
Rational Design of a Pan-Coronavirus Vaccine Based on Conserved CTL Epitopes

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Phylogenetic Analysis and Sequence Alignment for Coronavirus
2.2. Identification of Potential CTL Epitope
2.3. Mosaic Antigen Design
2.4. Phylogenetic and Sequence Analysis of Mosaic Antigen Cocktail
2.5. Computational Model of Three-Dimensional (3-D) for Mosaic Antigen
3. Results and Discussion
3.1. Phylogenetic Analysis of CoV Sequences in This Study
3.2. Analysis of Potential CTL Epitopes with Existing HCoV Sequences
3.3. Design of the Mosaic CoV Antigens Based on Conserved CTL Epitopes
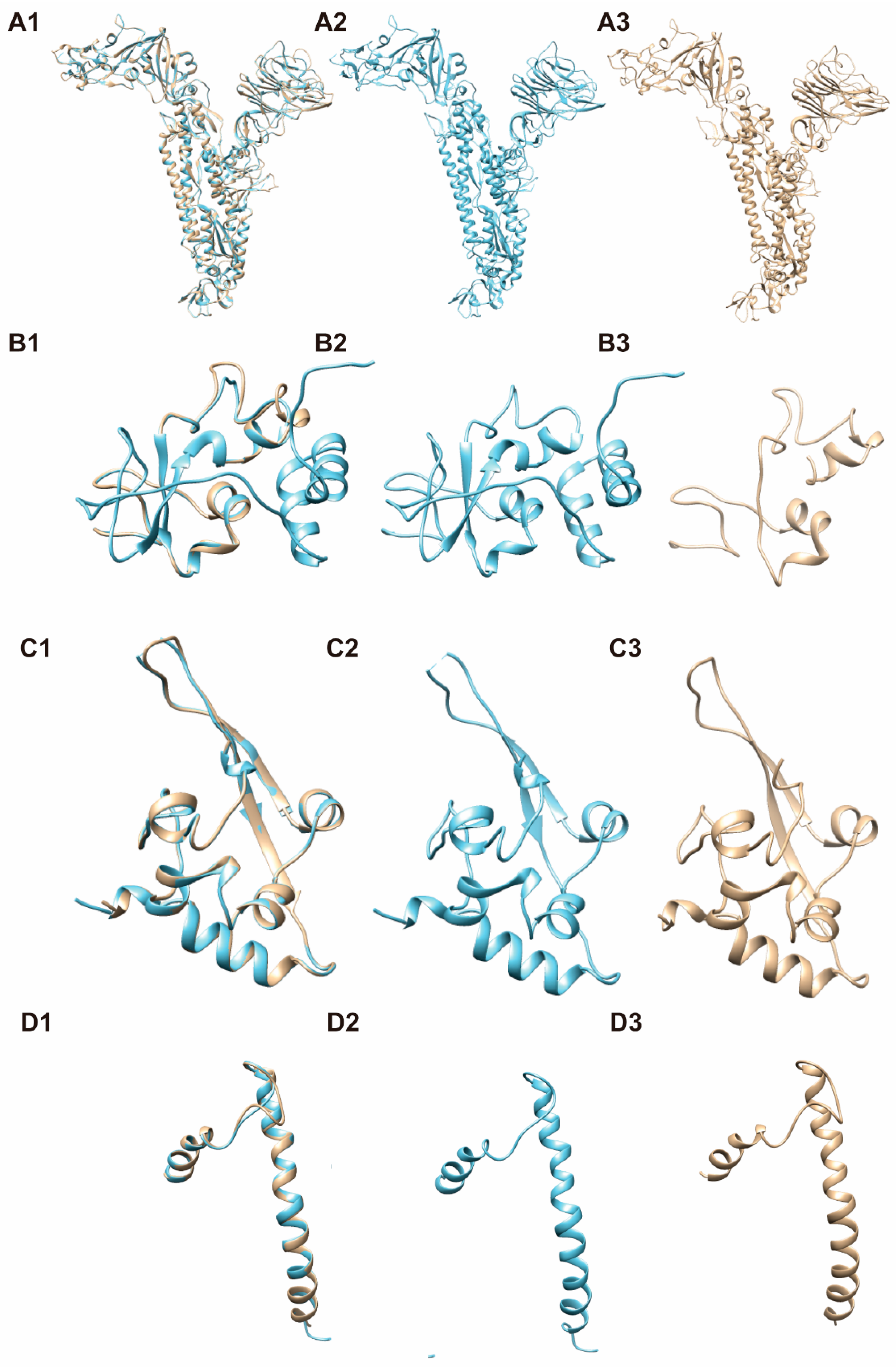
3.4. 3-D Structure Modeling for Mosaic CoV Antigen
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chinese Center for Disease Control and Prevention. Distribution of COVID-19. Available online: http://2019ncov.chinacdc.cn/2019-nCoV/global.html (accessed on 7 February 2021).
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Killerby, M.E.; Biggs, H.M.; Midgley, C.M.; Gerber, S.I.; Watson, J.T. Middle East Respiratory Syndrome Coronavirus Transmission. Emerg. Infect. Dis. 2020, 26, 191–198. [Google Scholar] [CrossRef]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- Zhou, F.; Cao, H.; Zuo, X.; Zhang, T.; Zhang, X.; Liu, X.; Xu, R.; Chen, G.; Zhang, Y.; Zheng, X.; et al. Deep sequencing of the MHC region in the Chinese population contributes to studies of complex disease. Nat. Genet. 2016, 48, 740–746. [Google Scholar] [CrossRef]
- Middleton, D.; Menchaca, L.; Rood, H.; Komerofsky, R. New allele frequency database: http://www.allelefrequencies.net. Tissue Antigens 2003, 61, 403–407. [Google Scholar] [CrossRef]
- Gonzalez-Galarza, F.F.; McCabe, A.; Santos, E.J.M.D.; Jones, J.; Takeshita, L.; Ortega-Rivera, N.D.; Cid-Pavon, G.M.D.; Ramsbottom, K.; Ghattaoraya, G.; Alfirevic, A.; et al. Allele frequency net database (AFND) 2020 update: Gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res. 2020, 48, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Limin, F.; Beifang, N.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar]
- Grifoni, A.; Sidney, J.; Zhang, Y.; Scheuermann, R.H.; Peters, B.; Sette, A.A. Sequence Homology and Bioinformatic Approach Can Predict Candidate Targets for Immune Responses to SARS-CoV-2. Cell Host Microbe 2020, 27, 671–680. [Google Scholar] [CrossRef]
- Fischer, W.; Perkins, S.; Theiler, J.; Bhattacharya, T.; Yusim, K.; Funkhouser, R.; Kuiken, C.; Haynes, B.; Letvin, N.L.; Walker, B.D.; et al. Polyvalent vaccines for optimal coverage of potential T-cell epitopes in global HIV-1 variants. Nat. Med. 2007, 13, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H.; Stephenson, K.E.; Borducchi, E.N.; Smith, K.; Stanley, K.; McNally, A.G.; Liu, J.; Abbink, P.; Maxfield, L.F.; Seaman, M.S.; et al. Protective efficacy of a global HIV-1 mosaic vaccine against heterologous SHIV challenges in rhesus monkeys. Cell 2013, 155, 531–539. [Google Scholar] [CrossRef]
- Kamlangdee, A.; Kingstad-Bakke, B.; Anderson, T.K.; Goldberg, T.L.; Osorio, J.E. Broad protection against avian influenza virus by using a modified vacciniaAnkara virus expressing a mosaic hemagglutinin gene. J. Virol. 2014, 88, 13300–13309. [Google Scholar] [CrossRef]
- Gao, Q.; Bao, L.; Mao, H.; Wang, L.; Xu, K.; Yang, M.; Li, Y.; Zhu, L.; Wang, N.; Lv, Z.; et al. Rapid development of an inactivated vaccine candidate for SARS-CoV-2. Science 2020, 369, 77–81. [Google Scholar] [CrossRef]
- Zhu, F.; Wurie, A.H.; Hou, L.; Liang, Q.; Li, Y.; Russell, J.B.W.; Wu, S.; Li, J.; Hu, Y.; Guo, Q.; et al. Safety and immunogenicity of a recombinant adenovirus type-5 vector-based Ebola vaccine in healthy adults in Sierra Leone: A single-centre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2017, 389, 621–628. [Google Scholar] [CrossRef]
- Coleman, C.M.; Liu, Y.V.; Mu, H.; Taylor, J.K.; Massare, M.; Flyer, D.C.; Glenn, G.M.; Smith, G.E.; Frieman, M.B. Purified coronavirus spike protein nanoparticles induce coronavirus neutralizing antibodies in mice. Vaccine 2014, 32, 3169–3174. [Google Scholar] [CrossRef] [PubMed]
- Weingartl, H.; Czub, M.; Czub, S.; Neufeld, J.; Marszal, P.; Gren, J.; Smith, G.; Jones, S.; Proulx, R.; Deschambault, Y.; et al. Immunization with modified vaccinia virus Ankara-based recombinant vaccine against severe acute respiratory syndrome is associated with enhanced hepatitis in ferrets. J. Virol. 2004, 78, 12672–12676. [Google Scholar] [CrossRef]
- Liu, L.; Wei, Q.; Lin, Q.; Fang, J.; Wang, H.; Kwok, H.; Tang, H.; Nishiura, K.; Peng, J.; Tan, Z.; et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight 2019, 4, e123158. [Google Scholar] [CrossRef] [PubMed]
- Ng, O.; Chia, A.; Tan, A.T.; Jadi, R.S.; Leong, H.N.; Bertoletti, A.; Tan, Y.-J. Memory T cell responses targeting the SARS coronavirus persist up to 11 yearspost-infection. Vaccine 2016, 34, 2008–2014. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhao, J.; Mangalam, A.K.; Channappanavar, R.; Fett, C.; Meyerholz, D.K.; Agnihothram, S.; Baric, R.S.; David, C.S.; Perlman, S. Airway Memory CD4(+) T Cells Mediate Protective Immunity against Emerging Respiratory Coronaviruses. Immunity 2016, 44, 1379–1391. [Google Scholar] [CrossRef]
- Le Bert, N.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.L.; Hafezi, M.; Chia, A.; Chng, M.H.Y.; Lin, M.; Tan, N.; Linster, M.; et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 2020, 584, 457–462. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501. [Google Scholar] [CrossRef]
- Peng, Y.; Mentzer, A.J.; Liu, G.; Yao, X.; Yin, Z.; Dong, D.; Dejnirattisai, W.; Rostron, T.; Supasa, P.; Liu, C.; et al. Broad and strong memory CD4(+) and CD8(+) T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020, 21, 1336–1345. [Google Scholar] [CrossRef]
- Poran, A.; Harjanto, D.; Malloy, M.; Arieta, C.M.; Rothenberg, D.A.; Lenkala, D.; van Buuren, M.M.; Addona, T.A.; Rooney, M.S.; Srinivasan, L.; et al. Sequence-based prediction of SARS-CoV-2 vaccine targets using a mass spectrometry-based bioinformatics predictor identifies immunogenic T cell epitopes. Genome Med. 2020, 12, 70. [Google Scholar] [CrossRef] [PubMed]
- Mateus, J.; Grifoni, A.; Tarke, A.; Sidney, J.; Ramirez, S.I.; Dan, J.M.; Burger, Z.C.; Rawlings, S.A.; Smith, D.M.; Phillips, E.; et al. Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science 2020, 370, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Martina, B.; Domenico, B.; Marta, G.; Silvia, A.; Massimo, C.; Stefano, P. SARS-CoV-2 Envelope and Membrane Proteins: Structural Differences Linked to Virus Characteristics? Biomed. Res. Int. 2020, 2020, 4389089. [Google Scholar]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| HLA Alleles | Frequency in Chinese Population |
|---|---|
| HLA-A*11:01 | 18.57% |
| HLA-A*24:02 | 15.73% |
| HLA-A*02:01 | 13.79% |
| HLA-A*33:03 | 8.03% |
| HLA-A*30:01 | 6.67% |
| HLA-B*40:01 | 9.59% |
| HLA-B*46:01 | 7.83% |
| HLA-B*13:02 | 7.11% |
| HLA-C*07:02 | 14.46% |
| HLA-C*01:02 | 14.10% |
| HLA-C*06:02 | 10.50% |
| HLA-C*03:04 | 8.90% |
| HLA-C*08:01 | 8.23% |
| HLA-C*03:03 | 7.90% |
| HLA-C*04:01 | 6.02% |
| Virus Name | S Protein | M Protein | N Protein | E Protein |
|---|---|---|---|---|
| SARS-CoV-2 | YP_009724390.1 | YP_009724393.1 | YP_009724397.2 | YP_009724392.1 |
| MERS-CoV | YP_009047204.1 | YP_009047210.1 | YP_009047211.1 | YP_009047209.1 |
| SARS-CoV | NP_828851.1 | NP_828855.1 | NP_828858.1 | NP_828854.1 |
| HCoV-229E | NP_073551.1 | NP_073555.1 | NP_073556.1 | NP_073554.1 |
| HCoV-OC43 | YP_009555241.1 | YP_009555244.1 | YP_009555245.1 | YP_009555243.1 |
| HCoV-NL63 | YP_173238.1 | YP_173241.1 | YP_173242.1 | YP_173240.1 |
| HCoV-HKU1 | YP_003767.1 | YP_003770.1 | YP_003771.1 | YP_003769.1 |
| Name | S Protein | M Protein | N Protein | E Protein | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HLA-A | HLA-B | HLA-C | HLA-A | HLA-B | HLA-C | HLA-A | HLA-B | HLA-C | HLA-A | HLA-B | HLA-C | |
| SARS-CoV-2 | 105 | 88 | 107 | 34 | 15 | 27 | 35 | 16 | 21 | 11 | 6 | 6 |
| MERS-CoV | 116 | 101 | 121 | 24 | 21 | 32 | 32 | 17 | 16 | 8 | 7 | 5 |
| SARS-CoV | 108 | 84 | 111 | 36 | 20 | 26 | 35 | 16 | 21 | 9 | 8 | 6 |
| HCoV-229E | 104 | 95 | 111 | 30 | 24 | 28 | 32 | 21 | 26 | 8 | 4 | 7 |
| HCoV-OC43 | 112 | 91 | 115 | 35 | 19 | 30 | 35 | 24 | 23 | 11 | 8 | 11 |
| HCoV-NL63 | 125 | 110 | 133 | 29 | 15 | 21 | 30 | 19 | 30 | 12 | 5 | 13 |
| HCoV-HKU1 | 122 | 97 | 129 | 28 | 21 | 25 | 30 | 20 | 29 | 13 | 6 | 11 |
| Mosaic1 | 122 | 93 | 120 | 26 | 23 | 33 | 32 | 17 | 16 | 11 | 8 | 12 |
| Mosaic2 | 105 | 88 | 107 | 29 | 15 | 20 | 34 | 22 | 27 | 8 | 7 | 5 |
| Mosaic3 | 123 | 111 | 134 | 30 | 16 | 32 | 35 | 16 | 21 | 12 | 5 | 13 |
| Mosaic4 | 117 | 101 | 121 | 37 | 15 | 24 | 31 | 23 | 27 | 11 | 6 | 6 |
| Mosaic Antigen Set (n = 4) | Subset Counts | Exact Match | Off-By-1 Match |
|---|---|---|---|
| S protein | 534 | 0.8901 | 0.9339 |
| M protein | 485 | 0.8777 | 0.9505 |
| N protein | 410 | 0.9196 | 0.9553 |
| E protein | 478 | 0.9286 | 0.9447 |
| Protein Name | Epitope Verified by Experiment | CD4+/CD8+ T Cell Response | HLA Restriction(s) | Position | Mosaic Sequence |
|---|---|---|---|---|---|
| S protein | EYVSQPFLM [22] | CD8+ | A*11:01; C*07:02 | 169–177 | Seq2 |
| KSTNLVKNK [22] | CD8+ | A*02:01; B*40:01 | 529–537 | Seq2 | |
| N protein | QRNAPRITF [22] | CD8+ | A*11:01; B*40:01; C*07:02 | 9–17 | Seq3 |
| SPRWYFYYL [22] | CD8+ | A*02:01; C*07:02 | 105–113 | Seq3 | |
| YLGTGPEAGL [23] | CD8+ | A*02:01; C*07:02 | 112–121 | Seq3 | |
| KTFPPTEPK [22] | CD8+ | A*11:01; C*07:02 | 361–369 | Seq3 | |
| M protein | PKEITVATSRTLSYYKL [22] | CD4+ | DRB1*07:01; | 164–180 | Seq1 |
| FLWLLWPVTL [23] | CD8+ | A*02:01 | 53–62 | Seq4 | |
| FLFLTWICL [23] | CD8+ | A*02:01 | 26–34 | Seq4 | |
| KLLEQWNL [23] | CD8+ | A*02:01 | 15–22 | Seq4 | |
| E protein | FLLVTLAIL [23] | CD8+ | A*02:01 | 26–34 | Seq4 |
| SEETGTLIVNSVLLF [24] | CD4+ | DQA1*0501; DQB1*0301 | 6–20 | Seq4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Zeng, J.; Li, R.; Wen, Z.; Cai, Y.; Wallin, J.; Shu, Y.; Du, X.; Sun, C. Rational Design of a Pan-Coronavirus Vaccine Based on Conserved CTL Epitopes. Viruses 2021, 13, 333. https://doi.org/10.3390/v13020333
Li M, Zeng J, Li R, Wen Z, Cai Y, Wallin J, Shu Y, Du X, Sun C. Rational Design of a Pan-Coronavirus Vaccine Based on Conserved CTL Epitopes. Viruses. 2021; 13(2):333. https://doi.org/10.3390/v13020333
Chicago/Turabian StyleLi, Minchao, Jinfeng Zeng, Ruiting Li, Ziyu Wen, Yanhui Cai, Jeffrey Wallin, Yuelong Shu, Xiangjun Du, and Caijun Sun. 2021. "Rational Design of a Pan-Coronavirus Vaccine Based on Conserved CTL Epitopes" Viruses 13, no. 2: 333. https://doi.org/10.3390/v13020333
APA StyleLi, M., Zeng, J., Li, R., Wen, Z., Cai, Y., Wallin, J., Shu, Y., Du, X., & Sun, C. (2021). Rational Design of a Pan-Coronavirus Vaccine Based on Conserved CTL Epitopes. Viruses, 13(2), 333. https://doi.org/10.3390/v13020333