
Common Oral Medications Lead to Prophage Induction in Bacterial Isolates from the Human Gut


Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Isolates
2.2. Estimation of Medication Concentrations in the Human Gut
2.3. Preparation of Medication
2.4. In Vitro Treatments
2.5. VLP Enumeration
2.6. Prophage Induction of C. beijerinckii for DNA Sequencing and PCR
2.7. Purification of Viral DNA from VLPs
2.8. Extraction of Genomic DNA from Gut Bacterial Isolates
2.9. Shotgun Sequencing of Purified Viral DNA & Processing of Sequencing Data
3. Results
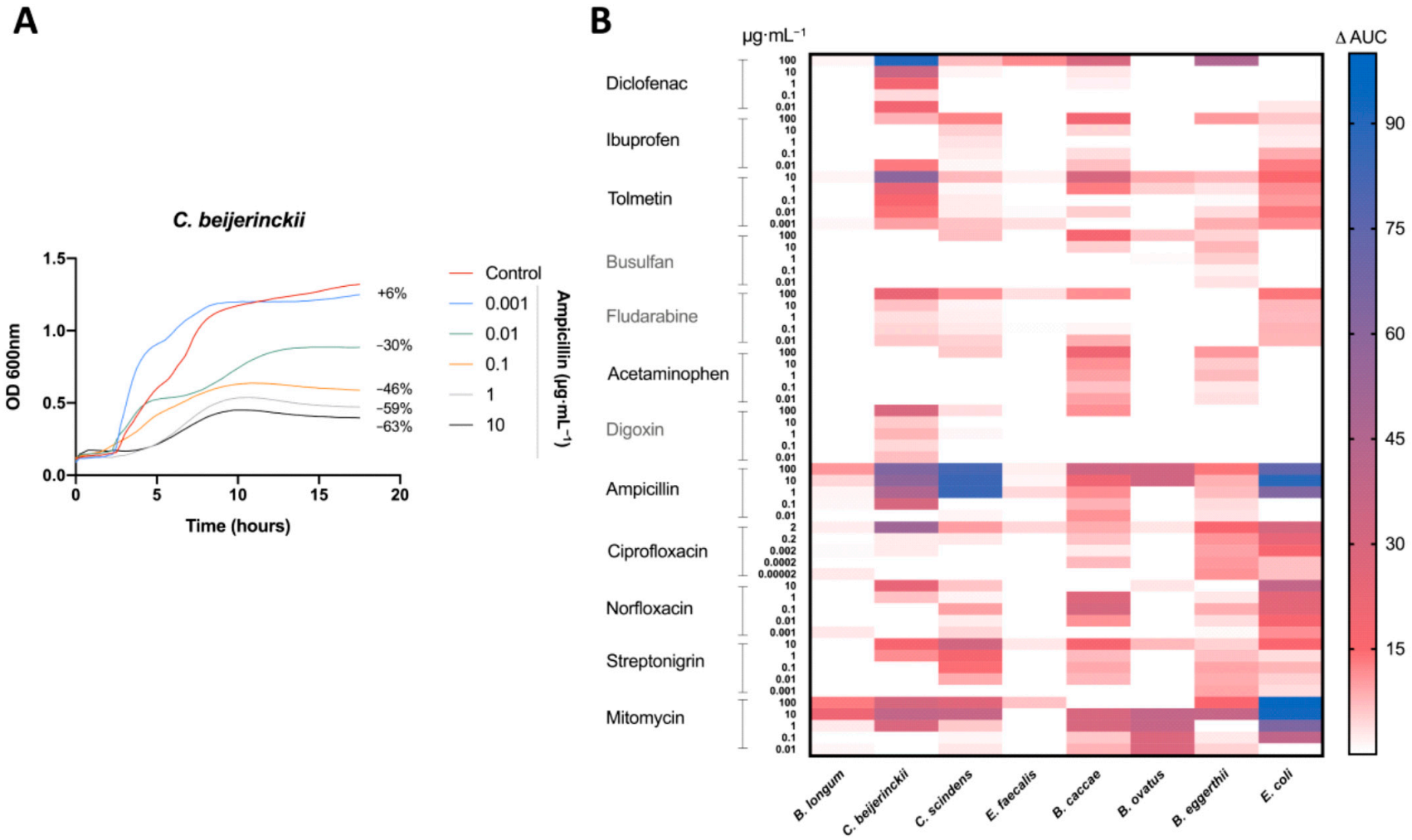
3.1. In Vitro Model to Study Prophage Induction of Human Gut Bacteria
3.2. Antibacterial Activity of Medications on Human Gut Isolates In Vitro
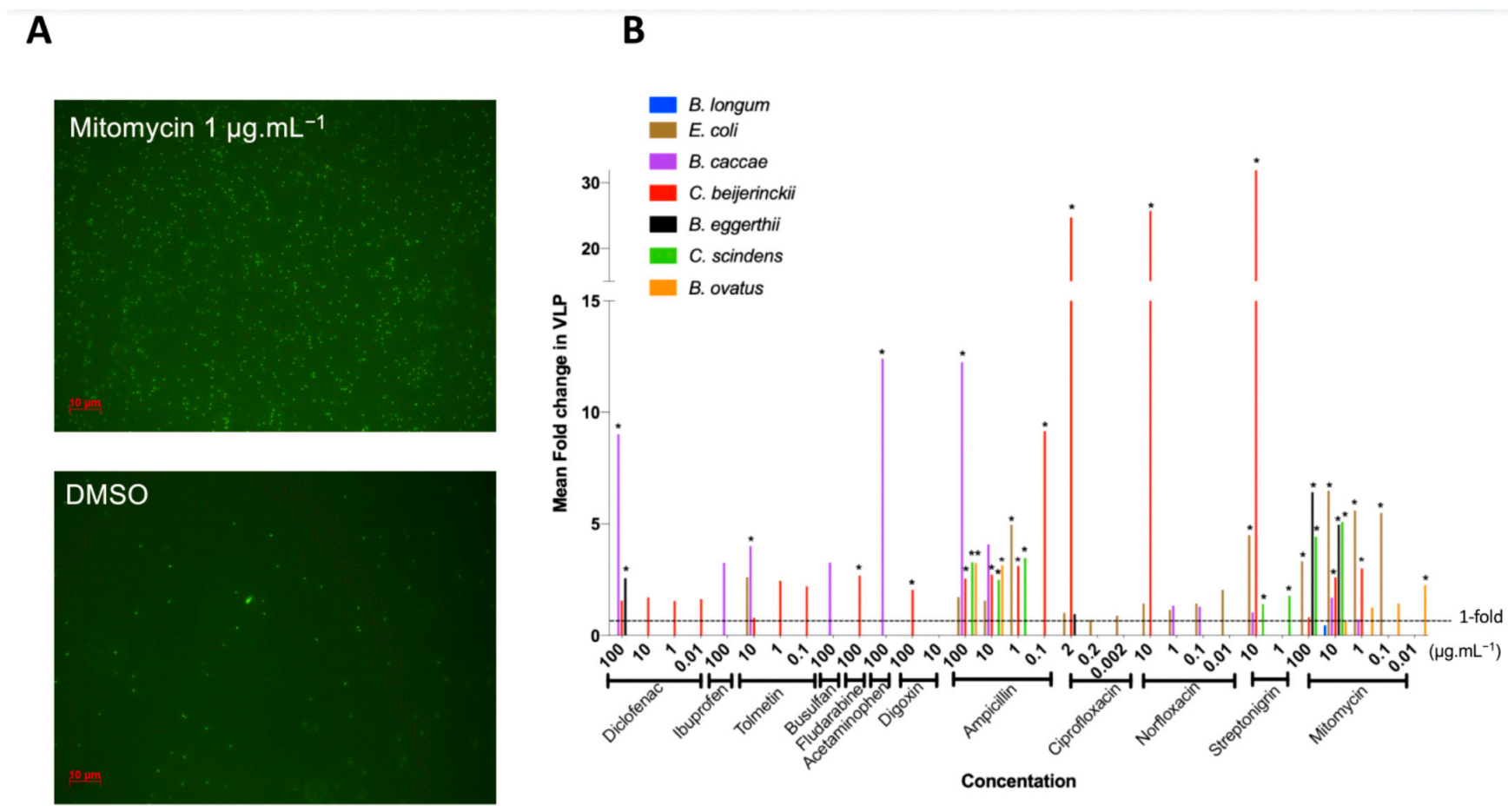
3.3. Medication Caused Prophage Induction of Human Gut Lysogens
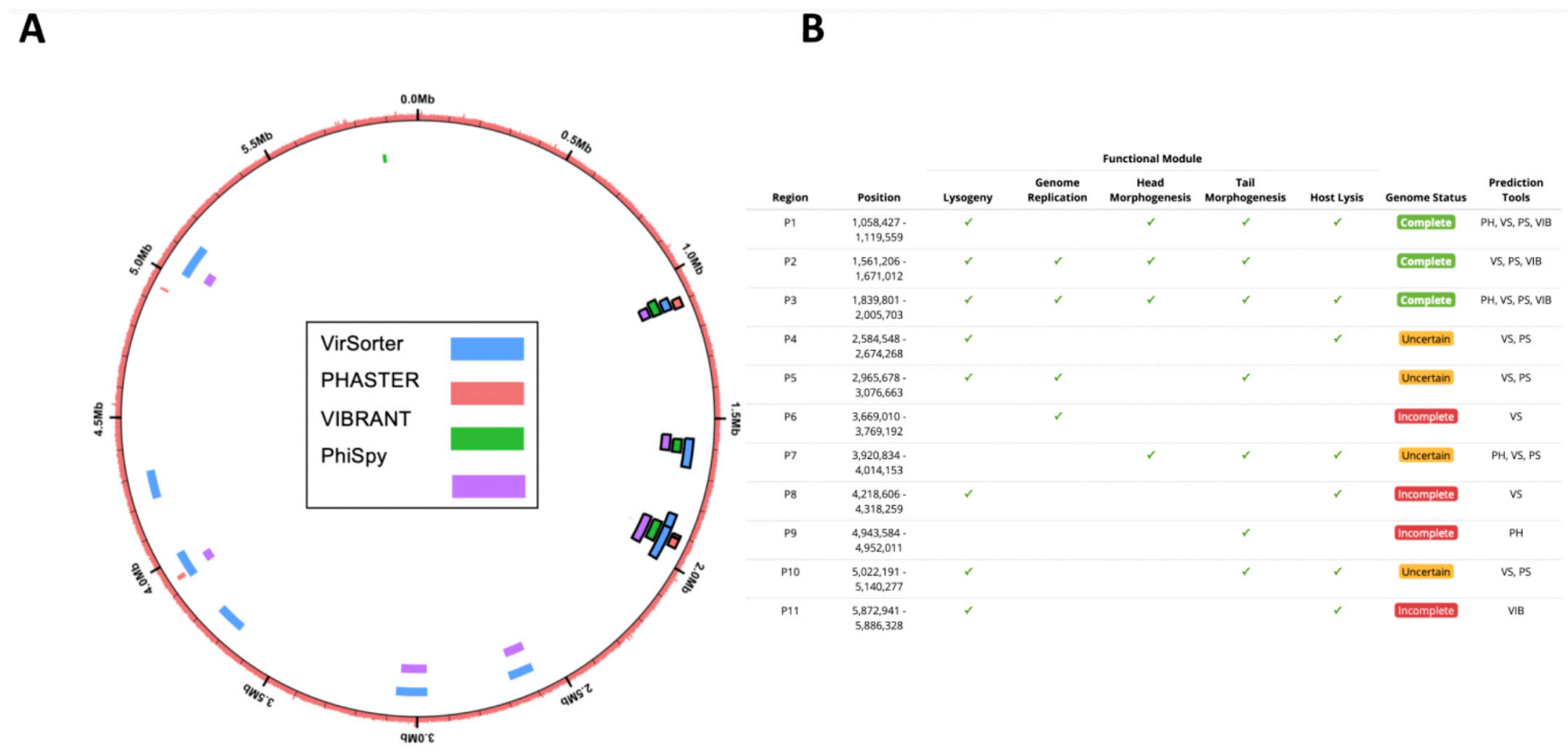
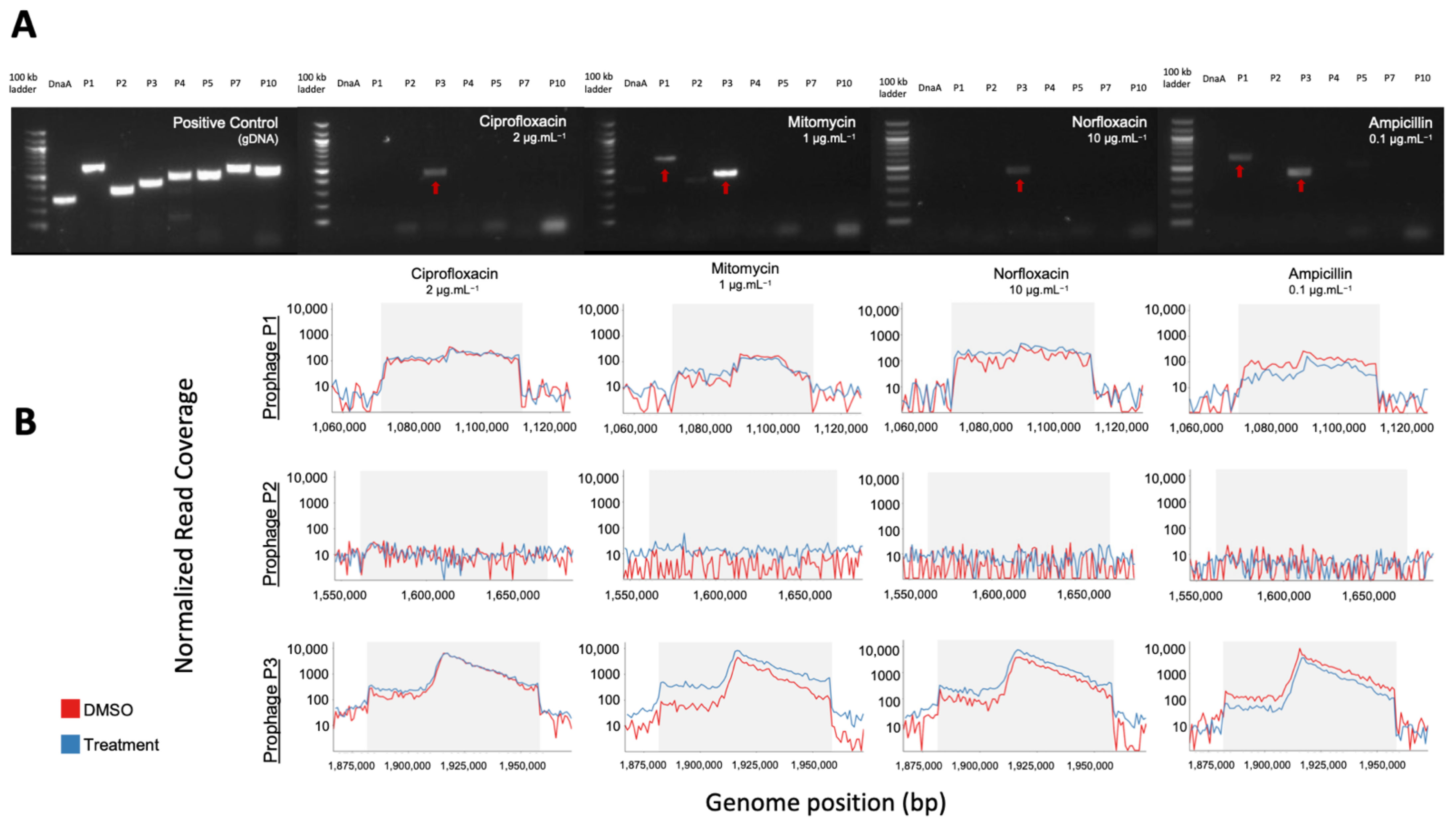
3.4. Confirmation of In Silico Predicted Prophages Induced in C. beijerinckii
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koppel, N.; Maini Rekdal, V.; Balskus, E.P. Chemical transformation of xenobiotics by the human gut microbiota. Science 2017, 356. [Google Scholar] [CrossRef]
- Kantor, E.D.; Rehm, C.D.; Haas, J.S.; Chan, A.T.; Giovannucci, E.L. Trends in Prescription Drug Use Among Adults in the United States From 1999–2012. J. Am. Med. Assoc. 2015, 314, 1818–1831. [Google Scholar] [CrossRef]
- World Health Organization. The World Medicines Situation Report; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Panda, S.; El khader, I.; Casellas, F.; Lopez Vivancos, J.; Garcia Cors, M.; Santiago, A.; Cuenca, S.; Guarner, F.; Manichanh, C. Short-term effect of antibiotics on human gut microbiota. PLoS ONE 2014, 9, e95476. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, L.; Huse, S.; Sogin, M.L.; Relman, D.A. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008, 6, e280. [Google Scholar] [CrossRef] [PubMed]
- Jernberg, C.; Lofmark, S.; Edlund, C.; Jansson, J.K. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J. 2007, 1, 56–66. [Google Scholar] [CrossRef]
- Jakobsson, H.E.; Jernberg, C.; Andersson, A.F.; Sjolund-Karlsson, M.; Jansson, J.K.; Engstrand, L. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS ONE 2010, 5, e9836. [Google Scholar] [CrossRef] [PubMed]
- Perez-Cobas, A.E.; Artacho, A.; Knecht, H.; Ferrus, M.L.; Friedrichs, A.; Ott, S.J.; Moya, A.; Latorre, A.; Gosalbes, M.J. Differential effects of antibiotic therapy on the structure and function of human gut microbiota. PLoS ONE 2013, 8, e80201. [Google Scholar] [CrossRef] [PubMed]
- Le Bastard, Q.; Al-Ghalith, G.A.; Gregoire, M.; Chapelet, G.; Javaudin, F.; Dailly, E.; Batard, E.; Knights, D.; Montassier, E. Systematic review: Human gut dysbiosis induced by non-antibiotic prescription medications. Aliment. Pharmacol. Ther. 2018, 47, 332–345. [Google Scholar] [CrossRef]
- Rogers, M.A.M.; Aronoff, D.M. The influence of non-steroidal anti-inflammatory drugs on the gut microbiome. Clin. Microbiol. Infect. 2016, 22, 178.e171–178.e179. [Google Scholar] [CrossRef] [PubMed]
- Montassier, E.; Gastinne, T.; Vangay, P.; Al-Ghalith, G.A.; Bruley des Varannes, S.; Massart, S.; Moreau, P.; Potel, G.; de La Cochetiere, M.F.; Batard, E.; et al. Chemotherapy-driven dysbiosis in the intestinal microbiome. Aliment. Pharmacol. Ther. 2015, 42, 515–528. [Google Scholar] [CrossRef]
- Maurice, C.F.; Haiser, H.J.; Turnbaugh, P.J. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 2013, 152, 39–50. [Google Scholar] [CrossRef]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 2018, 555, 623–628. [Google Scholar] [CrossRef]
- Zuo, T.; Sun, Y.; Wan, Y.; Yeoh, Y.K.; Zhang, F.; Cheung, C.P.; Chen, N.; Luo, J.; Wang, W.; Sung, J.J.Y.; et al. Human-Gut-DNA Virome Variations across Geography, Ethnicity, and Urbanization. Cell Host Microbe 2020. [Google Scholar] [CrossRef]
- Moreno-Gallego, J.L.; Chou, S.P.; Di Rienzi, S.C.; Goodrich, J.K.; Spector, T.D.; Bell, J.T.; Youngblut, N.D.; Hewson, I.; Reyes, A.; Ley, R.E. Virome Diversity Correlates with Intestinal Microbiome Diversity in Adult Monozygotic Twins. Cell Host Microbe 2019, 25, 261–272.e265. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, M.K.; Maurice, C.F. Ménage à trois in the human gut: Interactions between host, bacteria and phages. Nat. Rev. Microbiol. 2017, 15, 397. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Hewson, I.; Felts, B.; Mahaffy, J.M.; Nulton, J.; Salamon, P.; Rohwer, F. Metagenomic analyses of an uncultured viral community from human feces. J. Bacteriol. 2003, 185, 6220–6223. [Google Scholar] [CrossRef]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef]
- Lwoff, A. Lysogeny. Bacteriol. Rev. 1953, 17, 269–337. [Google Scholar] [CrossRef]
- Touchon, M.; Bernheim, A.; Rocha, E.P.C. Genetic and life-history traits associated with the distribution of prophages in bacteria. ISME J. 2016. [Google Scholar] [CrossRef] [PubMed]
- Chibani-Chennoufi, S.; Bruttin, A.; Dillmann, M.L.; Brussow, H. Phage-host interaction: An ecological perspective. J. Bacteriol. 2004, 186, 3677–3686. [Google Scholar] [CrossRef]
- Kim, M.S.; Bae, J.W. Lysogeny is prevalent and widely distributed in the murine gut microbiota. ISME J. 2018, 12, 1127–1141. [Google Scholar] [CrossRef]
- Fujimoto, K.; Kimura, Y.; Shimohigoshi, M.; Satoh, T.; Sato, S.; Tremmel, G.; Uematsu, M.; Kawaguchi, Y.; Usui, Y.; Nakano, Y.; et al. Metagenome Data on Intestinal Phage-Bacteria Associations Aids the Development of Phage Therapy against Pathobionts. Cell Host Microbe 2020. [Google Scholar] [CrossRef] [PubMed]
- Bondy-Denomy, J.; Qian, J.; Westra, E.R.; Buckling, A.; Guttman, D.S.; Davidson, A.R.; Maxwell, K.L. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 2016, 10, 2854–2866. [Google Scholar] [CrossRef]
- Brüssow, H.; Canchaya, C.; Hardt, W.-D. Phages and the Evolution of Bacterial Pathogens: From Genomic Rearrangements to Lysogenic Conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed]
- Clooney, A.G.; Sutton, T.D.S.; Shkoporov, A.N.; Holohan, R.K.; Daly, K.M.; O’Regan, O.; Ryan, F.J.; Draper, L.A.; Plevy, S.E.; Ross, R.P.; et al. Whole-Virome Analysis Sheds Light on Viral Dark Matter in Inflammatory Bowel Disease. Cell Host Microbe 2019, 26, 764–778.e765. [Google Scholar] [CrossRef]
- Wagner, P.L.; Waldor, M.K. Bacteriophage control of bacterial virulence. Infect. Immun. 2002, 70, 3985–3993. [Google Scholar] [CrossRef] [PubMed]
- Boling, L.; Cuevas, D.A.; Grasis, J.A.; Kang, H.S.; Knowles, B.; Levi, K.; Maughan, H.; McNair, K.; Rojas, M.I.; Sanchez, S.E.; et al. Dietary prophage inducers and antimicrobials: Toward landscaping the human gut microbiome. Gut Microbes 2020, 1–14. [Google Scholar] [CrossRef]
- Oh, J.H.; Alexander, L.M.; Pan, M.; Schueler, K.L.; Keller, M.P.; Attie, A.D.; Walter, J.; van Pijkeren, J.P. Dietary Fructose and Microbiota-Derived Short-Chain Fatty Acids Promote Bacteriophage Production in the Gut Symbiont Lactobacillus reuteri. Cell Host Microbe 2019, 25, 273–284.e276. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef] [PubMed]
- Haltalin, K.C.; Nelson, J.D.; Kusmiesz, H.T.; Hinton, L.V. Comparison of intramuscular and oral ampicillin therapy for shigellosis. J. Pediatrics 1968, 73, 617–622. [Google Scholar] [CrossRef]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed]
- Fraher, M.H.; O’Toole, P.W.; Quigley, E.M.M. Techniques used to characterize the gut microbiota: A guide for the clinician. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 2020, 8, 90. [Google Scholar] [CrossRef]
- Akhter, S.; Aziz, R.K.; Edwards, R.A. PhiSpy: A novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucleic Acids Res. 2012, 40, e126. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic Virus Orthologous Groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017, 45, D491–D498. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Roux, S.; Emerson, J.B.; Eloe-Fadrosh, E.A.; Sullivan, M.B. Benchmarking viromics: An in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ 2017, 5, e3817. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Crabtree, J.; Agrawal, S.; Mahurkar, A.; Myers, G.S.; Rasko, D.A.; White, O. Circleator: Flexible circular visualization of genome-associated data with BioPerl and SVG. Bioinformatics 2014, 30, 3125–3127. [Google Scholar] [CrossRef]
- Blieden, M.; Paramore, L.C.; Shah, D.; Ben-Joseph, R. A perspective on the epidemiology of acetaminophen exposure and toxicity in the United States. Expert Rev. Clin. Pharm. 2014, 7, 341–348. [Google Scholar] [CrossRef]
- Mazumdar, K.; Dutta, N.K.; Dastidar, S.G.; Motohashi, N.; Shirataki, Y. Diclofenac in the management of E. coli urinary tract infections. In Vivo 2006, 20, 613–619. [Google Scholar]
- Dastidar, S.G.; Ganguly, K.; Chaudhuri, K.; Chakrabarty, A.N. The anti-bacterial action of diclofenac shown by inhibition of DNA synthesis. Int. J. Antimicrob. Agents 2000, 14, 249–251. [Google Scholar] [CrossRef]
- Elvers, K.T.; Wright, S.J. Antibacterial activity of the anti-inflammatory compound ibuprofen. Lett. Appl. Microbiol. 1995, 20, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Lehouritis, P.; Cummins, J.; Stanton, M.; Murphy, C.T.; McCarthy, F.O.; Reid, G.; Urbaniak, C.; Byrne, W.L.; Tangney, M. Local bacteria affect the efficacy of chemotherapeutic drugs. Sci. Rep. 2015, 5, 14554. [Google Scholar] [CrossRef] [PubMed]
- Maiques, E.; Ubeda, C.; Campoy, S.; Salvador, N.; Lasa, I.; Novick, R.P.; Barbe, J.; Penades, J.R. beta-lactam antibiotics induce the SOS response and horizontal transfer of virulence factors in Staphylococcus aureus. J. Bacteriol. 2006, 188, 2726–2729. [Google Scholar] [CrossRef] [PubMed]
- Elespuru, R.K.; White, R.J. Biochemical prophage induction assay: A rapid test for antitumor agents that interact with DNA. Cancer Res. 1983, 43, 2819–2830. [Google Scholar] [PubMed]
- Goerke, C.; Koller, J.; Wolz, C. Ciprofloxacin and trimethoprim cause phage induction and virulence modulation in Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 171–177. [Google Scholar] [CrossRef]
- Matsushiro, A.; Sato, K.; Miyamoto, H.; Yamamura, T.; Honda, T. Induction of prophages of enterohemorrhagic Escherichia coli O157:H7 with norfloxacin. J. Bacteriol. 1999, 181, 2257–2260. [Google Scholar] [CrossRef] [PubMed]
- Otsuji, N.; Sekiguchi, M.; Iijima, T.; Takagi, Y. Induction of phage formation in the lysogenic Escherichia coli K-12 by mitomycin C. Nature 1959, 184 (Suppl. 14), 1079–1080. [Google Scholar] [CrossRef]
- Borek, E.; Ryan, A. Lysogenic induction. Prog. Nucleic Acid. Res. Mol. Biol. 1973, 13, 249–300. [Google Scholar] [CrossRef]
- Jiang, S.C.; Paul, J.H. Seasonal and Diel Abundance of Viruses and Occurrence of Lysogeny/Bacteriocinogeny in the Marine-Environment. Mar. Ecol. Prog. Ser. 1994, 104, 163–172. [Google Scholar] [CrossRef]
- Ventura, M.; Turroni, F.; Lima-Mendez, G.; Foroni, E.; Zomer, A.; Duranti, S.; Giubellini, V.; Bottacini, F.; Horvath, P.; Barrangou, R.; et al. Comparative analyses of prophage-like elements present in bifidobacterial genomes. Appl. Environ. Microbiol. 2009, 75, 6929–6936. [Google Scholar] [CrossRef]
- Knowles, B.; Bailey, B.; Boling, L.; Breitbart, M.; Cobian-Guemes, A.; Del Campo, J.; Edwards, R.; Felts, B.; Grasis, J.; Haas, A.F.; et al. Variability and host density independence in inductions-based estimates of environmental lysogeny. Nat. Microbiol. 2017, 2, 17064. [Google Scholar] [CrossRef]
- Amirkia, V.D.; Qiubao, P. The Antimicrobial Index: A comprehensive literature-based antimicrobial database and reference work. Bioinformation 2011, 5, 365–366. [Google Scholar] [CrossRef]
- Borobio, M.V.; Conejo, M.; Ramirez, E.; Suarez, A.I.; Perea, E.J. Comparative activities of eight quinolones against members of the Bacteroides fragilis group. Antimicrob. Agents Chemother. 1994, 38, 1442–1445. [Google Scholar] [CrossRef]
- Forterre, P.; Soler, N.; Krupovic, M.; Marguet, E.; Ackermann, H.W. Fake virus particles generated by fluorescence microscopy. Trends Microbiol. 2013, 21, 1–5. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phylum | Bacteria | Gram | Accession/Assembly | Isolated | Media |
|---|---|---|---|---|---|
| Actinobacteria | Bifidobacterium longum subsp. infantis ATCC 15697 | + | NC_011593 | Infant Intestine | BHI w/hemin |
| Firmicutes | Clostridium beijerinckii ATCC 51743 | + | GCA_000016965.1 | Likely Soil | ABB |
| Clostridium scindens 32-6-S 4 CNA AN | + | N/A | Human Feces | ABB w/hemin | |
| Enterococcus faecalis TUSoD Ef11 | + | NZ_ACOX02000011 | Human Oral | BHI | |
| Bacteroidetes | Bacteroides caccae ATCC 43185 | − | AAVM00000000 | Human Feces | TSB |
| Bacteroides ovatus 3_8_47 | − | ACWH00000000 | Human Colon biopsy | TSB | |
| Bacteroides eggerthii 1_2_48 | − | ACWG00000000 | Human Colon biopsy | BHI w/hemin | |
| Proteobacteria | Escherichia coli K12 ATCC 25404 | − | N/A | Human Feces | BHI |
| Type of Agent | Drug | Mechanism of Action | Estimated Colon Concentration (µg/mL) | Tested Concentrations (µg/mL) |
|---|---|---|---|---|
| Antibiotic | Ampicillin | β-lactam: Cell wall synthesis inhibition | 44.56–3565.06 | 100, 10, 1, 0.1, 0.01 |
| Ciprofloxacin | Fluoroquinolone: Bacterial DNA gyrase and topoisomerase | 106.95–1247.77 | 2, 0.2, 0.02, 0.002, 0.0002 | |
| Norfloxacin | Fluoroquinolone: Bacterial DNA gyrase and topoisomerase | 427.81–998.22 | 10, 1, 0.1, 0.01, 0.001 | |
| Streptonigrin | Aminoquinone: Bacterial DNA and topoisomerase | 0.10–0.19 | 10, 1, 0.1, 0.01, 0.001 | |
| Mitomycin | DNA Cross Linker | - | 100, 10, 1, 0.1, 0.01 | |
| NSAID | Diclofenac | Analgesic, antipyretic, and anti-inflammatory | 44.56–66.84 | 100, 10, 1, 0.1, 0.01 |
| Ibuprofen | Inhibitor of COX | 106.95–427.81 | 100, 10, 1, 0.1, 0.01 | |
| Tolmetin | tNSAID heteroaryl acetic acid derivative | 35.65–1048.13 | 10, 1, 0.1, 0.01, 0.001 | |
| Chemotherapy | Busulfan | Alkylating agent-Alkyl sulfonate | 1069.52 | 100, 10, 1, 0.1, 0.01 |
| Fludarabine | Inhibits DNA Synthesis | 7–7.49 | 100, 10, 1, 0.1, 0.01 | |
| Mild Analgesic | Acetaminophen | Not well known | 0.00–312.83 | 100, 10, 1, 0.1, 0.01 |
| Cardiac | Digoxin | Na+/K+ pumps | 0.07–0.13 | 100, 10, 1, 0.1, 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sutcliffe, S.G.; Shamash, M.; Hynes, A.P.; Maurice, C.F. Common Oral Medications Lead to Prophage Induction in Bacterial Isolates from the Human Gut. Viruses 2021, 13, 455. https://doi.org/10.3390/v13030455
Sutcliffe SG, Shamash M, Hynes AP, Maurice CF. Common Oral Medications Lead to Prophage Induction in Bacterial Isolates from the Human Gut. Viruses. 2021; 13(3):455. https://doi.org/10.3390/v13030455
Chicago/Turabian StyleSutcliffe, Steven G., Michael Shamash, Alexander P. Hynes, and Corinne F. Maurice. 2021. "Common Oral Medications Lead to Prophage Induction in Bacterial Isolates from the Human Gut" Viruses 13, no. 3: 455. https://doi.org/10.3390/v13030455
APA StyleSutcliffe, S. G., Shamash, M., Hynes, A. P., & Maurice, C. F. (2021). Common Oral Medications Lead to Prophage Induction in Bacterial Isolates from the Human Gut. Viruses, 13(3), 455. https://doi.org/10.3390/v13030455