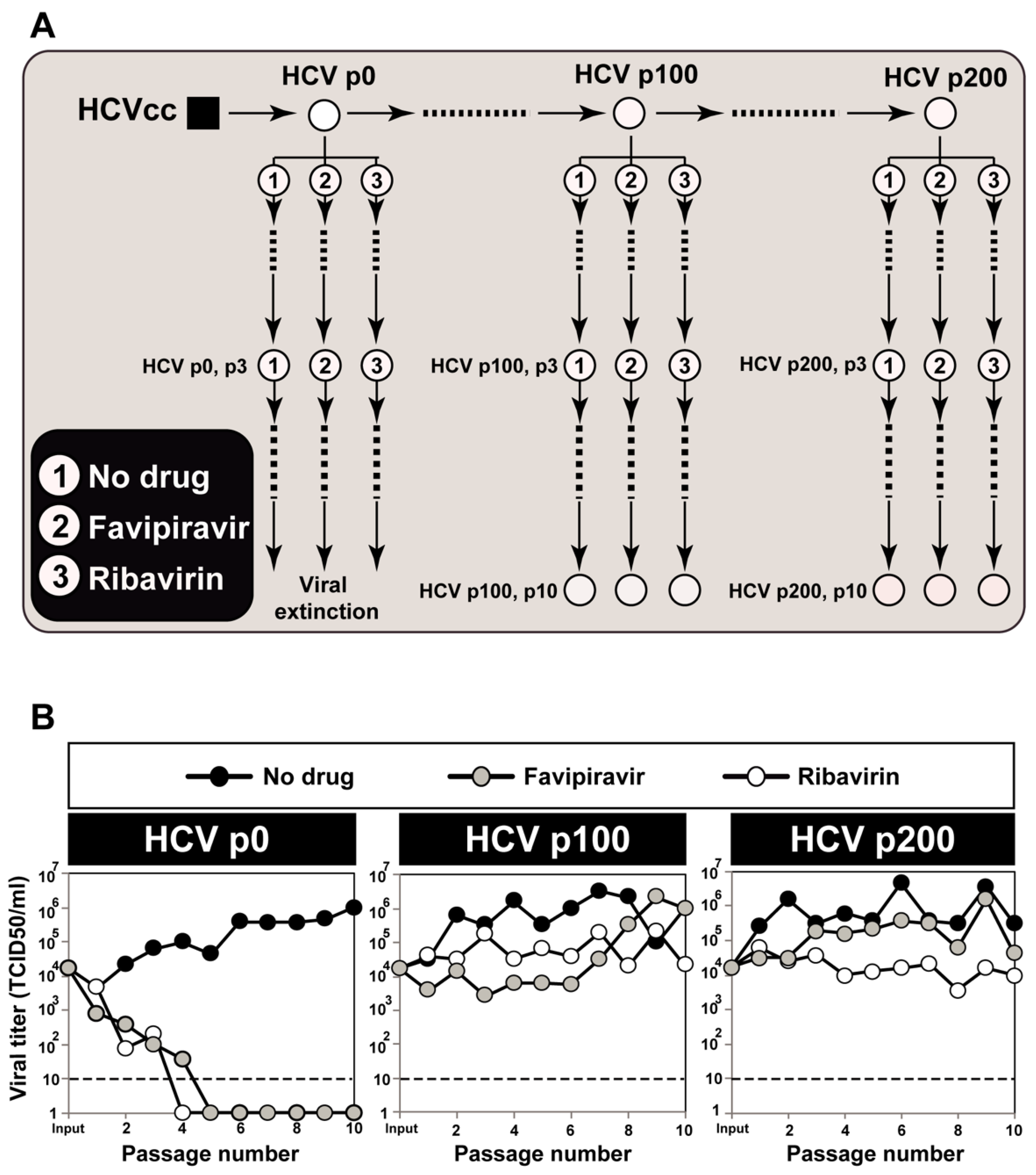
2.1. Origin of the Hepatitis C Virus Populations, and Infection of Huh-7.5 Cells
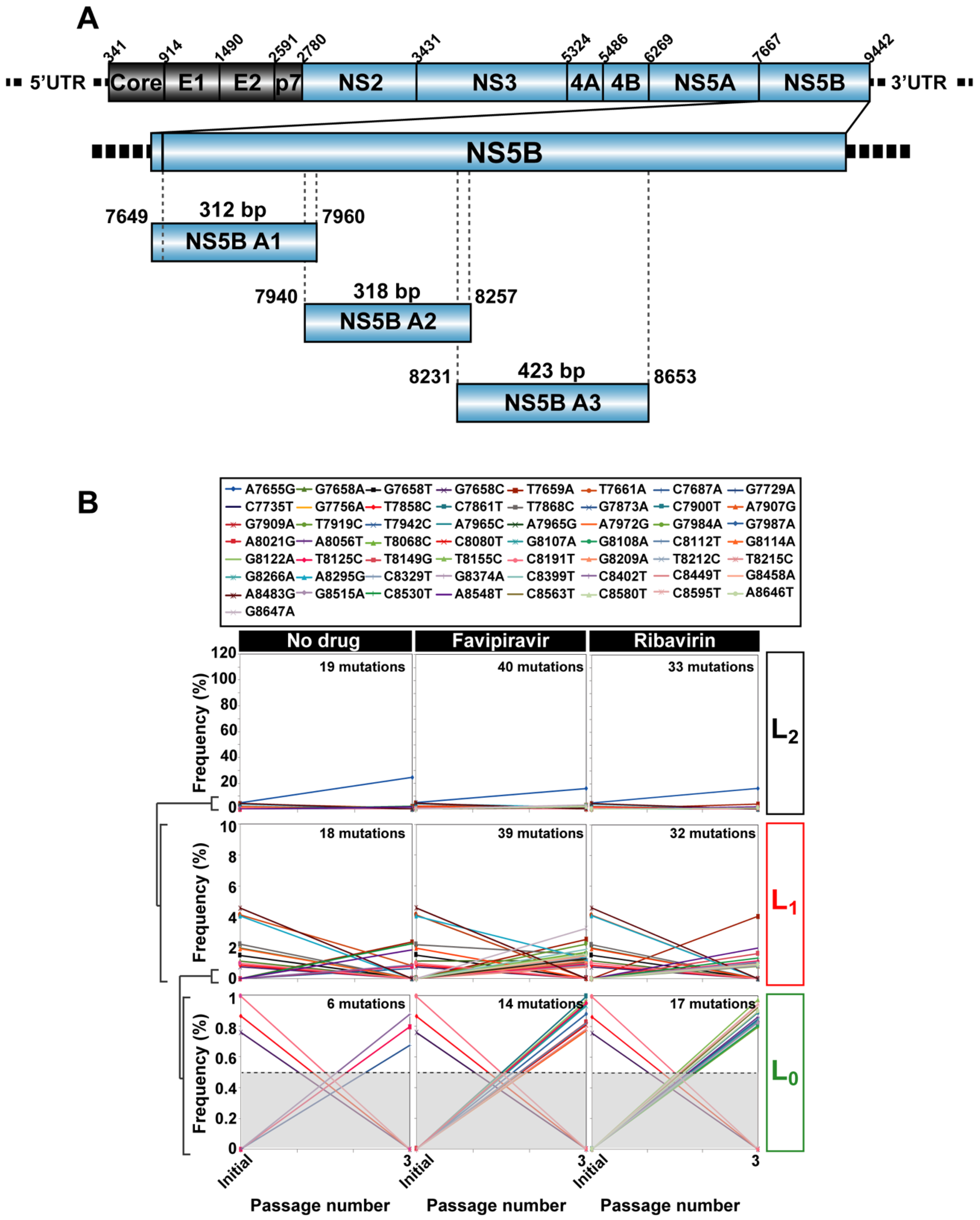
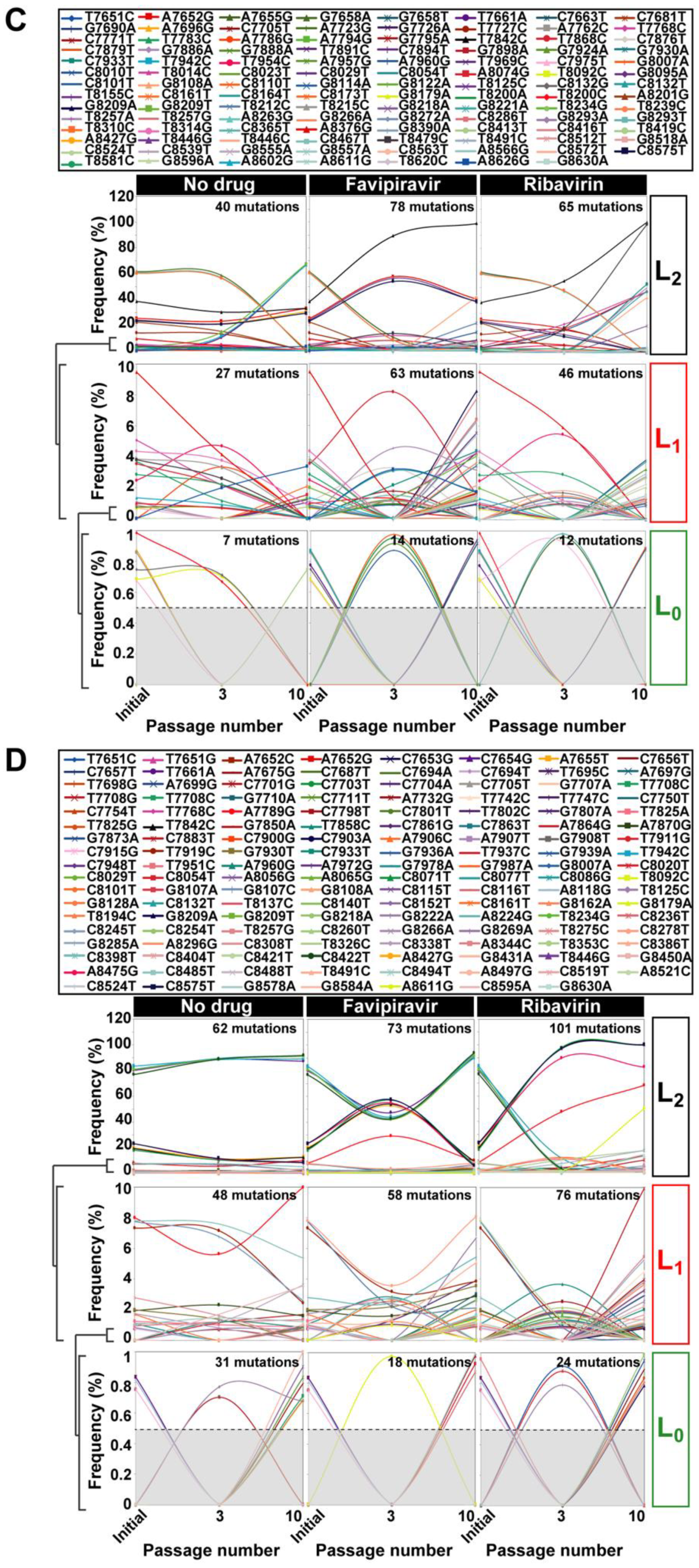
2.2. RNA Extraction, Viral RNA Amplification, and Ultra-Deep Sequencing of Cell Culture Populations
2.3. RNA Extraction, Viral RNA Amplification, and Ultra-Deep Sequencing of HCV from Infected Patients
2.4. Controls in the Ultra-Deep Sequencing Analyses
2.5. Amino Acid Conservation in Infected Patients versus Los Alamos Database (LANL)
2.6. Statistics
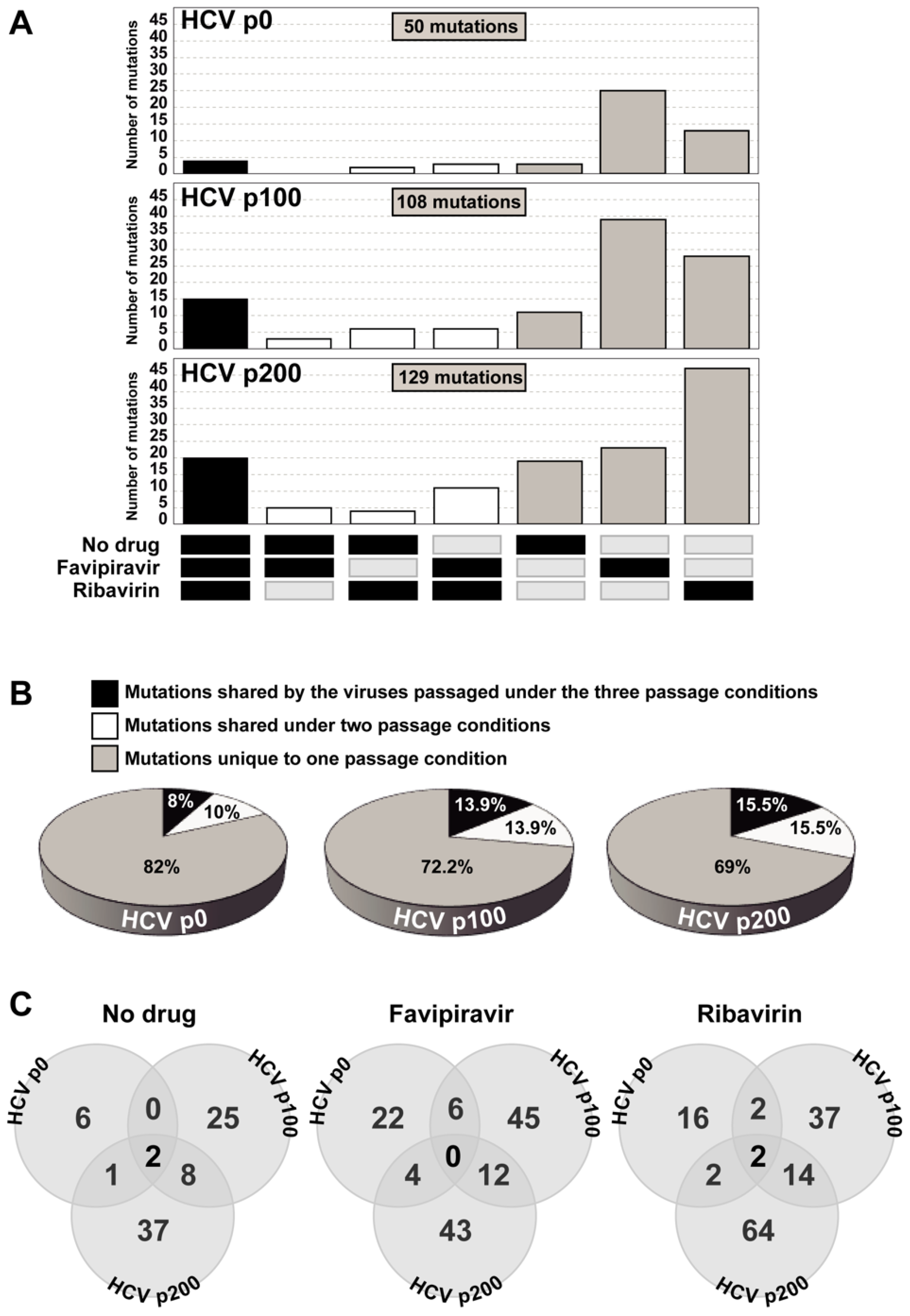
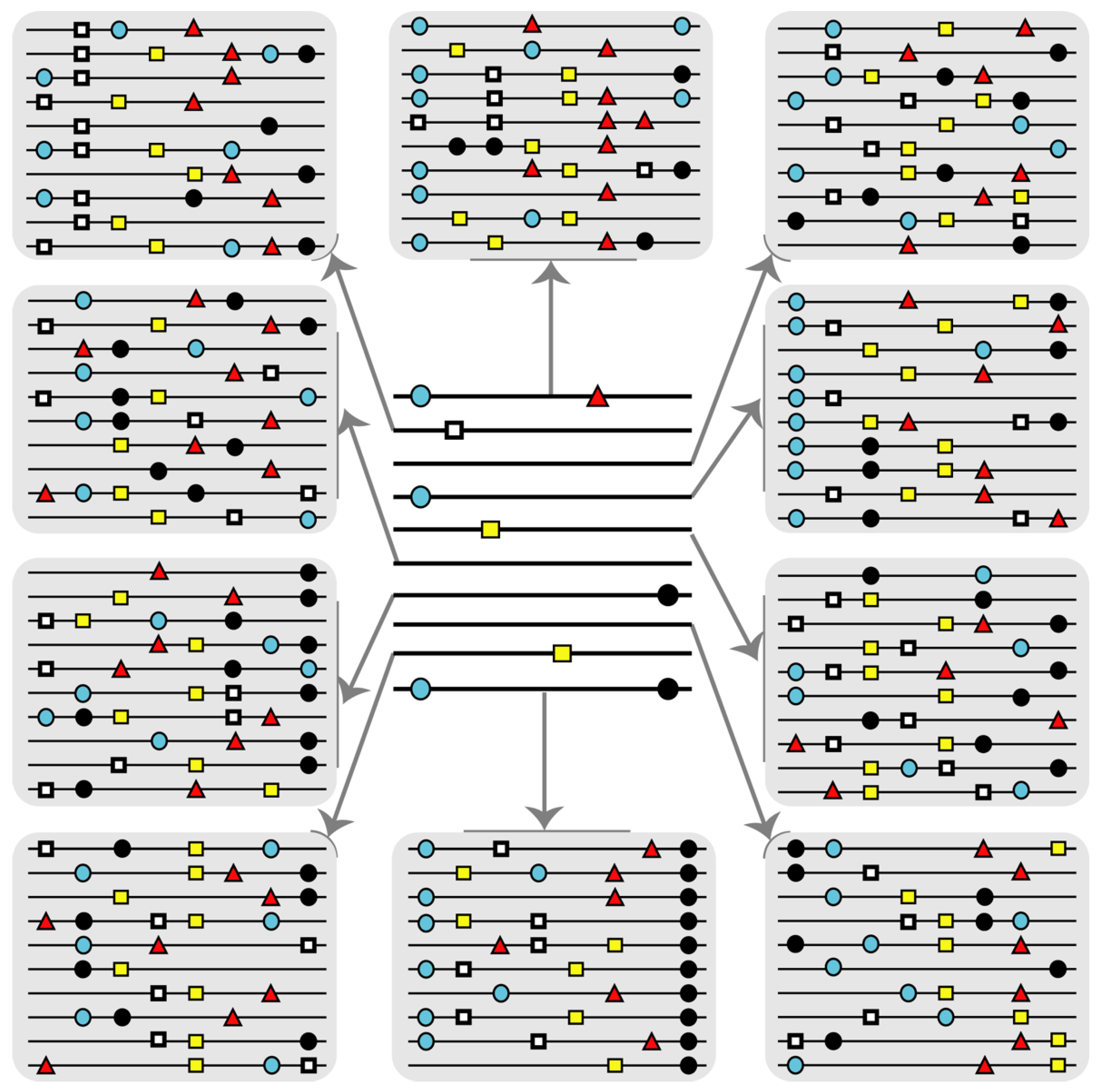
3. HCV Mutational Waves in the Presence of Mutagenic Nucleotide Analogues
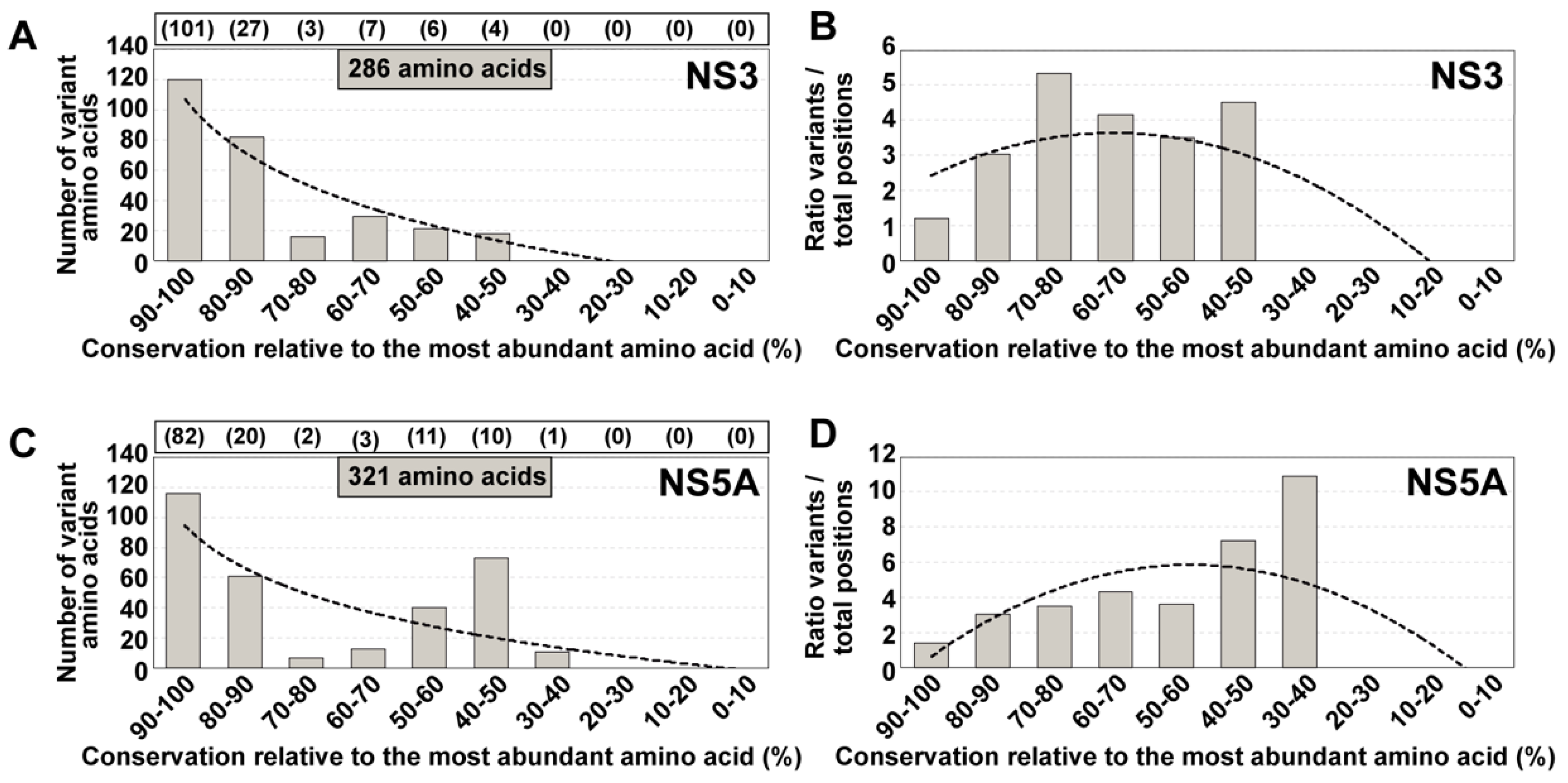
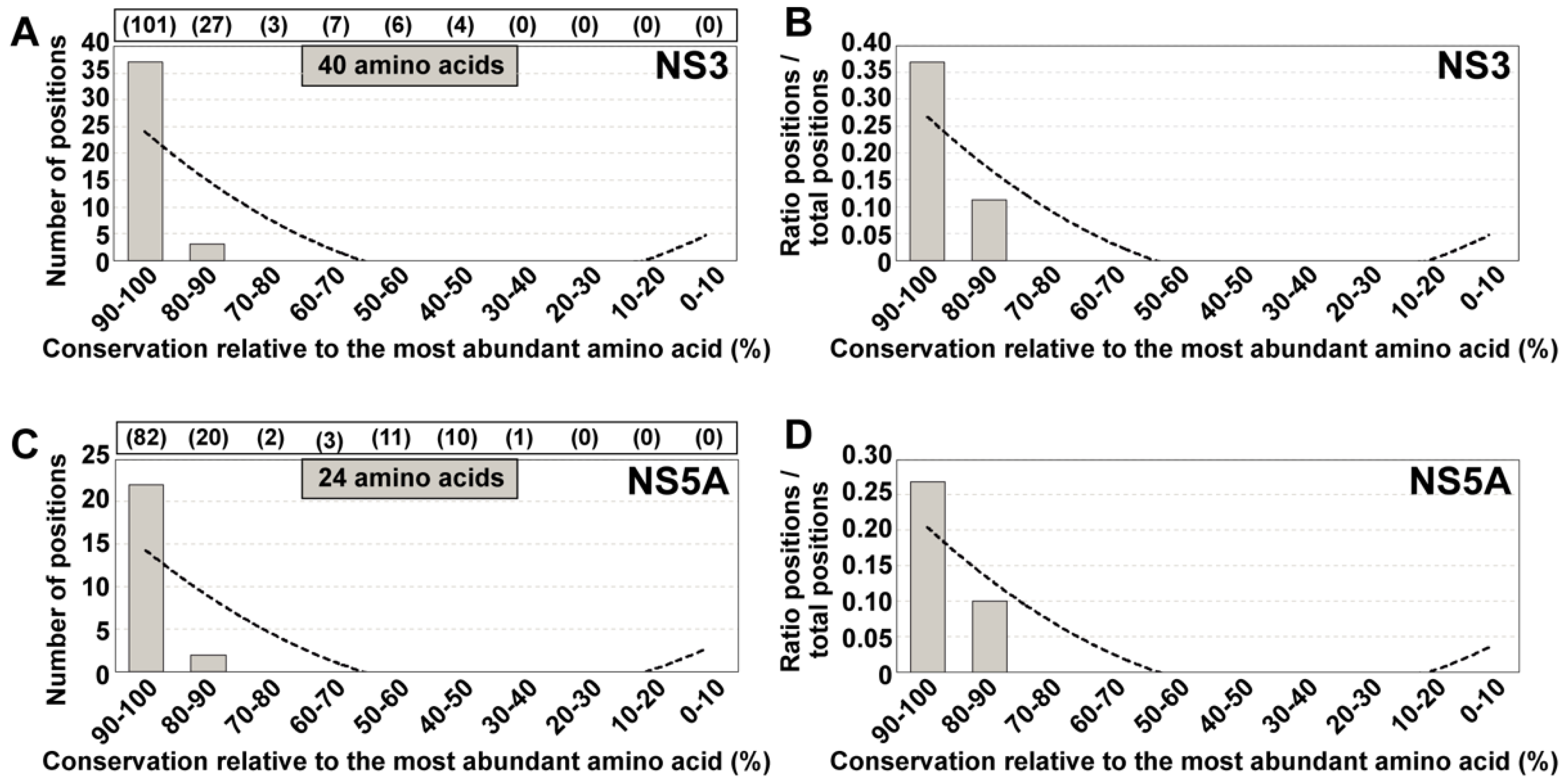
4. Amino Acid Conservation in HCV Quasispecies from Infected Patients
5. Antiviral Resistance Mechanisms during Hepatitis C Virus Infections
6. A Unifying Disequilibrium Model for Expanded Adaptive Potential
7. Concluding Remarks: Remembering John Holland in COVID-19 Times
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Eigen, M.; Schuster, P. The hypercycle. In A Principle of Natural Self-Organization; Springer: Berlin, Germany, 1979. [Google Scholar]
- Eigen, M.; Schuster, P. Stages of emerging life—Five principles of early organization. J. Mol. Evol. 1982, 19, 47–61. [Google Scholar] [CrossRef]
- Domingo, E.; Sabo, D.; Taniguchi, T.; Weissmann, C. Nucleotide sequence heterogeneity of an RNA phage population. Cell 1978, 13, 735–744. [Google Scholar] [CrossRef]
- Domingo, E.; Perales, C. Viral quasispecies. PLoS Genet. 2019, 15, e1008271. [Google Scholar] [CrossRef]
- Acevedo, A.; Brodsky, L.; Andino, R. Mutational and fitness landscapes of an RNA virus revealed through population se-quencing. Nature 2014, 505, 686–690. [Google Scholar] [CrossRef]
- Seifert, D.; Beerenwinkel, N. Estimating Fitness of Viral Quasispecies from Next-Generation Sequencing Data. Curr. Top. Microbiol. Immunol. 2015, 392, 181–200. [Google Scholar] [CrossRef]
- Braun, T.; Bordería, A.V.; Barbezange, C.; Vignuzzi, M.; Louzoun, Y. Long-term context-dependent genetic adaptation of the viral genetic cloud. Bioinformatics 2019, 35, 1907–1915. [Google Scholar] [CrossRef]
- Huang, S.-W.; Hung, S.-J.; Wang, J.-R. Application of deep sequencing methods for inferring viral population diversity. J. Virol. Methods 2019, 266, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Gregori, J.; Esteban, J.I.; Cubero, M.; Garcia-Cehic, D.; Perales, C.; Casillas, R.; Alvarez-Tejado, M.; Rodríguez-Frías, F.; Guardia, J.; Domingo, E.; et al. Ultra-Deep Pyrosequencing (UDPS) Data Treatment to Study Amplicon HCV Minor Variants. PLoS ONE 2013, 8, e83361. [Google Scholar] [CrossRef]
- Gregori, J.; Perales, C.; Rodriguez-Frias, F.; Esteban, J.I.; Quer, J.; Domingo, E. Viral quasispecies complexity measures. Virology 2016, 493, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Soria, M.E.; Gregori, J.; Chen, Q.; García-Cehic, D.; Llorens, M.; De Ávila, A.I.; Beach, N.M.; Domingo, E.; Rodríguez-Frías, F.; Buti, M.; et al. Pipeline for specific subtype amplification and drug resistance detection in hepatitis C virus. BMC Infect. Dis. 2018, 18, 446. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Gallego, I.; Gregori, J.; Lucía-Sanz, A.; Soria, M.E.; Castro, V.; Beach, N.M.; Manrubia, S.; Quer, J.; Esteban, J.I.; et al. Internal Disequilibria and Phenotypic Diversification during Replication of Hepatitis C Virus in a Noncoevolving Cellular Environment. J. Virol. 2017, 91, e02505. [Google Scholar] [CrossRef]
- Chen, Q.; Perales, C.; Soria, M.E.; García-Cehic, D.; Gregori, J.; Rodríguez-Frías, F.; Buti, M.; Crespo, J.; Calleja, J.L.; Tabernero, D.; et al. Deep-sequencing reveals broad subtype-specific HCV resistance mutations associated with treatment failure. Antivir. Res. 2020, 174, 104694. [Google Scholar] [CrossRef] [PubMed]
- Gallego, I.; Soria, M.E.; García-Crespo, C.; Chen, Q.; Martínez-Barragán, P.; Khalfaoui, S.; Martínez-González, B.; Sanchez-Martin, I.; Palacios-Blanco, I.; De Ávila, A.I.; et al. Broad and Dynamic Diversification of Infectious Hepatitis C Virus in a Cell Culture Environment. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Soria, M.E.; García-Crespo, C.; Martínez-González, B.; Vázquez-Sirvent, L.; Lobo-Vega, R.; De Ávila, A.I.; Gallego, I.; Chen, Q.; García-Cehic, D.; Llorens-Revull, M.; et al. Amino Acid Substitutions Associated with Treatment Failure for Hepatitis C Virus Infection. J. Clin. Microbiol. 2020, 58, 1985. [Google Scholar] [CrossRef] [PubMed]
- García-Crespo, C.; Soria, M.E.; Gallego, I.; De Ávila, A.I.; Martínez-González, B.; Vázquez-Sirvent, L.; Gómez, J.; Briones, C.; Gregori, J.; Quer, J.; et al. Dissimilar Conservation Pattern in Hepatitis C Virus Mutant Spectra, Consensus Sequences and Data Banks. J. Clin. Med. 2020, 9, 3450. [Google Scholar] [CrossRef]
- Perales, C.; Beach, N.M.; Gallego, I.; Soria, M.E.; Quer, J.; Esteban, J.I.; Rice, C.; Domingo, E.; Sheldon, J. Response of Hepatitis C Virus to Long-Term Passage in the Presence of Alpha Interferon: Multiple Mutations and a Common Phenotype. J. Virol. 2013, 87, 7593–7607. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, J.; Beach, N.M.; Moreno, E.; Gallego, I.; Piñeiro, D.; Martínez-Salas, E.; Gregori, J.; Quer, J.; Esteban, J.I.; Rice, C.M.; et al. Increased Replicative Fitness Can Lead to Decreased Drug Sensitivity of Hepatitis C Virus. J. Virol. 2014, 88, 12098–12111. [Google Scholar] [CrossRef] [PubMed]
- Gallego, I.; Gregori, J.; Soria, M.E.; García-Crespo, C.; García-Álvarez, M.; Gómez-González, A.; Valiergue, R.; Gómez, J.; Esteban, J.I.; Quer, J.; et al. Resistance of high fitness hepatitis C virus to lethal mutagenesis. Virology 2018, 523, 100–109. [Google Scholar] [CrossRef]
- Heiny, A.T.; Miotto, O.; Srinivasan, K.N.; Khan, A.M.; Zhang, G.L.; Brusic, V.; Tan, T.W.; August, J.T. Evolutionarily Conserved Protein Sequences of Influenza A Viruses, Avian and Human, as Vaccine Targets. PLoS ONE 2007, 2, e1190. [Google Scholar] [CrossRef]
- Wijesundara, D.K.; Gummow, J.; Li, Y.; Yu, W.; Quah, B.J.; Ranasinghe, C.; Torresi, J.; Gowans, E.J.; Grubor-Bauk, B. Induction of Genotype Cross-Reactive, Hepatitis C Virus-Specific, Cell-Mediated Immunity in DNA-Vaccinated Mice. J. Virol. 2018, 92, e02133. [Google Scholar] [CrossRef]
- Hart, G.R.; Ferguson, A.L. Computational design of hepatitis C virus immunogens from host-pathogen dynamics over empirical viral fitness landscapes. Phys. Biol. 2018, 16, 016004. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.M. Sofosbuvir–velpatasvir: A single-tablet treatment for hepatitis C infection of all genotypes. Am. J. Health Pharm. 2017, 74, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- McLean, G.R. Vaccine strategies to induce broadly protective immunity to rhinoviruses. Hum. Vaccines Immunother. 2020, 16, 684–686. [Google Scholar] [CrossRef] [PubMed]
- Vogel, O.A.; Manicassamy, B. Broadly Protective Strategies Against Influenza Viruses: Universal Vaccines and Therapeutics. Front. Microbiol. 2020, 11, 135. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Deng, L.; Kang, S.-M.; Wang, B.-Z. Universal influenza vaccines: From viruses to nanoparticles. Expert Rev. Vaccines 2018, 17, 967–976. [Google Scholar] [CrossRef]
- Sautto, G.A.; Kirchenbaum, G.A.; Ross, T.M. Towards a universal influenza vaccine: Different approaches for one goal. Virol. J. 2018, 15, 1–12. [Google Scholar] [CrossRef]
- Qiu, X.; Duvvuri, V.R.; Bahl, J. Computational Approaches and Challenges to Developing Universal Influenza Vaccines. Vaccines 2019, 7, 45. [Google Scholar] [CrossRef]
- Robson, B. COVID-19 Coronavirus spike protein analysis for synthetic vaccines, a peptidomimetic antagonist, and therapeutic drugs, and analysis of a proposed achille’s heel conserved region to minimize probability of escape mutations and drug re-sistance. Comput. Biol. Med. 2020, 121, 103749. [Google Scholar] [CrossRef] [PubMed]
- Marukian, S.; Jones, C.T.; Andrus, L.; Evans, M.J.; Ritola, K.D.; Charles, E.D.; Rice, C.M.; Dustin, L.B. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology 2008, 48, 1843–1850. [Google Scholar] [CrossRef]
- Reed, L.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Ortega-Prieto, A.M.; Sheldon, J.; Grande-Perez, A.; Tejero, H.; Gregori, J.; Quer, J.; Esteban, J.I.; Domingo, E.; Perales, C. Ex-tinction of hepatitis C virus by ribavirin in hepatoma cells involves lethal mutagenesis. PLoS ONE 2013, 8, e71039. [Google Scholar] [CrossRef] [PubMed]
- De Ávila, A.I.; Gallego, I.; Soria, M.E.; Gregori, J.; Quer, J.; Esteban, J.I.; Rice, C.M.; Domingo, E.; Perales, C. Lethal Mutagenesis of Hepatitis C Virus Induced by Favipiravir. PLoS ONE 2016, 11, e0164691. [Google Scholar] [CrossRef] [PubMed]
- Gallego, I.; Soria, M.E.; Gregori, J.; De Ávila, A.I.; García-Crespo, C.; Moreno, E.; Gadea, I.; Esteban, J.; Fernández-Roblas, R.; Esteban, J.I.; et al. Synergistic Lethal Mutagenesis of Hepatitis C Virus. Antimicrob. Agents Chemother. 2019, 63, e01653-19. [Google Scholar] [CrossRef] [PubMed]
- Gregori, J.; Salicrú, M.; Domingo, E.; Sanchez, A.; Esteban, J.I.; Rodríguez-Frías, F.; Quer, J. Inference with viral quasispecies diversity indices: Clonal and NGS approaches. Bioinformatics 2014, 30, 1104–1111. [Google Scholar] [CrossRef]
- Asahina, Y.; Izumi, N.; Enomoto, N.; Uchihara, M.; Kurosaki, M.; Onuki, Y.; Nishimura, Y.; Ueda, K.; Tsuchiya, K.; Nakanishi, H.; et al. Mutagenic effects of ribavirin and response to interferon/ribavirin combination therapy in chronic hepatitis C. J. Hepatol. 2005, 43, 623–629. [Google Scholar] [CrossRef]
- Dietz, J.; Schelhorn, S.-E.; Fitting, D.; Mihm, U.; Susser, S.; Welker, M.-W.; Füller, C.; Däumer, M.; Teuber, G.; Wedemeyer, H.; et al. Deep Sequencing Reveals Mutagenic Effects of Ribavirin during Monotherapy of Hepatitis C Virus Genotype 1-Infected Patients. J. Virol. 2013, 87, 6172–6181. [Google Scholar] [CrossRef]
- Perales, C.; Gallego, I.; De Ávila, A.I.; Soria, M.E.; Gregori, J.; Quer, J.; Domingo, E. The increasing impact of lethal mutagenesis of viruses. Future Med. Chem. 2019, 11, 1645–1657. [Google Scholar] [CrossRef]
- Baranovich, T.; Wong, S.-S.; Armstrong, J.; Marjuki, H.; Webby, R.J.; Webster, R.G.; Govorkova, E.A. T-705 (Favipiravir) Induces Lethal Mutagenesis in Influenza A H1N1 Viruses In Vitro. J. Virol. 2013, 87, 3741–3751. [Google Scholar] [CrossRef]
- Arias, A.; Thorne, L.; Goodfellow, I. Favipiravir elicits antiviral mutagenesis during virus replication in vivo. Elife 2014, 3, e03679. [Google Scholar] [CrossRef]
- Shannon, A.; Selisko, B.; Le, N.T.; Huchting, J.; Touret, F.; Piorkowski, G.; Fattorini, V.; Ferron, F.; Decroly, E.; Meier, C.; et al. Rapid incorporation of Favipiravir by the fast and permissive viral RNA polymerase complex results in SARS-CoV-2 lethal mutagenesis. Nat. Commun. 2020, 11, 4682. [Google Scholar] [CrossRef]
- Domingo, E.; Soria, M.E.; Gallego, I.; De Ávila, A.I.; García-Crespo, C.; Martínez-González, B.; Gómez, J.; Briones, C.; Gregori, J.; Quer, J.; et al. A new implication of quasispecies dynamics: Broad virus diversification in absence of external perturbations. Infect. Genet. Evol. 2020, 82, 104278. [Google Scholar] [CrossRef]
- Gallego, I.; Sheldon, J.; Moreno, E.; Gregori, J.; Quer, J.; Esteban, J.I.; Rice, C.M.; Domingo, E.; Perales, C. Barrier-Independent, Fitness-Associated Differences in Sofosbuvir Efficacy against Hepatitis C Virus. Antimicrob. Agents Chemother. 2016, 60, 3786–3793. [Google Scholar] [CrossRef]
- Domingo, E. Quasispecies: Concepts and Implications for Virology. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2006; Volume 299, Available online: https://www.springer.com/gp/book/9783540263951 (accessed on 4 March 2021).
- Domingo, E.; Sheldon, J.; Perales, C. Viral Quasispecies Evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Perales, C. The time for COVID-19 vaccination. J. Virol. 2021. [Google Scholar] [CrossRef]
- Ong, A.T.; Tay, E.E.; Dwyer, D.; George, J.; Douglas, M.W. Pre-treatment antiviral resistance in Australians with chronic hepatitis C: Prevalence of NS3 and NS5A resistance data in the state of New South Wales. Antivir. Ther. 2019, 24, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.P.; García, G.; Ridruejo, E.; Culasso, A.C.; Pérez, P.S.; Pereson, M.J.; Neukam, K.; Flichman, D.; Di Lello, F.A. Hepatitis C virus genotype 1 infection: Prevalence of NS5A and NS5B resistance-associated substitutions in naïve patients from Argentina. J. Med. Virol. 2019, 91, 1970–1978. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.A.; Franco, S. Therapy Implications of Hepatitis C Virus Genetic Diversity. Viruses 2020, 13, 41. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver; Clinical Practice Guidelines Panel: Chair; EASL Governing Board Representative. EASL recommendations on treatment of hepatitis C: Final update of the series. J. Hepatol. 2020, 73, 1170–1218. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E. Virus as Populations, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Di Maio, V.C.; Cento, V.; Lenci, I.; Aragri, M.; Rossi, P.; Barbaliscia, S.; Melis, M.; Verucchi, G.; Magni, C.F.; Teti, E.; et al. Multiclass HCV resistance to direct-acting antiviral failure in real-life patients advocates for tailored second-line therapies. Liver Int. 2017, 37, 514–528. [Google Scholar] [CrossRef]
- Dietz, J.; Susser, S.; Vermehren, J.; Peiffer, K.H.; Grammatikos, G.; Berger, A.; Ferenci, P.; Buti, M.; Mullhaupt, B.; Hunyady, B.; et al. Patterns of Resistance-Associated Substitutions in Patients with Chronic HCV Infection Following Treatment with Direct-Acting Antivirals. Gastroenterology 2018, 154, 976–988. [Google Scholar] [CrossRef]
- Gozlan, Y.; Bucris, E.; Shirazi, R.; Rakovsky, A.; Ben-Ari, Z.; Davidov, Y.; Veizman, E.; Saadi, T.; Braun, M.; Cohen-Naftaly, M.; et al. High frequency of multiclass HCV resistance-associated mutations in patients failing direct-acting antivirals: Real-life data. Antivir. Ther. 2019, 24, 221–228. [Google Scholar] [CrossRef]
- Perales, C. Quasispecies dynamics and clinical significance of hepatitis C virus (HCV) antiviral resistance. Int. J. Antimicrob. Agents 2020, 56, 105562. [Google Scholar] [CrossRef]
- Uchida, Y.; Nakamura, S.; Kouyama, J.-I.; Naiki, K.; Motoya, D.; Sugawara, K.; Inao, M.; Imai, Y.; Nakayama, N.; Tomiya, T.; et al. Significance of NS5B Substitutions in Genotype 1b Hepatitis C Virus Evaluated by Bioinformatics Analysis. Sci. Rep. 2018, 8, 8818. [Google Scholar] [CrossRef] [PubMed]
- Bellocchi, M.C.; Aragri, M.; Carioti, L.; Fabeni, L.; Pipitone, R.M.; Brancaccio, G.; Sorbo, M.C.; Barbaliscia, S.; Di Maio, V.C.; Bronte, F.; et al. NS5A Gene Analysis by Next Generation Sequencing in HCV Nosocomial Transmission Clusters of HCV Genotype 1b Infected Patients. Cells 2019, 8, 666. [Google Scholar] [CrossRef]
- Nakamoto, S.; Kanda, T.; Wu, S.; Shirasawa, H.; Yokosuka, O. Hepatitis C virus NS5A inhibitors and drug resistance mutations. World J. Gastroenterol. 2014, 20, 2902–2912. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-W.; Lee, S.-A.; Kim, H.; Won, Y.-S.; Kim, B.-J. Naturally Occurring Mutations in the Nonstructural Region 5B of Hepatitis C Virus (HCV) from Treatment-Naïve Korean Patients Chronically Infected with HCV Genotype 1b. PLoS ONE 2014, 9, e87773. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zhang, E.Z.; Ardzinski, A.; Tigges, A.; Davis, A.; Sullivan, J.C.; Nelson, M.; Spanks, J.; Dorrian, J.; Nicolas, O.; et al. Genotypic and Phenotypic Analyses of Hepatitis C Virus Variants Observed in Clinical Studies of VX-222, a Nonnucleoside NS5B Polymerase Inhibitor. Antimicrob. Agents Chemother. 2014, 58, 5456–5465. [Google Scholar] [CrossRef] [PubMed]
- Sierra, S.; Dávila, M.; Lowenstein, P.R.; Domingo, E. Response of Foot-and-Mouth Disease Virus to Increased Mutagenesis: Influence of Viral Load and Fitness in Loss of Infectivity. J. Virol. 2000, 74, 8316–8323. [Google Scholar] [CrossRef]
- Pariente, N.; Sierra, S.; Lowenstein, P.R.; Domingo, E. Efficient virus extinction by combinations of a mutagen and antiviral inhibitors. J. Virol. 2001, 75, 9723–9730. [Google Scholar] [CrossRef] [PubMed]
- Tapia, N.; Fernández, G.; Parera, M.; Gómez-Mariano, G.; Clotet, B.; Quinones-Mateu, M.; Domingo, E.; Martínez, M.A. Combination of a mutagenic agent with a reverse transcriptase inhibitor results in systematic inhibition of HIV-1 infection. Virology 2005, 338, 1–8. [Google Scholar] [CrossRef]
- Wang, C.; Sun, J.H.; O’Boyle, D.R., 2nd; Nower, P.; Valera, L.; Roberts, S.; Fridell, R.A.; Gao, M. Persistence of resistant variants in hepatitis C virus-infected patients treated with the NS5A replication complex inhibitor daclatasvir. Antimicrob. Agents Chemother. 2013, 57, 2054–2065. [Google Scholar] [CrossRef]
- Küppers, B.-O. The Nucleation of Semantic Information in Prebiotic Matter. Curr. Top. Microbiol. Immunol. 2015, 392, 23–42. [Google Scholar] [CrossRef]
- Domingo, E. RNA virus evolution and the control of viral disease. Prog. Drug Res. 1989, 33, 93–133. [Google Scholar] [PubMed]
- Perales, C.; Iranzo, J.; Manrubia, S.C.; Domingo, E. The impact of quasispecies dynamics on the use of therapeutics. Trends Microbiol. 2012, 20, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Trypsteen, W.; Van Cleemput, J.; Snippenberg, W.V.; Gerlo, S.; Vandekerckhove, L. On the whereabouts of SARS-CoV-2 in the human body: A systematic review. PLoS Pathog. 2020, 16, e1009037. [Google Scholar] [CrossRef] [PubMed]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Holland, J.J.; De La Torre, J.C.; Steinhauer, D.A. RNA Virus Populations as Quasispecies. Curr. Top. Microbiol. Immunol. 1992, 176, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, C.E. Explaining Epidemics and Other Studies in the History of Medicine; Cambridge University Press: Cambridge, UK, 1992. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
|
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Crespo, C.; Gallego, I.; Soria, M.E.; de Ávila, A.I.; Martínez-González, B.; Vázquez-Sirvent, L.; Lobo-Vega, R.; Moreno, E.; Gómez, J.; Briones, C.; et al. Population Disequilibrium as Promoter of Adaptive Explorations in Hepatitis C Virus. Viruses 2021, 13, 616. https://doi.org/10.3390/v13040616
García-Crespo C, Gallego I, Soria ME, de Ávila AI, Martínez-González B, Vázquez-Sirvent L, Lobo-Vega R, Moreno E, Gómez J, Briones C, et al. Population Disequilibrium as Promoter of Adaptive Explorations in Hepatitis C Virus. Viruses. 2021; 13(4):616. https://doi.org/10.3390/v13040616
Chicago/Turabian StyleGarcía-Crespo, Carlos, Isabel Gallego, María Eugenia Soria, Ana Isabel de Ávila, Brenda Martínez-González, Lucía Vázquez-Sirvent, Rebeca Lobo-Vega, Elena Moreno, Jordi Gómez, Carlos Briones, and et al. 2021. "Population Disequilibrium as Promoter of Adaptive Explorations in Hepatitis C Virus" Viruses 13, no. 4: 616. https://doi.org/10.3390/v13040616
APA StyleGarcía-Crespo, C., Gallego, I., Soria, M. E., de Ávila, A. I., Martínez-González, B., Vázquez-Sirvent, L., Lobo-Vega, R., Moreno, E., Gómez, J., Briones, C., Gregori, J., Quer, J., Domingo, E., & Perales, C. (2021). Population Disequilibrium as Promoter of Adaptive Explorations in Hepatitis C Virus. Viruses, 13(4), 616. https://doi.org/10.3390/v13040616