
Multiscale Electron Microscopy for the Study of Viral Replication Organelles


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
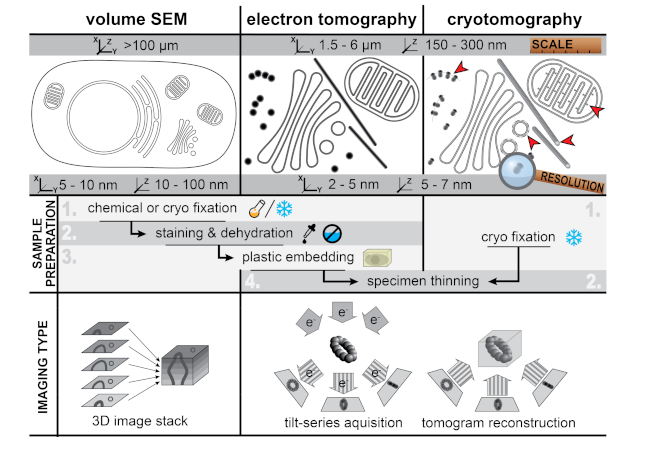
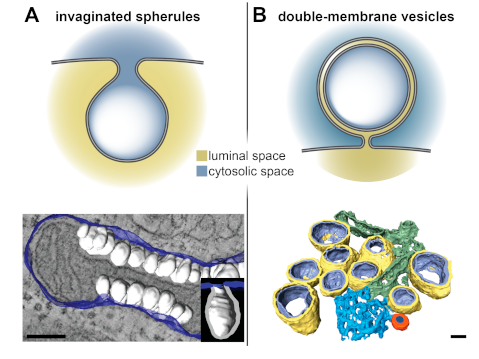
2. Viral ROs by Conventional 2D and 3D Transmission Electron Microscopy
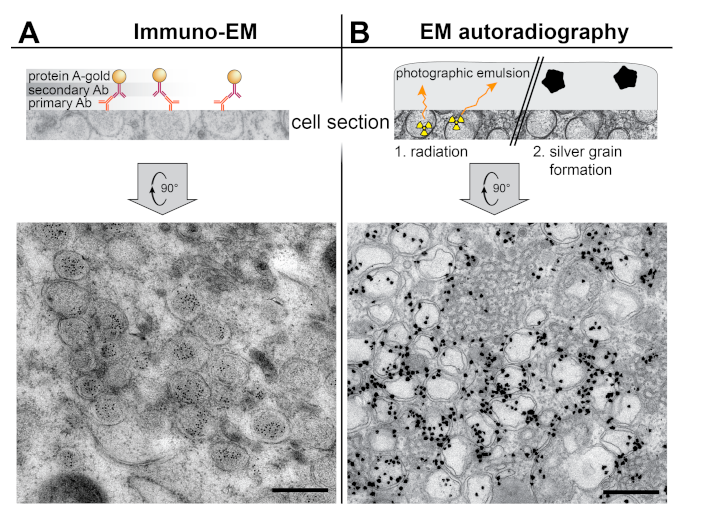
3. Localization of Molecular Factors by Labeling
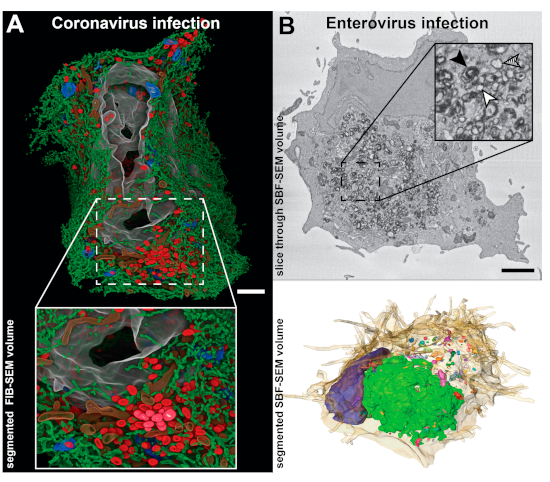
4. Zooming Out—Volume Scanning Electron Microscopy
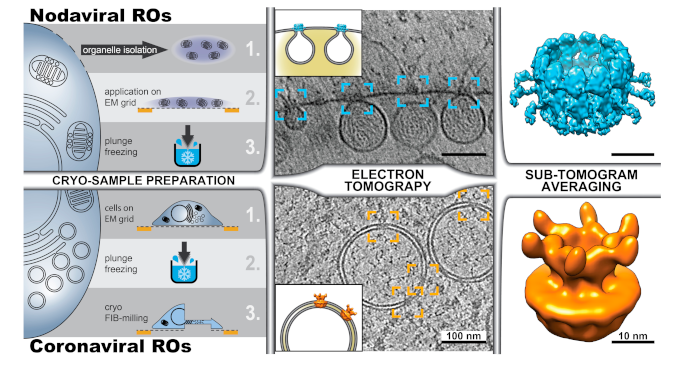
5. Zooming In—Cryo-Electron Microscopy
6. Concluding Remarks and Outlook
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kruger, D.H.; Schneck, P.; Gelderblom, H.R. Helmut Ruska and the visualisation of viruses. Lancet 2000, 355, 1713–1717. [Google Scholar] [CrossRef]
- Masters, B.R. History of the Electron Microscope in Cell Biology. In Enciclopedia of Life Sciences (ELS); John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2009. [Google Scholar]
- Kaelber, J.T.; Hryc, C.F.; Chiu, W. Electron Cryomicroscopy of Viruses at Near-Atomic Resolutions. Annu. Rev. Virol. 2017, 4, 287–308. [Google Scholar] [CrossRef] [PubMed]
- Bozzola, J.J.; Russell, L.D. Electron Microscopy: Principles and Techniques for Biologists; Jones and Bartlett Publishers, Inc.: Sudbury, MA, USA, 1999. [Google Scholar]
- Koning, R.I.; Koster, A.J.; Sharp, T.H. Advances in cryo-electron tomography for biology and medicine. Ann. Anat. 2018, 217, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Dales, S.; Franklin, R.M. A comparison of the changes in fine structure of L cells during single cycles of viral multiplication, following their infection with the viruses of Mengo and encephalomyocarditis. J. Cell Biol. 1962, 14, 281–302. [Google Scholar] [CrossRef] [PubMed]
- Godman, G.C.; Rifkind, R.A.; Howe, C.; Rose, H.M. A Description of Echo-9 Virus Infection in Cultured Cells. I. The Cytopathic Effect. Am. J. Pathol. 1964, 44, 1–27. [Google Scholar] [PubMed]
- Dales, S.; Eggers, H.J.; Tamm, I.; Palade, G.E. Electron Microscopic Study of the Formation of Poliovirus. Virology 1965, 26, 379–389. [Google Scholar] [CrossRef]
- David-Ferreira, J.F.; Manaker, R.A. An Electron Microscope Study of the Development of a Mouse Hepatitis Virus in Tissue Culture Cells. J. Cell Biol. 1965, 24, 57–78. [Google Scholar] [CrossRef]
- Moor, H.; Riehle, U. Snap-Freezing under High Pressure: A New Fixation Technique for Freeze-Etching. In Proceedings of the Fourth European Regional Conference of Electron Microscopy, Rome, Italy, 1–7 September 1968; pp. 33–34. [Google Scholar]
- McDonald, K. Cryopreparation methods for electron microscopy of selected model systems. Methods Cell Biol. 2007, 79, 23–56. [Google Scholar]
- Sosinsky, G.E.; Crum, J.; Jones, Y.Z.; Lanman, J.; Smarr, B.; Terada, M.; Martone, M.E.; Deerinck, T.J.; Johnson, J.E.; Ellisman, M.H. The combination of chemical fixation procedures with high pressure freezing and freeze substitution preserves highly labile tissue ultrastructure for electron tomography applications. J. Struct. Biol. 2008, 161, 359–371. [Google Scholar] [CrossRef]
- Murk, J.L.; Posthuma, G.; Koster, A.J.; Geuze, H.J.; Verkleij, A.J.; Kleijmeer, M.J.; Humbel, B.M. Influence of aldehyde fixation on the morphology of endosomes and lysosomes: Quantitative analysis and electron tomography. J. Microsc. 2003, 212 Pt 1, 81–90. [Google Scholar] [CrossRef]
- Kopek, B.G.; Perkins, G.; Miller, D.J.; Ellisman, M.H.; Ahlquist, P. Three-dimensional analysis of a viral RNA replication complex reveals a virus-induced mini-organelle. PLoS Biol. 2007, 5, e220. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Kikkert, M.; Worm, S.H.; Zevenhoven-Dobbe, J.C.; Van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The endoplasmic reticulum provides the membrane platform for biogenesis of the flavivirus replication complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef]
- Offerdahl, D.K.; Dorward, D.W.; Hansen, B.T.; Bloom, M.E. A three-dimensional comparison of tick-borne flavivirus infection in mammalian and tick cell lines. PLoS ONE 2012, 7, e47912. [Google Scholar] [CrossRef]
- Miorin, L.; Romero-Brey, I.; Maiuri, P.; Hoppe, S.; Krijnse-Locker, J.; Bartenschlager, R.; Marcello, A. Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. J. Virol. 2013, 87, 6469–6481. [Google Scholar] [CrossRef]
- Schwartz, M.; Chen, J.; Janda, M.; Sullivan, M.; Den Boon, J.; Ahlquist, P. A positive-strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol. Cell 2002, 9, 505–514. [Google Scholar] [CrossRef]
- Cao, X.; Jin, X.; Zhang, X.; Li, Y.; Wang, C.; Wang, X.; Hong, J.; Wang, X.; Li, D.; Zhang, Y. Morphogenesis of Endoplasmic Reticulum Membrane-Invaginated Vesicles during Beet Black Scorch Virus Infection: Role of Auxiliary Replication Protein and New Implications of Three-Dimensional Architecture. J. Virol. 2015, 89, 6184–6195. [Google Scholar] [CrossRef]
- De Castro, I.F.; Fernandez, J.J.; Barajas, D.; Nagy, P.D.; Risco, C. Three-dimensional imaging of the intracellular assembly of a functional viral RNA replicase complex. J. Cell Sci. 2017, 130, 260–268. [Google Scholar] [CrossRef]
- Grimley, P.M.; Berezesky, I.K.; Friedman, R.M. Cytoplasmic structures associated with an arbovirus infection: Loci of viral ribonucleic acid synthesis. J. Virol. 1968, 2, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Froshauer, S.; Kartenbeck, J.; Helenius, A. Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. J. Cell Biol. 1988, 107, 2075–2086. [Google Scholar] [CrossRef] [PubMed]
- Kujala, P.; Ikaheimonen, A.; Ehsani, N.; Vihinen, H.; Auvinen, P.; Kaariainen, L. Biogenesis of the Semliki Forest virus RNA replication complex. J. Virol. 2001, 75, 3873–3884. [Google Scholar] [CrossRef] [PubMed]
- Fontana, J.; Lopez-Iglesias, C.; Tzeng, W.P.; Frey, T.K.; Fernandez, J.J.; Risco, C. Three-dimensional structure of Rubella virus factories. Virology 2010, 405, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Frolova, E.I.; Gorchakov, R.; Pereboeva, L.; Atasheva, S.; Frolov, I. Functional Sindbis virus replicative complexes are formed at the plasma membrane. J. Virol. 2010, 84, 11679–11695. [Google Scholar] [CrossRef]
- Schlegel, A.; Giddings, T.H., Jr.; Ladinsky, M.S.; Kirkegaard, K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol. 1996, 70, 6576–6588. [Google Scholar] [CrossRef]
- Limpens, R.W.; Van der Schaar, H.M.; Kumar, D.; Koster, A.J.; Snijder, E.J.; Van Kuppeveld, F.J.; Bárcena, M. The transformation of enterovirus replication structures: A three-dimensional study of single- and double-membrane compartments. mBio 2011, 2. [Google Scholar] [CrossRef]
- Belov, G.A.; Nair, V.; Hansen, B.T.; Hoyt, F.H.; Fischer, E.R.; Ehrenfeld, E. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 2012, 86, 302–312. [Google Scholar] [CrossRef]
- Melia, C.E.; Van der Schaar, H.M.; De Jong, A.W.M.; Lyoo, H.R.; Snijder, E.J.; Koster, A.J.; Van Kuppeveld, F.J.M.; Bárcena, M. The Origin, Dynamic Morphology, and PI4P-Independent Formation of Encephalomyocarditis Virus Replication Organelles. mBio 2018, 9, e00420-18. [Google Scholar] [CrossRef]
- Hyde, J.L.; Sosnovtsev, S.V.; Green, K.Y.; Wobus, C.; Virgin, H.W.; Mackenzie, J.M. Mouse norovirus replication is associated with virus-induced vesicle clusters originating from membranes derived from the secretory pathway. J. Virol. 2009, 83, 9709–9719. [Google Scholar] [CrossRef]
- Doerflinger, S.Y.; Cortese, M.; Romero-Brey, I.; Menne, Z.; Tubiana, T.; Schenk, C.; White, P.A.; Bartenschlager, R.; Bressanelli, S.; Hansman, G.S.; et al. Membrane alterations induced by nonstructural proteins of human norovirus. PLoS Pathog. 2017, 13, e1006705. [Google Scholar] [CrossRef]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.W.; Van der Meer, Y.; Roos, N.; Snijder, E.J. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J. Virol. 1999, 73, 2016–2026. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Bárcena, M.; Limpens, R.W.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. Ultrastructural characterization of arterivirus replication structures: Reshaping the endoplasmic reticulum to accommodate viral RNA synthesis. J. Virol. 2012, 86, 2474–2487. [Google Scholar] [CrossRef]
- Gosert, R.; Kanjanahaluethai, A.; Egger, D.; Bienz, K.; Baker, S.C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 2002, 76, 3697–3708. [Google Scholar] [CrossRef] [PubMed]
- Maier, H.J.; Hawes, P.C.; Cottam, E.M.; Mantell, J.; Verkade, P.; Monaghan, P.; Wileman, T.; Britton, P. Infectious bronchitis virus generates spherules from zippered endoplasmic reticulum membranes. mBio 2013, 4, e00801-13. [Google Scholar] [CrossRef]
- Snijder, E.J.; Limpens, R.W.A.L.; De Wilde, A.H.; De Jong, A.W.M.; Zevenhoven-Dobbe, J.C.; Maier, H.J.; Faas, F.; Koster, A.J.; Bárcena, M. A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol. 2020, 18, e3000715. [Google Scholar] [CrossRef]
- Cortese, M.; Lee, J.Y.; Cerikan, B.; Neufeldt, C.J.; Oorschot, V.M.J.; Kohrer, S.; Hennies, J.; Schieber, N.L.; Ronchi, P.; Mizzon, G.; et al. Integrative Imaging Reveals SARS-CoV-2-Induced Reshaping of Subcellular Morphologies. Cell Host Microbe 2020, 28, 853–866. [Google Scholar] [CrossRef]
- Wolff, G.; Melia, C.E.; Snijder, E.J.; Bárcena, M. Double-Membrane Vesicles as Platforms for Viral Replication. Trends Microbiol. 2020, 28, 1022–1033. [Google Scholar] [CrossRef]
- Van der Hoeven, B.; Oudshoorn, D.; Koster, A.J.; Snijder, E.J.; Kikkert, M.; Bárcena, M. Biogenesis and architecture of arterivirus replication organelles. Virus Res. 2016, 220, 70–90. [Google Scholar] [CrossRef]
- Bykov, Y.S.; Cortese, M.; Briggs, J.A.; Bartenschlager, R. Correlative light and electron microscopy methods for the study of virus-cell interactions. FEBS Lett. 2016, 590, 1877–1895. [Google Scholar] [CrossRef]
- Winey, M.; Meehl, J.B.; O’Toole, E.T.; Giddings, T.H., Jr. Conventional transmission electron microscopy. Mol. Biol. Cell 2014, 25, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Möbius, W.; Posthuma, G. Sugar and ice: Immunoelectron microscopy using cryosections according to the Tokuyasu method. Tissue Cell 2019, 57, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Bienz, K.; Egger, D.; Pasamontes, L. Association of polioviral proteins of the P2 genomic region with the viral replication complex and virus-induced membrane synthesis as visualized by electron microscopic immunocytochemistry and autoradiography. Virology 1987, 160, 220–226. [Google Scholar] [CrossRef]
- Romero-Brey, I.; Bartenschlager, R. Membranous replication factories induced by plus-strand RNA viruses. Viruses 2014, 6, 2826–2857. [Google Scholar] [CrossRef]
- Quinkert, D.; Bartenschlager, R.; Lohmann, V. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 2005, 79, 13594–13605. [Google Scholar] [CrossRef]
- Bienz, K.; Egger, D.; Rasser, Y.; Bossart, W. Kinetics and location of poliovirus macromolecular synthesis in correlation to virus-induced cytopathology. Virology 1980, 100, 390–399. [Google Scholar] [CrossRef]
- Melia, C.E.; Van der Schaar, H.M.; Lyoo, H.; Limpens, R.; Feng, Q.; Wahedi, M.; Overheul, G.J.; Van Rij, R.P.; Snijder, E.J.; Koster, A.J.; et al. Escaping Host Factor PI4KB Inhibition: Enterovirus Genomic RNA Replication in the Absence of Replication Organelles. Cell Rep. 2017, 21, 587–599. [Google Scholar] [CrossRef]
- Bienz, K.A. Techniques and applications of autoradiography in the light and electron microscope. Microsc. Acta 1977, 79, 1–22. [Google Scholar]
- Ulasli, M.; Verheije, M.H.; De Haan, C.A.; Reggiori, F. Qualitative and quantitative ultrastructural analysis of the membrane rearrangements induced by coronavirus. Cell. Microbiol. 2010, 12, 844–861. [Google Scholar] [CrossRef]
- De Wilde, A.H.; Raj, V.S.; Oudshoorn, D.; Bestebroer, T.M.; Van Nieuwkoop, S.; Limpens, R.W.; Posthuma, C.C.; Van der Meer, Y.; Bárcena, M.; Haagmans, B.L.; et al. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-alpha treatment. J. Gen. Virol. 2013, 94 Pt 8, 1749–1760. [Google Scholar] [CrossRef]
- Zhou, X.; Cong, Y.; Veenendaal, T.; Klumperman, J.; Shi, D.; Mari, M.; Reggiori, F. Ultrastructural Characterization of Membrane Rearrangements Induced by Porcine Epidemic Diarrhea Virus Infection. Viruses 2017, 9, 251. [Google Scholar] [CrossRef] [PubMed]
- Doyle, N.; Hawes, P.C.; Simpson, J.; Adams, L.H.; Maier, H.J. The Porcine Deltacoronavirus Replication Organelle Comprises Double-Membrane Vesicles and Zippered Endoplasmic Reticulum with Double-Membrane Spherules. Viruses 2019, 11, 1030. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Hoppe, S.; Saher, G.; Krijnse-Locker, J.; Bartenschlager, R. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J. Virol. 2013, 87, 10612–10627. [Google Scholar] [CrossRef] [PubMed]
- Marsh, B.J. Reconstructing mammalian membrane architecture by large area cellular tomography. Methods Cell Biol. 2007, 79, 193–220. [Google Scholar] [PubMed]
- Peddie, C.J.; Collinson, L.M. Exploring the third dimension: Volume electron microscopy comes of age. Micron 2014, 61, 9–19. [Google Scholar] [CrossRef]
- Melia, C.E.; Peddie, C.J.; De Jong, A.W.M.; Snijder, E.J.; Collinson, L.M.; Koster, A.J.; Van der Schaar, H.M.; Van Kuppeveld, F.J.M.; Bárcena, M. Origins of Enterovirus Replication Organelles Established by Whole-Cell Electron Microscopy. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Viktorova, E.G.; Nchoutmboube, J.A.; Ford-Siltz, L.A.; Iverson, E.; Belov, G.A. Phospholipid synthesis fueled by lipid droplets drives the structural development of poliovirus replication organelles. PLoS Pathog. 2018, 14, e1007280. [Google Scholar] [CrossRef]
- Laufman, O.; Perrino, J.; Andino, R. Viral Generated Inter-Organelle Contacts Redirect Lipid Flux for Genome Replication. Cell 2019, 178, 275–289. [Google Scholar] [CrossRef]
- Dubochet, J.; Mcdowall, A.W. Vitrification of Pure Water for Electron-Microscopy. J. Microsc. 1981, 124, 3–4. [Google Scholar] [CrossRef]
- Dubochet, J.; Adrian, M.; Chang, J.J.; Homo, J.C.; Lepault, J.; McDowall, A.W.; Schultz, P. Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys. 1988, 21, 129–228. [Google Scholar] [CrossRef]
- Cheng, Y. Single-particle cryo-EM-How did it get here and where will it go. Science 2018, 361, 876–880. [Google Scholar] [CrossRef]
- Callaway, E. Revolutionary cryo-EM is taking over structural biology. Nature 2020, 578, 201. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Cerikan, B.; et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020, 588, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Turonova, B.; Sikora, M.; Schurmann, C.; Hagen, W.J.H.; Welsch, S.; Blanc, F.E.C.; Von Bulow, S.; Gecht, M.; Bagola, K.; Horner, C.; et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208. [Google Scholar] [CrossRef]
- Yao, H.; Song, Y.; Chen, Y.; Wu, N.; Xu, J.; Sun, C.; Zhang, J.; Weng, T.; Zhang, Z.; Wu, Z.; et al. Molecular Architecture of the SARS-CoV-2 Virus. Cell 2020, 183, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Peukes, J.; Xiong, X.; Erlendsson, S.; Qu, K.; Wan, W.; Calder, L.J.; Schraidt, O.; Kummer, S.; Freund, S.M.V.; Krausslich, H.G.; et al. The native structure of the assembled matrix protein 1 of influenza A virus. Nature 2020, 587, 495–498. [Google Scholar] [CrossRef]
- Ertel, K.J.; Benefield, D.; Castano-Diez, D.; Pennington, J.G.; Horswill, M.; Den Boon, J.A.; Otegui, M.S.; Ahlquist, P. Cryo-electron tomography reveals novel features of a viral RNA replication compartment. Elife 2017, 6, e25940. [Google Scholar] [CrossRef]
- Unchwaniwala, N.; Zhan, H.; Pennington, J.; Horswill, M.; Den Boon, J.A.; Ahlquist, P. Subdomain cryo-EM structure of nodaviral replication protein A crown complex provides mechanistic insights into RNA genome replication. Proc. Natl. Acad. Sci. USA 2020, 117, 18680–18691. [Google Scholar] [CrossRef]
- Wolff, G.; Limpens, R.; Zevenhoven-Dobbe, J.C.; Laugks, U.; Zheng, S.; De Jong, A.W.M.; Koning, R.I.; Agard, D.A.; Grünewald, K.; Koster, A.J.; et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Science 2020, 369, 1395–1398. [Google Scholar] [CrossRef]
- Castaño-Diez, D.; Zanetti, G. In situ structure determination by subtomogram averaging. Curr. Opin. Struct. Biol. 2019, 58, 68–75. [Google Scholar] [CrossRef]
- McDowall, A.W.; Chang, J.J.; Freeman, R.; Lepault, J.; Walter, C.A.; Dubochet, J. Electron microscopy of frozen hydrated sections of vitreous ice and vitrified biological samples. J. Microsc. 1983, 131 Pt 1, 1–9. [Google Scholar] [CrossRef]
- Al-Amoudi, A.; Studer, D.; Dubochet, J. Cutting artefacts and cutting process in vitreous sections for cryo-electron microscopy. J. Struct. Biol. 2005, 150, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Marko, M.; Hsieh, C.; Moberlychan, W.; Mannella, C.; Frank, J. Focused ion beam milling of vitreous water: Prospects for an alternative to cryo-ultramicrotomy of frozen-hydrated biological samples. J. Microsc. 2006, 222, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Rigort, A.; Bäuerlein, F.J.; Villa, E.; Eibauer, M.; Laugks, T.; Baumeister, W.; Plitzko, J.M. Focused ion beam micromachining of eukaryotic cells for cryoelectron tomography. Proc. Natl. Acad. Sci. USA 2012, 109, 4449–4454. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Schaffer, M.; Fernandez-Busnadiego, R. Cryo-electron tomography-the cell biology that came in from the cold. FEBS Lett. 2017, 591, 2520–2533. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat. Commun. 2020, 11, 5885. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.C.; Graham, R.L.; Lu, X.; Peek, C.T.; Denison, M.R. Coronavirus replicase-reporter fusions provide quantitative analysis of replication and replication complex formation. J. Virol. 2014, 88, 5319–5327. [Google Scholar] [CrossRef]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 2013, 4, e00524-13. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Monastyrska, I.; Griffith, J.; Van der Sluijs, P.; Voortman, J.; Van Bergen en Henegouwen, P.M.; Vonk, A.M.; Rottier, P.J.; Reggiori, F.; De Haan, C.A. Membrane rearrangements mediated by coronavirus nonstructural proteins 3 and 4. Virology 2014, 458-459, 125–135. [Google Scholar] [CrossRef]
- Oudshoorn, D.; Rijs, K.; Limpens, R.; Groen, K.; Koster, A.J.; Snijder, E.J.; Kikkert, M.; Bárcena, M. Expression and Cleavage of Middle East Respiratory Syndrome Coronavirus nsp3-4 Polyprotein Induce the Formation of Double-Membrane Vesicles That Mimic Those Associated with Coronaviral RNA Replication. mBio 2017, 8, e01658-17. [Google Scholar] [CrossRef]
- Unchwaniwala, N.; Ahlquist, P. Coronavirus dons a new crown. Science 2020, 369, 1306–1307. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.; Bragagnolo, G.; Arranz, R.; Reguera, J. Capping pores of alphavirus nsP1 gate membranous viral replication factories. Nature 2020, 37, 4175. [Google Scholar]
- Felts, R.L.; Narayan, K.; Estes, J.D.; Shi, D.; Trubey, C.M.; Fu, J.; Hartnell, L.M.; Ruthel, G.T.; Schneider, D.K.; Nagashima, K.; et al. 3D visualization of HIV transfer at the virological synapse between dendritic cells and T cells. Proc. Natl. Acad. Sci. USA 2010, 107, 13336–13341. [Google Scholar] [CrossRef] [PubMed]
- Do, T.; Murphy, G.; Earl, L.A.; Del Prete, G.Q.; Grandinetti, G.; Li, G.H.; Estes, J.D.; Rao, P.; Trubey, C.M.; Thomas, J.; et al. Three-dimensional imaging of HIV-1 virological synapses reveals membrane architectures involved in virus transmission. J. Virol. 2014, 88, 10327–10339. [Google Scholar] [CrossRef] [PubMed]
- Dahm, T.; Adams, O.; Boettcher, S.; Diedrich, S.; Morozov, V.; Hansman, G.; Fallier-Becker, P.; Schadler, S.; Burkhardt, C.J.; Weiss, C.; et al. Strain-dependent effects of clinical echovirus 30 outbreak isolates at the blood-CSF barrier. J. Neuroinflammation 2018, 15, 50. [Google Scholar] [CrossRef] [PubMed]
- Kantor, A.M.; Grant, D.G.; Balaraman, V.; White, T.A.; Franz, A.W.E. Ultrastructural Analysis of Chikungunya Virus Dissemination from the Midgut of the Yellow Fever Mosquito, Aedes aegypti. Viruses 2018, 10, 571. [Google Scholar] [CrossRef]
- Schertel, A.; Snaidero, N.; Han, H.M.; Ruhwedel, T.; Laue, M.; Grabenbauer, M.; Mobius, W. Cryo FIB-SEM: Volume imaging of cellular ultrastructure in native frozen specimens. J. Struct. Biol. 2013, 184, 355–360. [Google Scholar] [CrossRef]
- Vidavsky, N.; Akiva, A.; Kaplan-Ashiri, I.; Rechav, K.; Addadi, L.; Weiner, S.; Schertel, A. Cryo-FIB-SEM serial milling and block face imaging: Large volume structural analysis of biological tissues preserved close to their native state. J. Struct. Biol. 2016, 196, 487–495. [Google Scholar] [CrossRef]
- Wu, G.H.; Mitchell, P.G.; Galaz-Montoya, J.G.; Hecksel, C.W.; Sontag, E.M.; Gangadharan, V.; Marshman, J.; Mankus, D.; Bisher, M.E.; Lytton-Jean, A.K.R.; et al. Multi-scale 3D Cryo-Correlative Microscopy for Vitrified Cells. Structure 2020, 28, 1231–1237. [Google Scholar] [CrossRef]
- Schaffer, M.; Pfeffer, S.; Mahamid, J.; Kleindiek, S.; Laugks, T.; Albert, S.; Engel, B.D.; Rummel, A.; Smith, A.J.; Baumeister, W.; et al. A cryo-FIB lift-out technique enables molecular-resolution cryo-ET within native Caenorhabditis elegans tissue. Nat. Methods 2019, 16, 757–762. [Google Scholar] [CrossRef]
- Buckley, G.; Gervinskas, G.; Taveneau, C.; Venugopal, H.; Whisstock, J.C.; De Marco, A. Automated cryo-lamella preparation for high-throughput in-situ structural biology. J. Struct. Biol. 2020, 210, 107488. [Google Scholar] [CrossRef] [PubMed]
- Zachs, T.; Schertel, A.; Medeiros, J.; Weiss, G.L.; Hugener, J.; Matos, J.; Pilhofer, M. Fully automated, sequential focused ion beam milling for cryo-electron tomography. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolff, G.; Bárcena, M. Multiscale Electron Microscopy for the Study of Viral Replication Organelles. Viruses 2021, 13, 197. https://doi.org/10.3390/v13020197
Wolff G, Bárcena M. Multiscale Electron Microscopy for the Study of Viral Replication Organelles. Viruses. 2021; 13(2):197. https://doi.org/10.3390/v13020197
Chicago/Turabian StyleWolff, Georg, and Montserrat Bárcena. 2021. "Multiscale Electron Microscopy for the Study of Viral Replication Organelles" Viruses 13, no. 2: 197. https://doi.org/10.3390/v13020197
APA StyleWolff, G., & Bárcena, M. (2021). Multiscale Electron Microscopy for the Study of Viral Replication Organelles. Viruses, 13(2), 197. https://doi.org/10.3390/v13020197