
The Novel Genetic Background of Infectious Bursal Disease Virus Strains Emerging from the Action of Positive Selection



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Dataset
2.2. Recombination Selection Analysis
2.3. Evolutionary History and Population Dynamics
2.4. Positive Selection Analysis
2.5. Functional Analysis
2.6. D Protein Visulization
2.7. Experimental Study
2.7.1. Ethical Statement
2.7.2. Viral Samples
2.7.3. Sequencing of the Full-Length A and B Segments of IBDV Strains Bug/03, 117/14, and 75/11
2.7.4. Pathogenicity Study in Chickens
2.7.5. Histopathology
2.7.6. Statistical Analysis
3. Results
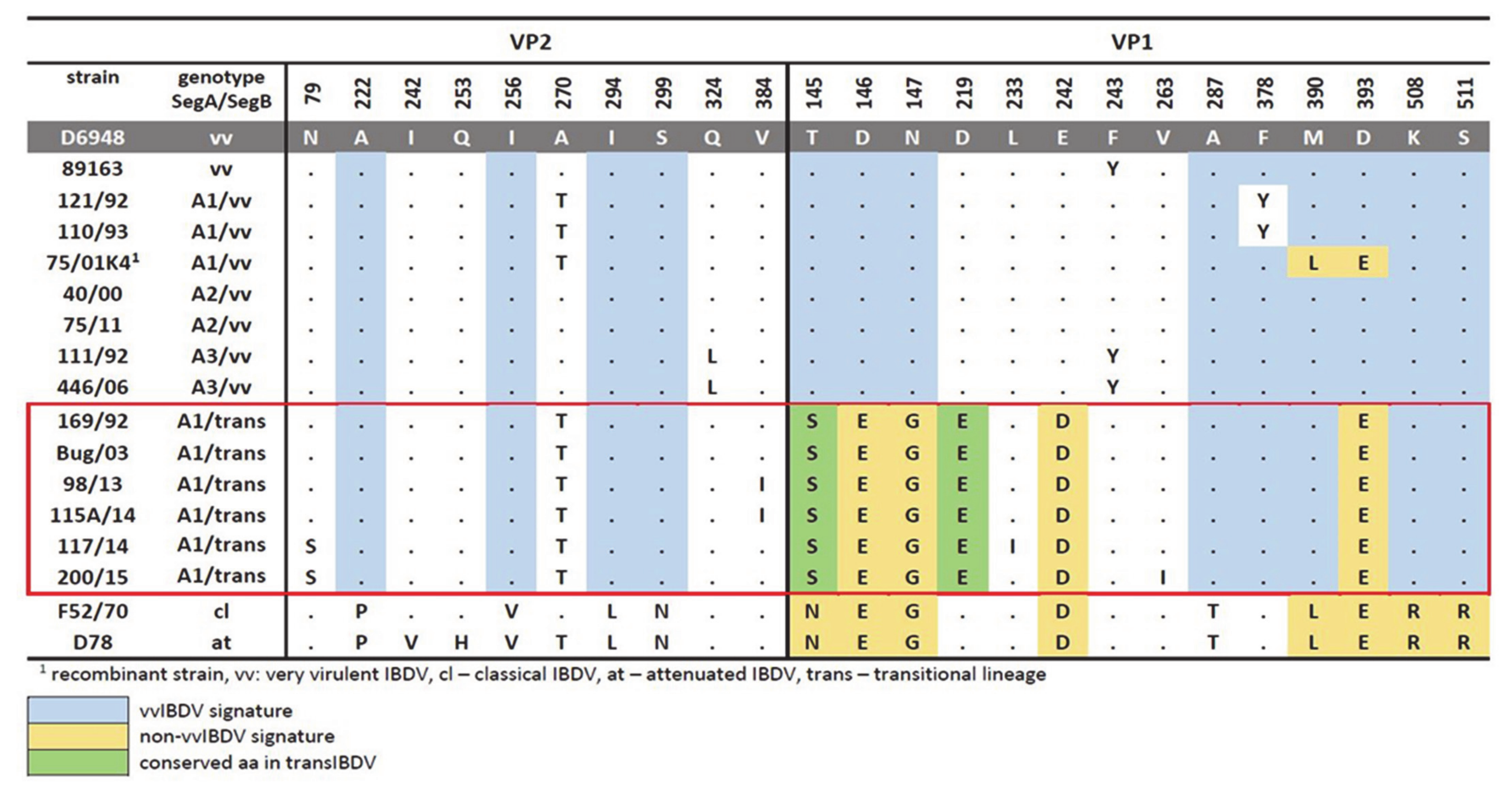
3.1. Molecular Characterization of VP2 and VP1 Proteins in All of the Polish IBDV Strains Studied
3.2. Recombination on the VP1 Gene of IBDV Is a Mechanism of Genetic Variability on Polish Strains
3.3. Temporal Reconstruction, Evolutionary Rates, and Population Dynamics of Polish IBDV Strains
3.4. Positive Selection and Functional Divergence Act on Segments A and B of the Polish IBDV Strains, Inducing Evolutionary Advantages
3.5. Evaluation of Virulence and Molecular Signatures Linked to the Biological Properties of the Novel Re-Assortant Strains vvIBDV-A/transIBDV-B
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Delmas, B.; Attoui, H.; Ghosh, S.; Malik, Y.S.; Mundt, E.; Vakharia, V.N.; Ictv Report, C. ICTV virus taxonomy profile: Birnaviridae. J. Gen. Virol. 2019, 100, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, A.S. An apparently new disease of chickens: Avian nephrosis. Avian. Dis. 1962, 6, 385–389. [Google Scholar] [CrossRef]
- Hirai, K.; Funakoshi, T.; Nakai, T.; Shimakura, S. Sequential-changes in the number of surface immunoglobulin-bearing lymphocytes-B in infectious bursal disease virus-infected chickens. Avian. Dis. 1981, 25, 484–496. [Google Scholar] [CrossRef]
- Kim, I.J.; You, S.K.; Kim, H.; Yeh, H.Y.; Sharma, J.M. Characteristics of bursal T lymphocytes induced by infectious bursal disease virus. J. Virol. 2000, 74, 8884–8892. [Google Scholar] [CrossRef]
- Brown Jordan, A.; Gongora, V.; Hartley, D.; Oura, C. A review of eight high-priority, economically important viral pathogens of poultry within the Caribbean region. Vet. Sci. 2018, 5, 14. [Google Scholar] [CrossRef]
- Zachar, T.; Popowich, S.; Goodhope, B.; Knezacek, T.; Ojkic, D.; Willson, P.; Ahmed, K.A.; Gomis, S. A 5-year study of the incidence and economic impact of variant infectious bursal disease viruses on broiler production in Saskatchewan, Canada. Can. J. Vet. Res. 2016, 80, 255–261. [Google Scholar]
- Spackman, E.; Stephens, C.B.; Pantin-Jackwood, M.J. The effect of infectious bursal disease virus–induced immunosuppression on vaccination against highly pathogenic avian influenza virus. Avian. Dis. 2017, 62, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Dobos, P.; Hill, B.J.; Hallett, R.; Kells, D.T.; Becht, H.; Teninges, D. Biophysical and biochemical characterization of five animal viruses with bisegmented double-stranded RNA genomes. J. Virol. 1979, 32, 593–605. [Google Scholar] [CrossRef]
- Hudson, P.J.; Mckern, N.M.; Power, B.E.; Azad, A.A. Genomic structure of the large rna segment of infectious bursal disease virus. Nucleic Acids Res. 1986, 14, 5001–5012. [Google Scholar] [CrossRef]
- Li, Z.H.; Wang, Y.Q.; Xue, Y.F.; Li, X.Q.; Cao, H.; Zheng, S.J.J. Critical role for voltage-dependent anion channel 2 in infectious bursal disease virus-induced apoptosis in host cells via interaction with VP5. J. Virol. 2012, 86, 1328–1338. [Google Scholar] [CrossRef] [PubMed]
- Méndez, F.; Romero, N.; Cubas, L.L.; Delgui, L.R.; Rodriguez, D.; Rodriguez, J.F. Non-lytic egression of infectious bursal disease virus (IBDV) particles from infected cells. PLoS ONE 2017, 12, e017008010. [Google Scholar] [CrossRef]
- Lejal, N.; Da Costa, B.; Huet, J.-C.; Delmas, B. Role of Ser-652 and Lys-692 in the protease activity of infectious bursal disease virus VP4 and identification of its substrate cleavage sites. J. Gen. Virol. 2000, 81, 983–992. [Google Scholar] [CrossRef] [PubMed]
- von Einem, U.I.; Gorbalenya, A.E.; Schirrmeier, H.; Behrens, S.E.; Letzel, T.; Mundt, E. VP1 of infectious bursal disease virus is an RNA-dependent RNA polymerase. J. Gen. Virol. 2004, 85, 2221–2229. [Google Scholar] [CrossRef]
- Ye, C.; Wang, Y.; Zhang, E.; Han, X.; Yu, Z.; Liu, H. VP1 and VP3 are required and sufficient for translation initiation of uncapped infectious bursal disease virus genomic double-stranded RNA. J. Viro.l 2018, 92. [Google Scholar] [CrossRef]
- Lee, C.-C.; Ko, T.-P.; Chou, C.-C.; Yoshimura, M.; Doong, S.-R.; Wang, M.-Y.; Wang, A.H.J. Crystal structure of infectious bursal disease virus VP2 subviral particle at 2.6Å resolution: Implications in virion assembly and immunogenicity. J. Struct. Biol. 2006, 155, 74–86. [Google Scholar] [CrossRef]
- Mundt, E. Tissue culture infectivity of different strains of infectious bursal disease virus is determined by distinct amino acids in VP2. J. Gen. Virol. 1999, 80, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Brandt, M.; Yao, K.; Liu, M.H.; Heckert, R.A.; Vakharia, V.N. Molecular determinants of virulence, cell tropism, and pathogenic phenotype of infectious bursal disease virus. J. Virol. 2001, 75, 11974–11982. [Google Scholar] [CrossRef] [PubMed]
- Le Nouen, C.; Toquin, D.; Muller, H.; Raue, R.; Kean, K.M.; Langlois, P.; Cherbonnel, M.; Eterradossi, N. Different domains of the RNA polymerase of infectious bursal disease virus contribute to virulence. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Escaffre, O.; Le Nouen, C.; Amelot, M.; Ambroggio, X.; Ogden, K.M.; Guionie, O.; Toquin, D.; Muller, H.; Islam, M.R.; Eterradossi, N. Both genome segments contribute to the pathogenicity of very virulent infectious bursal disease virus. J. Virol. 2013, 87, 2767–2780. [Google Scholar] [CrossRef]
- Boot, H.J.; Hoekman, A.J.W.; Gielkens, A.L.J. The enhanced virulence of very virulent infectious bursal disease virus is partly determined by its B-segment. Arch. Virol. 2005, 150, 137–144. [Google Scholar] [CrossRef]
- Le Nouën, C.; Rivallan, G.; Toquin, D.; Darlu, P.; Morin, Y.; Beven, V.; de Boisseson, C.; Cazaban, C.; Comte, S.; Gardin, Y.; et al. Very virulent infectious bursal disease virus: Reduced pathogenicity in a rare natural segment-B-reassorted isolate. J. Gen. Virol. 2006, 87, 209–216. [Google Scholar] [CrossRef]
- Stoute, S.T.; Jackwood, D.J.; Crossley, B.M.; Michel, L.O.; Blakey, J.R. Molecular epidemiology of endemic and very virulent infectious bursal disease virus genogroups in backyard chickens in California, 2009–2017. J. Vet. Diagn. Invest. 2019, 31, 371–377. [Google Scholar] [CrossRef]
- Wei, Y.; Li, J.; Zheng, J.; Xu, H.; Li, L.; Yu, L. Genetic reassortment of infectious bursal disease virus in nature. Biochem. Biophys. Res. Commun. 2006, 350, 277–287. [Google Scholar] [CrossRef]
- Pikuła, A.; Lisowska, A.; Jasik, A.; Śmietanka, K. Identification and assessment of virulence of a natural reassortant of infectious bursal disease virus. Vet. Res. 2018, 49, 89. [Google Scholar] [CrossRef] [PubMed]
- Hon, C.C.; Lam, T.Y.; Drummond, A.; Rambaut, A.; Lee, Y.F.; Yip, C.W.; Zeng, F.; Lam, P.Y.; Ng, P.T.; Leung, F.C. Phylogenetic analysis reveals a correlation between the expansion of very virulent infectious bursal disease virus and reassortment of its genome segment B. J. Virol. 2006, 80, 8503–8509. [Google Scholar] [CrossRef]
- Alfonso-Morales, A.; Rios, L.; Martinez-Perez, O.; Dolz, R.; Valle, R.; Perera, C.L.; Bertran, K.; Frias, M.T.; Ganges, L.; Diaz de Arce, H.; et al. Evaluation of a phylogenetic marker based on genomic segment B of infectious bursal disease virus: Facilitating a feasible incorporation of this segment to the molecular epidemiology studies for this viral agent. PLoS ONE 2015, 10, e0125853. [Google Scholar] [CrossRef] [PubMed]
- He, C.Q.; Ma, L.Y.; Wang, D.; Li, G.R.; Ding, N.Z. Homologous recombination is apparent in infectious bursal disease virus. Virology 2009, 384, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, D.J.; Sommer-Wagner, S.E.; Crossley, B.M.; Stoute, S.T.; Woolcock, P.R.; Charlton, B.R. Identification and pathogenicity of a natural reassortant between a very virulent serotype 1 infectious bursal disease virus (IBDV) and a serotype 2 IBDV. Virology 2011, 420, 98–105. [Google Scholar] [CrossRef]
- Michel, L.O.; Jackwood D., J. Classification of infectious bursal disease virus into genogroups. Arch. Virol. 2017, 162, 3661–3670. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Wu, T.; Hussain, A.; Gao, Y.; Zeng, X.; Wang, Y.; Gao, L.; Li, K.; Wang, Y.; Liu, C.; et al. Novel variant strains of infectious bursal disease virus isolated in China. Vet. Microbiol. 2019, 230, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Tomás, G.; Marandino, A.; Courtillon, C.; Amelot, M.; Keita, A.; Pikuła, A.; Hernández, M.; Hernández, D.; Vagnozzi, A.; Panzera, Y.; et al. Antigenicity, pathogenicity and immunosuppressive effect caused by a South American isolate of infectious bursal disease virus belonging to the “distinct” enetic lineage. Avian. Pathol. 2019, 48, 245–254. [Google Scholar] [CrossRef]
- Pikuła, A.; Śmietanka, K.; Perez, L.J. Emergence and expansion of novel pathogenic reassortant strains of infectious bursal disease virus causing acute outbreaks of the disease in Europe. Transbound. Emerg. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Khoosal, A.; Muhire, B. Detecting and analyzing genetic recombination using RDP4. Methods Mol. Biol. 2017, 1525, 433–460. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Morales, A.; Martínez-Pérez, O.; Dolz, R.; Valle, R.; Perera, C.L.; Bertran, K.; Frias, M.T.; Majo, N.; Ganges, L.; Peréz, L.J. Spatiotemporal phylogenetic analysis and molecular characterisation of infectious bursal disease viruses based on the VP2 hyper-variable region. PLoS ONE 2013, 8, e65999. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H.; Wong, W.S.W.; Nielsen, R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef]
- Rios, L.; Coronado, L.; Naranjo-Feliciano, D.; Martinez-Perez, O.; Perera, C.L.; Hernandez-Alvarez, L.; Diaz de Arce, H.; Nunez, J.I.; Ganges, L.; Perez, L.J. Deciphering the emergence, genetic diversity and evolution of classical swine fever virus. Sci. Rep. 2017, 7, 17887. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Yang, Z.H. Multiple hypothesis testing to detect lineages under positive selection that affects only a few sites. Mol. Biol. Evol. 2007, 24, 1219–1228. [Google Scholar] [CrossRef]
- Studer, R.A.; Robinson-Rechavi, M. Evidence for an episodic model of protein sequence evolution. Biochem. Soc. T 2009, 37, 783–786. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Domanska, K.; Mato, T.; Rivallan, G.; Smietanka, K.; Minta, Z.; de Boisseson, C.; Toquin, D.; Lomniczi, B.; Palya, V.; Eterradossi, N. Antigenic and genetic diversity of early European isolates of infectious bursal disease virus prior to the emergence of the very virulent viruses: Early European epidemiology of infectious bursal disease virus revisited? Arch. Virol. 2004, 149, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Eterradossi, N.; Arnauld, C.; Toquin, D.; Rivallan, G. Critical amino acid changes in VP2 variable domain are associated with typical and atypical antigenicity in very virulent infectious bursal disease viruses. Arch. Virol. 1998, 143, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- de Fraga, A.P.; Graf, T.; Coltro, V.P.; Ikuta, N.; Fonseca, A.S.K.; Majo, N.; Lunge, V.R. Phylodynamic analyses of Brazilian antigenic variants of infectious bursal disease virus. Infect. Genet. Evol. 2019, 73, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, B.; Rastogi, S.K. Structural and functional modeling of viral protein 5 of Infectious Bursal Disease Virus. Virus Res. 2018, 247, 55–60. [Google Scholar] [CrossRef]
- Dey, S.; Pathak, D.C.; Ramamurthy, N.; Maity, H.K.; Chellappa, M.M. Infectious bursal disease virus in chickens: Prevalence, impact, and management strategies. Vet. Med. 2019, 10, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Minta, Z.; Daniel, A. Infectious bursal disease virus in Poland: Current situation and vaccinal control. In Proceedings of the International Symposium on Infectious Bursal Disease and Chicken Infectious Anaemia; Kaleta, E.F., Heffels-Redmann, U., Lange-Herbst, H., Eds.; World Veterinary Poultry Association: Rauischholzhausen, Germany, 1994; Volume 7. [Google Scholar]
- Silva, F.M.F.; Vidigal, P.M.P.; Myrrha, L.W.; Fietto, J.L.R.; Silva, A.; Almeida, M.R. Tracking the molecular epidemiology of Brazilian infectious bursal disease virus (IBDV) isolates. Infect. Genet. Evol. 2013, 13, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Hon, C.C.; Lam, T.T.; Yip, C.W.; Wong, R.T.; Shi, M.; Jiang, J.; Zeng, F.; Leung, F.C. Phylogenetic evidence for homologous recombination within the family Birnaviridae. J. Gen. Virol. 2008, 89, 3156–3164. [Google Scholar] [CrossRef]
- Jackwood, D.J.; Schat, K.A.; Michel, L.O.; de Wit, S. A proposed nomenclature for infectious bursal disease virus isolates. Avian. Pathol. 2018, 47, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, D.J.; Sommer, S.E. Identification of infectious bursal disease virus quasispecies in commercial vaccines and field isolates of this double-stranded RNA virus. Virology 2002, 304, 105–113. [Google Scholar] [CrossRef][Green Version]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Perales, C. Viral quasispecies. PLoS Genet. 2019, 15, e1008271. [Google Scholar] [CrossRef] [PubMed]
- Domanska, K.; Smietanka, K.; Minta, Z. Characterization of Polish infectious bursal disease virus strains by reverse transcription polymerase chain reaction combined with restriction enzyme analysis. B Vet. I Pulawy 2003, 47, 349–356. [Google Scholar]
- Agol, V.I.; Gmyl, A.P. Emergency services of viral RNAs: Repair and remodeling. Microbiol. Mol. Biol. Rev. 2018, 82, e00067-17. [Google Scholar] [CrossRef] [PubMed]
- Mató, T.; Tatár-Kis, T.; Felföldi, B.; Jansson, D.S.; Homonnay, Z.; Bányai, K.; Palya, V. Occurrence and spread of reassortant very virulent genotype of infectious bursal disease virus altered VP2 amino acid profile and pathogenicity in some European countries. Vet. Microbiol. 2020, 245, 108663. [Google Scholar] [CrossRef]
- Coronado, L.; Rios, L.; Frias, M.T.; Amaran, L.; Naranjo, P.; Percedo, M.I.; Perera, C.L.; Prieto, F.; Fonseca-Rodriguez, O.; Perez, L.J. Positive selection pressure on E2 protein of classical swine fever virus drives variations in virulence, pathogenesis and antigenicity: Implication for epidemiological surveillance in endemic areas. Transbound. Emerg. Dis. 2019, 66, 2362–2382. [Google Scholar] [CrossRef] [PubMed]
- Perez, L.J.; Diaz de Arce, H.; Perera, C.L.; Rosell, R.; Frias, M.T.; Percedo, M.I.; Tarradas, J.; Dominguez, P.; Nunez, J.I.; Ganges, L. Positive selection pressure on the B/C domains of the E2-gene of classical swine fever virus in endemic areas under C-strain vaccination. Infect. Genet. Evol. 2012, 12, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Escarmís, C.; Perales, C.; Domingo, E. Biological effect of Muller’s ratchet: Distant capsid site can affect picornavirus protein processing. J. Virol. 2009, 83, 6748. [Google Scholar] [CrossRef]
- Pan, J.H.; Vakharia, V.N.; Tao, Y.J. The structure of a birnavirus polymerase reveals a distinct active site topology. Proc. Natl. Acad. Sci. USA 2007, 104, 7385–7390. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.A.; Reddy, V.R.A.P.; Gray, A.G.; Wells, J.; Simpson, J.; Skinner, M.A.; Hawes, P.C.; Broadbent, A.J. Discrete virus factories form in the cytoplasm of cells co-infected with two replication competent tagged reporter birnaviruses, that subsequently coalesce over time. J. Virol. 2020, 02107–02119. [Google Scholar] [CrossRef]
- Schulte, M.B.; Draghi, J.A.; Plotkin, J.B.; Andino, R. Experimentally guided models reveal replication principles that shape the mutation distribution of RNA viruses. eLife 2015, 4, e03753. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.L.; Wang, X.M.; Gao, Y.L.; Fu, C.Y. Direct evidence of reassortment and mutant spectrum analysis of a very virulent infectious bursal disease virus. Avian. Dis. 2007, 51, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Drissi Touzani, C.; Fellahi, S.; Gaboun, F.; Fassi Fihri, O.; Baschieri, S.; Mentag, R.; El Houadfi, M. Molecular characterization and phylogenetic analysis of very virulent infectious bursal disease virus circulating in Morocco during 2016–2017. Arch. Virol. 2019, 164, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Escarmis, C.; Davila, M.; Domingo, E. Multiple molecular pathways for fitness recovery of an RNA virus debilitated by operation of Muller’s ratchet. J. Mol. Biol. 1999, 285, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.M.; Omar, A.R.; Chong, L.K.; Hair-Bejo, M.; Aini, I. Pathogenicity of SspI-positive infectious bursal disease virus and molecular characterization of the VP2 hypervariable region. Avian. Pathol. 2001, 30, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.L.; Zhang, L.Z.; Chen, Y.M.; Gao, L.; Wu, G.; Qin, L.T.; Wang, Y.Q.; Ren, X.G.; Gao, Y.L.; Gao, H.L.; et al. Mutations of residues 249 and 256 in VP2 are involved in the replication and virulence of infectious bursal disease virus. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Tammiranta, N.; Ek-Kommonen, C.E.; Rossow, L.; Huovilainen, A. Circulation of very virulent avian infectious bursal disease virus in Finland. Avian. Pathol. 2018, 47, 520–525. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pikuła, A.; Lisowska, A.; Jasik, A.; Perez, L.J. The Novel Genetic Background of Infectious Bursal Disease Virus Strains Emerging from the Action of Positive Selection. Viruses 2021, 13, 396. https://doi.org/10.3390/v13030396
Pikuła A, Lisowska A, Jasik A, Perez LJ. The Novel Genetic Background of Infectious Bursal Disease Virus Strains Emerging from the Action of Positive Selection. Viruses. 2021; 13(3):396. https://doi.org/10.3390/v13030396
Chicago/Turabian StylePikuła, Anna, Anna Lisowska, Agnieszka Jasik, and Lester J. Perez. 2021. "The Novel Genetic Background of Infectious Bursal Disease Virus Strains Emerging from the Action of Positive Selection" Viruses 13, no. 3: 396. https://doi.org/10.3390/v13030396
APA StylePikuła, A., Lisowska, A., Jasik, A., & Perez, L. J. (2021). The Novel Genetic Background of Infectious Bursal Disease Virus Strains Emerging from the Action of Positive Selection. Viruses, 13(3), 396. https://doi.org/10.3390/v13030396