
HIV-1 Infection Transcriptomics: Meta-Analysis of CD4+ T Cells Gene Expression Profiles

 , , , , ,
, , , , ,  , and
, and
Abstract
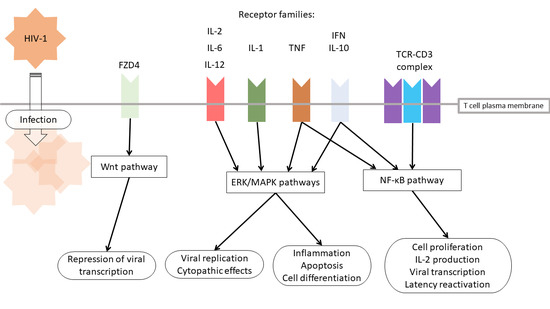
1. Introduction
2. Materials and Methods
2.1. Study Search Strategy
2.2. RNA-Seq Data Collection, Processing and Meta-Analysis
2.3. Gene Ontology Enrichment Analysis
2.4. Cross-Referencing and Set Analysis of DEGs
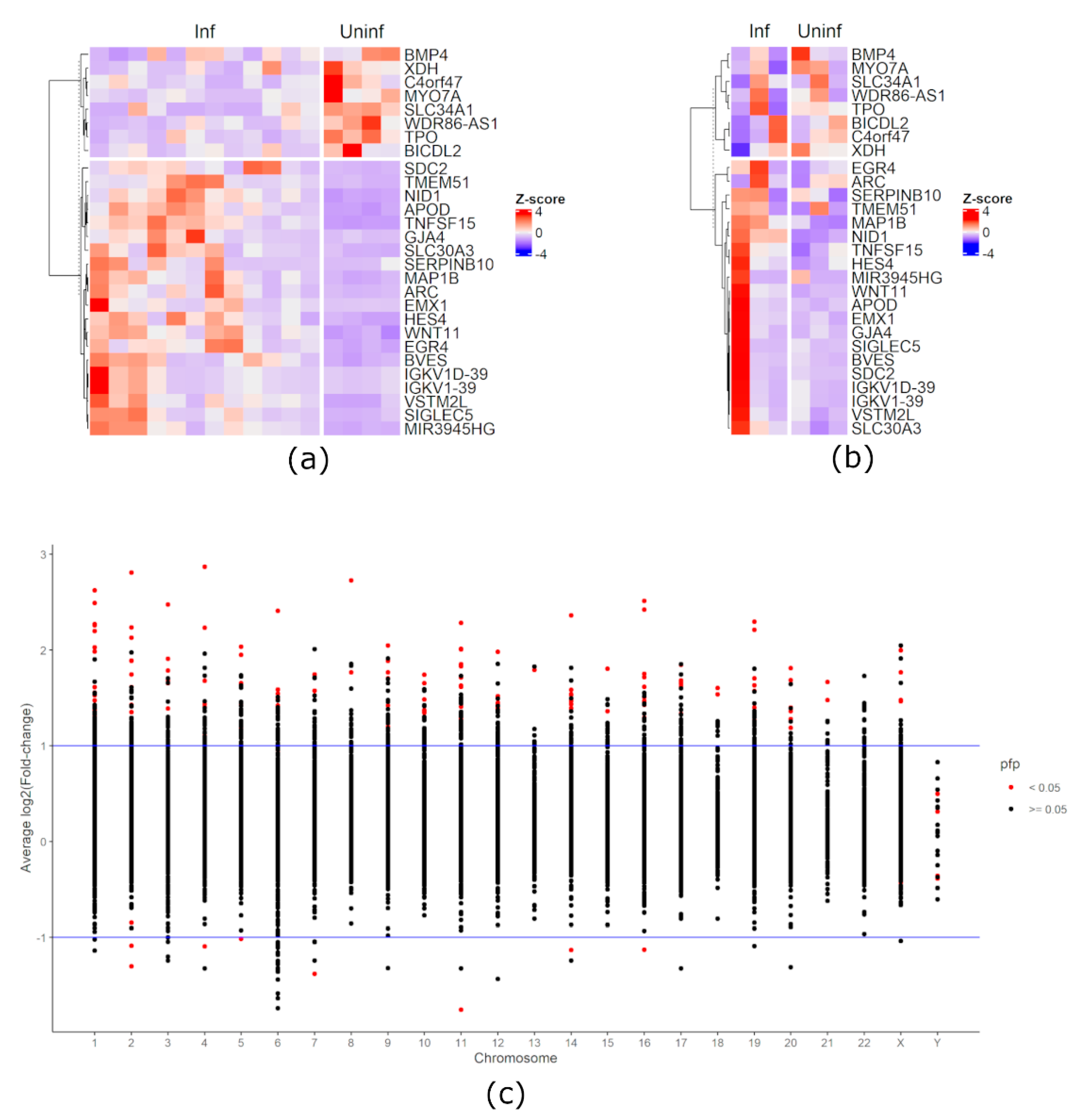
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Summary of the Global HIV Epidemic. 2019. Available online: https://www.who.int/hiv/data/en/ (accessed on 10 September 2020).
- Le Clerc, S.; Limou, S.; Zagury, J.F. Large-Scale “OMICS” Studies to Explore the Physiopatholgy of HIV-1 Infection. Front. Genet. 2019, 10, 799. [Google Scholar] [CrossRef]
- Saraiva-Agostinho, N.; Barbosa-Morais, N.L. Psichomics: Graphical application for alternative splicing quantification and analysis. Nucleic Acids Res. 2019, 47, e7. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 3 July 2019).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Lee, S.Y.; Park, Y.K.; Yoon, C.H.; Kim, K.; Kim, K.C. Meta-analysis of gene expression profiles in long-term non-progressors infected with HIV-1. BMC Med. Genom. 2019, 12, 3. [Google Scholar] [CrossRef]
- Gentleman, R. Annotate: Annotation for Microarrays; R Package Version 1.62.0; Bioconductor: Seattle, WA, USA, 2019. [Google Scholar]
- Carlson, M. Org.Hs.eg.db: Genome Wide Annotation for Human; R Package Version 3.8.2; Bioconductor: Seattle, WA, USA, 2019. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Konig, R.; Zhou, Y.; Elleder, D.; Diamond, T.L.; Bonamy, G.M.; Irelan, J.T.; Chiang, C.Y.; Tu, B.P.; De Jesus, P.D.; Lilley, C.E.; et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 2008, 135, 49–60. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, M.; Huang, Q.; Gates, A.T.; Zhang, X.D.; Castle, J.C.; Stec, E.; Ferrer, M.; Strulovici, B.; Hazuda, D.J.; et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008, 4, 495–504. [Google Scholar] [CrossRef]
- Yeung, M.L.; Houzet, L.; Yedavalli, V.S.; Jeang, K.T. A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. J. Biol. Chem. 2009, 284, 19463–19473. [Google Scholar] [CrossRef]
- Ptak, R.G.; Fu, W.; Sanders-Beer, B.E.; Dickerson, J.E.; Pinney, J.W.; Robertson, D.L.; Rozanov, M.N.; Katz, K.S.; Maglott, D.R.; Pruitt, K.D.; et al. Cataloguing the HIV type 1 human protein interaction network. AIDS Res. Hum. Retrovir. 2008, 24, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Sanders-Beer, B.E.; Katz, K.S.; Maglott, D.R.; Pruitt, K.D.; Ptak, R.G. Human immunodeficiency virus type 1, human protein interaction database at NCBI. Nucleic Acids Res. 2009, 37, D417–D422. [Google Scholar] [CrossRef] [PubMed]
- Pinney, J.W.; Dickerson, J.E.; Fu, W.; Sanders-Beer, B.E.; Ptak, R.G.; Robertson, D.L. HIV-host interactions: A map of viral perturbation of the host system. AIDS 2009, 23, 549–554. [Google Scholar] [CrossRef] [PubMed]
- The RNAcentral Consortium. RNAcentral: A hub of information for non-coding RNA sequences. Nucleic Acids Res. 2019, 47, D221–D229. [Google Scholar] [CrossRef] [PubMed]
- FitzJohn, R. Ids: Generate Random Identifiers; R Package Version 1.0.1; R Foundation for Statistical Computing: London, UK, 2017. [Google Scholar]
- Langer, S.; Hammer, C.; Hopfensperger, K.; Klein, L.; Hotter, D.; De Jesus, P.D.; Herbert, K.M.; Pache, L.; Smith, N.; van der Merwe, J.A.; et al. HIV-1 Vpu is a potent transcriptional suppressor of NF-kappaB-elicited antiviral immune responses. eLife 2019, 8. [Google Scholar] [CrossRef]
- Shytaj, I.L.; Lucic, B.; Forcato, M.; Penzo, C.; Billingsley, J.; Laketa, V.; Bosinger, S.; Stanic, M.; Gregoretti, F.; Antonelli, L.; et al. Alterations of redox and iron metabolism accompany the development of HIV latency. EMBO J. 2020, 39, e102209. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Veloso Carvalho-Silva, W.H.; Andrade-Santos, J.L.; Dos Santos Guedes, M.C.; Guimaraes, R.L. Genetics and immunological recovery with antiretroviral treatment for HIV. Pharmacogenomics 2020, 21, 979–983. [Google Scholar] [CrossRef]
- Sherrill-Mix, S.; Ocwieja, K.E.; Bushman, F.D. Gene activity in primary T cells infected with HIV89.6: Intron retention and induction of genomic repeats. Retrovirology 2015, 12, 79. [Google Scholar] [CrossRef] [PubMed]
- Meas, H.Z.; Haug, M.; Beckwith, M.S.; Louet, C.; Ryan, L.; Hu, Z.; Landskron, J.; Nordbo, S.A.; Tasken, K.; Yin, H.; et al. Sensing of HIV-1 by TLR8 activates human T cells and reverses latency. Nat. Commun. 2020, 11, 147. [Google Scholar] [CrossRef] [PubMed]
- Nisole, S.; Stoye, J.P.; Saib, A. TRIM family proteins: Retroviral restriction and antiviral defence. Nat. Rev. Microbiol. 2005, 3, 799–808. [Google Scholar] [CrossRef] [PubMed]
- van Gent, M.; Sparrer, K.M.J.; Gack, M.U. TRIM Proteins and Their Roles in Antiviral Host Defenses. Annu. Rev. Virol. 2018, 5, 385–405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Jouanguy, E.; Sancho-Shimizu, V.; von Bernuth, H.; Yang, K.; Abel, L.; Picard, C.; Puel, A.; Casanova, J.L. Human Toll-like receptor-dependent induction of interferons in protective immunity to viruses. Immunol. Rev. 2007, 220, 225–236. [Google Scholar] [CrossRef]
- Schlaepfer, E.; Speck, R.F. TLR8 activates HIV from latently infected cells of myeloid-monocytic origin directly via the MAPK pathway and from latently infected CD4+ T cells indirectly via TNF-alpha. J. Immunol. 2011, 186, 4314–4324. [Google Scholar] [CrossRef]
- Atluri, V.S.; Pilakka-Kanthikeel, S.; Garcia, G.; Jayant, R.D.; Sagar, V.; Samikkannu, T.; Yndart, A.; Nair, M. Effect of Cocaine on HIV Infection and Inflammasome Gene Expression Profile in HIV Infected Macrophages. Sci. Rep. 2016, 6, 27864. [Google Scholar] [CrossRef]
- Carrol, E.D.; Mankhambo, L.A.; Balmer, P.; Nkhoma, S.; Banda, D.L.; Guiver, M.; Jeffers, G.; Makwana, N.; Molyneux, E.M.; Molyneux, M.E.; et al. Chemokine responses are increased in HIV-infected Malawian children with invasive pneumococcal disease. J. Acquir. Immune Defic. Syndr. 2007, 44, 443–450. [Google Scholar] [CrossRef]
- Chang, C.C.; Omarjee, S.; Lim, A.; Spelman, T.; Gosnell, B.I.; Carr, W.H.; Elliott, J.H.; Moosa, M.Y.; Ndung’u, T.; French, M.A.; et al. Chemokine levels and chemokine receptor expression in the blood and the cerebrospinal fluid of HIV-infected patients with cryptococcal meningitis and cryptococcosis-associated immune reconstitution inflammatory syndrome. J. Infect. Dis. 2013, 208, 1604–1612. [Google Scholar] [CrossRef]
- Furler, R.L.; Uittenbogaart, C.H. Signaling through the P38 and ERK pathways: A common link between HIV replication and the immune response. Immunol. Res. 2010, 48, 99–109. [Google Scholar] [CrossRef]
- Medders, K.E.; Kaul, M. Mitogen-activated protein kinase p38 in HIV infection and associated brain injury. J. Neuroimmune Pharmacol. 2011, 6, 202–215. [Google Scholar] [CrossRef]
- Kumar, A.; Zloza, A.; Moon, R.T.; Watts, J.; Tenorio, A.R.; Al-Harthi, L. Active beta-catenin signaling is an inhibitory pathway for human immunodeficiency virus replication in peripheral blood mononuclear cells. J. Virol. 2008, 82, 2813–2820. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Mukerjee, R.; Ferrante, P.; Khalili, K.; Amini, S.; Sawaya, B.E. Human immunodeficiency virus type 1 Tat prevents dephosphorylation of Sp1 by TCF-4 in astrocytes. J. Gen. Virol. 2006, 87, 1613–1623. [Google Scholar] [CrossRef]
- Henderson, L.J.; Al-Harthi, L. Role of beta-catenin/TCF-4 signaling in HIV replication and pathogenesis: Insights to informing novel anti-HIV molecular therapeutics. J. Neuroimmune Pharmacol. 2011, 6, 247–259. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kulpa, D.A.; Brehm, J.H.; Fromentin, R.; Cooper, A.; Cooper, C.; Ahlers, J.; Chomont, N.; Sekaly, R.P. The immunological synapse: The gateway to the HIV reservoir. Immunol. Rev. 2013, 254, 305–325. [Google Scholar] [CrossRef] [PubMed]
- Salgado, M.; Lopez-Romero, P.; Callejas, S.; Lopez, M.; Labarga, P.; Dopazo, A.; Soriano, V.; Rodes, B. Characterization of host genetic expression patterns in HIV-infected individuals with divergent disease progression. Virology 2011, 411, 103–112. [Google Scholar] [CrossRef]
- Seetharaman, S.; Etienne-Manneville, S. Cytoskeletal Crosstalk in Cell Migration. Trends Cell Biol. 2020, 30, 720–735. [Google Scholar] [CrossRef] [PubMed]
- Ospina Stella, A.; Turville, S. All-Round Manipulation of the Actin Cytoskeleton by HIV. Viruses 2018, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Huertas, M.R.; Callejas, S.; Abia, D.; Mateos, E.; Dopazo, A.; Alcami, J.; Coiras, M. Modifications in host cell cytoskeleton structure and function mediated by intracellular HIV-1 Tat protein are greatly dependent on the second coding exon. Nucleic Acids Res. 2010, 38, 3287–3307. [Google Scholar] [CrossRef]
- Puri, R.V.; Yerrathota, S.; Home, T.; Idowu, J.Y.; Chakravarthi, V.P.; Ward, C.J.; Singhal, P.C.; Vanden Heuvel, G.B.; Fields, T.A.; Sharma, M. Notch4 activation aggravates NF-kappaB-mediated inflammation in HIV-1-associated nephropathy. Dis. Models Mech. 2019, 12. [Google Scholar] [CrossRef]
- Stroud, J.C.; Oltman, A.; Han, A.; Bates, D.L.; Chen, L. Structural basis of HIV-1 activation by NF-kappaB—A higher-order complex of p50:RelA bound to the HIV-1 LTR. J. Mol. Biol. 2009, 393, 98–112. [Google Scholar] [CrossRef]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef]
- Khoury, G.; Darcis, G.; Lee, M.Y.; Bouchat, S.; Van Driessche, B.; Purcell, D.F.J.; Van Lint, C. The Molecular Biology of HIV Latency. Adv. Exp. Med. Biol. 2018, 1075, 187–212. [Google Scholar]
- Campos Coelho, A.V.; Moura, R.R.; Crovella, S. Reanalysis of Gene Expression Profiles of CD4+ T Cells Treated with HIV-1 Latency Reversal Agents. Microorganisms 2020, 8, 1505. [Google Scholar] [CrossRef]
- Hataye, J.M.; Casazza, J.P.; Best, K.; Liang, C.J.; Immonen, T.T.; Ambrozak, D.R.; Darko, S.; Henry, A.R.; Laboune, F.; Maldarelli, F.; et al. Principles Governing Establishment versus Collapse of HIV-1 Cellular Spread. Cell Host Microbe 2019, 26, 748–763.e20. [Google Scholar] [CrossRef]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Telwatte, S.; Moron-Lopez, S.; Aran, D.; Kim, P.; Hsieh, C.; Joshi, S.; Montano, M.; Greene, W.C.; Butte, A.J.; Wong, J.K.; et al. Heterogeneity in HIV and cellular transcription profiles in cell line models of latent and productive infection: Implications for HIV latency. Retrovirology 2019, 16, 32. [Google Scholar] [CrossRef] [PubMed]
- Spector, C.; Mele, A.R.; Wigdahl, B.; Nonnemacher, M.R. Genetic variation and function of the HIV-1 Tat protein. Med. Microbiol. Immunol. 2019, 208, 131–169. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Study | PRJ, GSE IDs | Selected Samples | Control:Infected Ratio |
|---|---|---|---|
| Langer et al., 2019 [24] | PRJNA482835, GSE117655 | 16 | 1:3 |
| Shytaj et al., 2020 [25] | PRJNA524856, GSE127468 | 6 | 1:1 |
| Reference or Database | Unique Genes | Observed Intersection | Expected Intersection | p-Value |
|---|---|---|---|---|
| Konig et al., 2008 [16] a | 406 | 4 | 3 | 0.35 |
| Zhou et al., 2008 [17] | 264 | 2 | 2 | 0.60 |
| Yeung et al., 2009 [18] | 262 | 3 | 2 | 0.32 |
| HIV interaction database [19,20,21] b | 4667 | 64 | 34 | <0.001 |
| RNAcentral [22] b | 7972 | 3 | 58 | 1.00 |
| Pathways and Ranks | GO ID | Term | FDR-Adjusted p-Value |
|---|---|---|---|
| Immune response | |||
| 1 | GO:0006958 | complement activation, classical pathway | 6.4 × 10−46 |
| 9 | GO:0002449 | lymphocyte mediated immunity | 1.2 × 10−41 |
| 12 | GO:0006955 | immune response | 7.8 × 10−39 |
| Cell proliferation | |||
| 97 | GO:0008283 | cell population proliferation | 2.9 × 10−14 |
| 116 | GO:0042127 | regulation of cell population proliferation | 5.0 × 10−12 |
| 351 | GO:0046651 | lymphocyte proliferation | 0.00004 |
| Cell adhesion | |||
| 151 | GO:0007155 | cell adhesion | 5.9 × 10−9 |
| 826 | GO:0007159 | leukocyte cell–cell adhesion | 0.0136 |
| Cell migration | |||
| 23 | GO:0016477 | cell migration | 1.2 × 10−31 |
| 39 | GO:0050900 | leukocyte migration | 2.1 × 10−28 |
| Apoptosis | |||
| 216 | GO:0012501 | programmed cell death | 1.1 × 10−6 |
| 225 | GO:0008219 | cell death | 1.7 × 10−6 |
| 234 | GO:0042981 | regulation of apoptotic process | 2.5 × 10−6 |
| Inflammation | |||
| 259 | GO:0050727 | regulation of inflammatory response | 5.1 × 10−6 |
| 611 | GO:0002526 | acute inflammatory response | 0.001 |
| Wnt signaling | |||
| 441 | GO:0016055 | Wnt signaling pathway | 0.0002 |
| 444 | GO:0198738 | cell–cell signaling by wnt | 0.0002 |
| 580 | GO:0060070 | canonical Wnt signaling pathway | 0.0010 |
| Notch signaling | |||
| 591 | GO:0008593 | regulation of Notch signaling pathway | 0.001 |
| 688 | GO:0007219 | Notch signaling pathway | 0.003 |
| ERK/MAPK signaling | |||
| 648 | GO:0000165 | MAPK cascade | 0.002 |
| 691 | GO:0043406 | positive regulation of MAP kinase activity | 0.003 |
| 886 | GO:0070371 | ERK1 and ERK2 cascade | 0.031 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coelho, A.V.C.; Gratton, R.; Melo, J.P.B.d.; Andrade-Santos, J.L.; Guimarães, R.L.; Crovella, S.; Tricarico, P.M.; Brandão, L.A.C. HIV-1 Infection Transcriptomics: Meta-Analysis of CD4+ T Cells Gene Expression Profiles. Viruses 2021, 13, 244. https://doi.org/10.3390/v13020244
Coelho AVC, Gratton R, Melo JPBd, Andrade-Santos JL, Guimarães RL, Crovella S, Tricarico PM, Brandão LAC. HIV-1 Infection Transcriptomics: Meta-Analysis of CD4+ T Cells Gene Expression Profiles. Viruses. 2021; 13(2):244. https://doi.org/10.3390/v13020244
Chicago/Turabian StyleCoelho, Antonio Victor Campos, Rossella Gratton, João Paulo Britto de Melo, José Leandro Andrade-Santos, Rafael Lima Guimarães, Sergio Crovella, Paola Maura Tricarico, and Lucas André Cavalcanti Brandão. 2021. "HIV-1 Infection Transcriptomics: Meta-Analysis of CD4+ T Cells Gene Expression Profiles" Viruses 13, no. 2: 244. https://doi.org/10.3390/v13020244
APA StyleCoelho, A. V. C., Gratton, R., Melo, J. P. B. d., Andrade-Santos, J. L., Guimarães, R. L., Crovella, S., Tricarico, P. M., & Brandão, L. A. C. (2021). HIV-1 Infection Transcriptomics: Meta-Analysis of CD4+ T Cells Gene Expression Profiles. Viruses, 13(2), 244. https://doi.org/10.3390/v13020244