
The Autophagy Cargo Receptor SQSTM1 Inhibits Infectious Bursal Disease Virus Infection through Selective Autophagic Degradation of Double-Stranded Viral RNA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Virus, and Reagents
2.2. Antibodies
2.3. Plasmids and Transfection
2.4. Construction of SQSTM1 Knockout Cell Lines
2.5. Construction of STable 293T Cells Expressing SQSTM1
2.6. Cytotoxicity Assay
2.7. IBDV Genomic dsRNA Extraction
2.8. IBDV dsRNA Degradation Assay
2.9. Detection of IFN-β and IFN-Stimulated Gene
2.10. Primer Pairs for qRT-PCR and PCR
2.11. Indirect Immunofluorescence Assay
2.12. Confocal Microscopy and Structured Illumination Microscopy
2.13. Western Blot
2.14. RNA Binding Protein Immunoprecipitation Assay
2.15. RNA Pull-Down
2.16. DsRNA Transcription
2.17. Statistical Analysis
3. Results
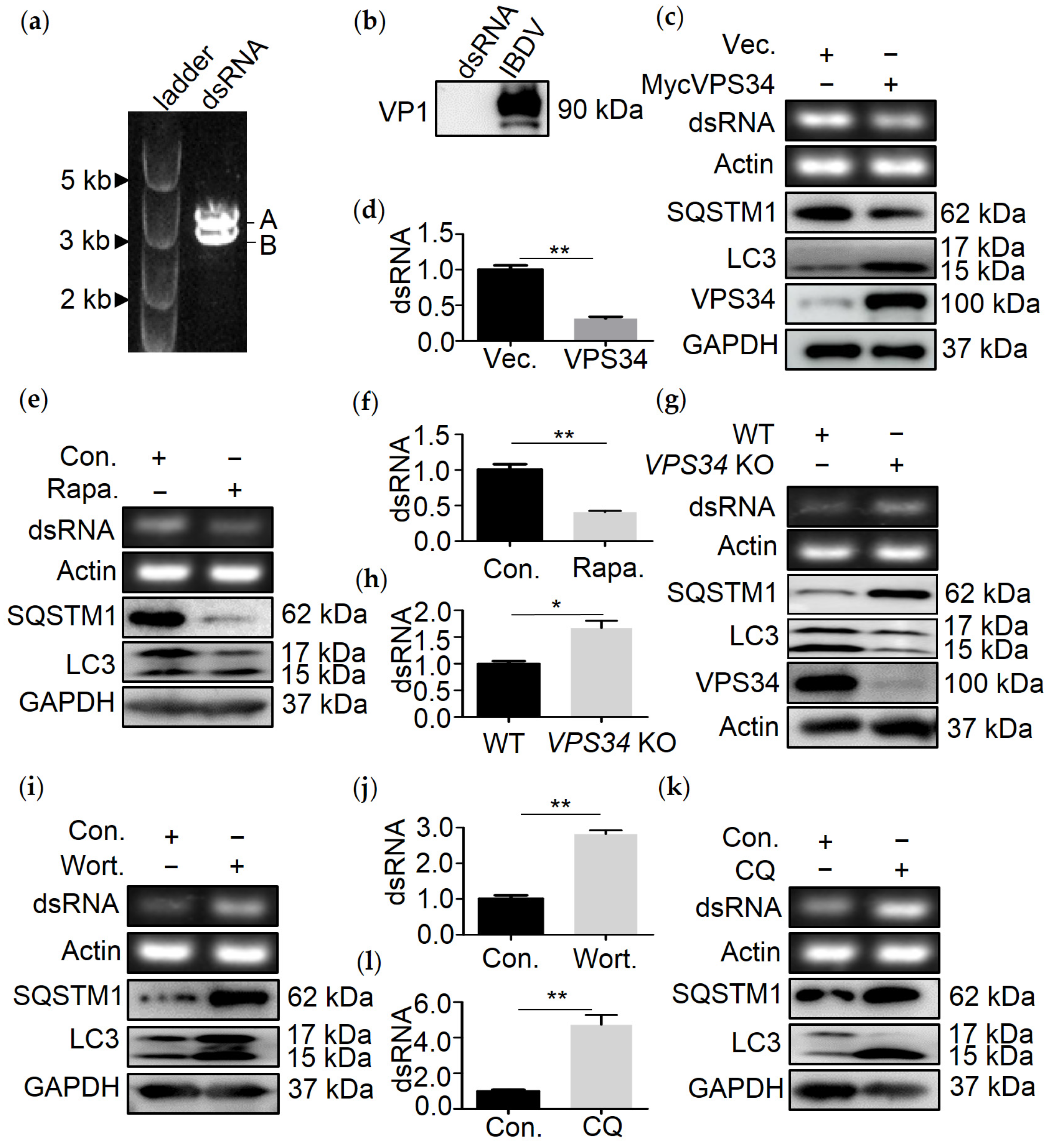
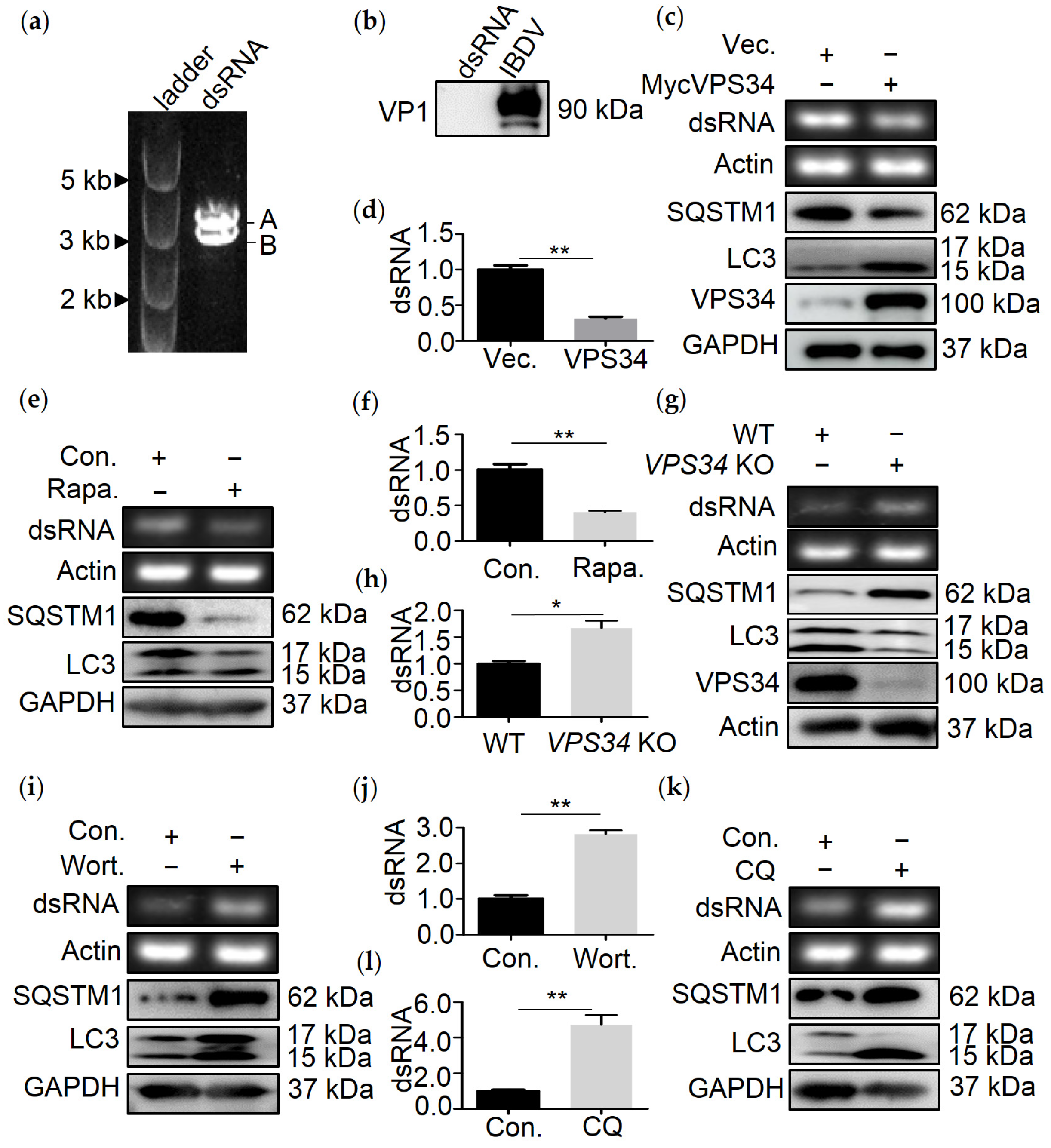
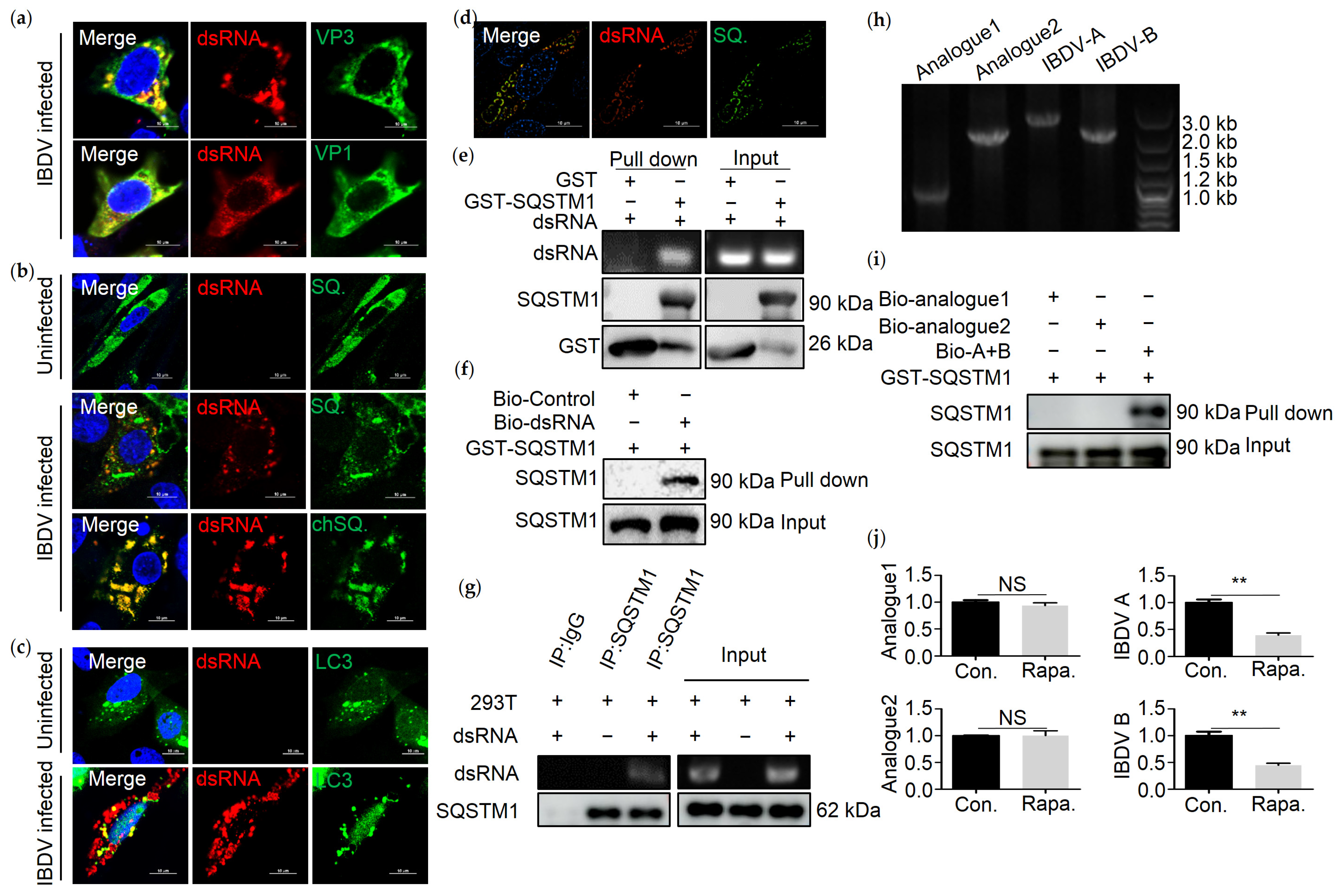
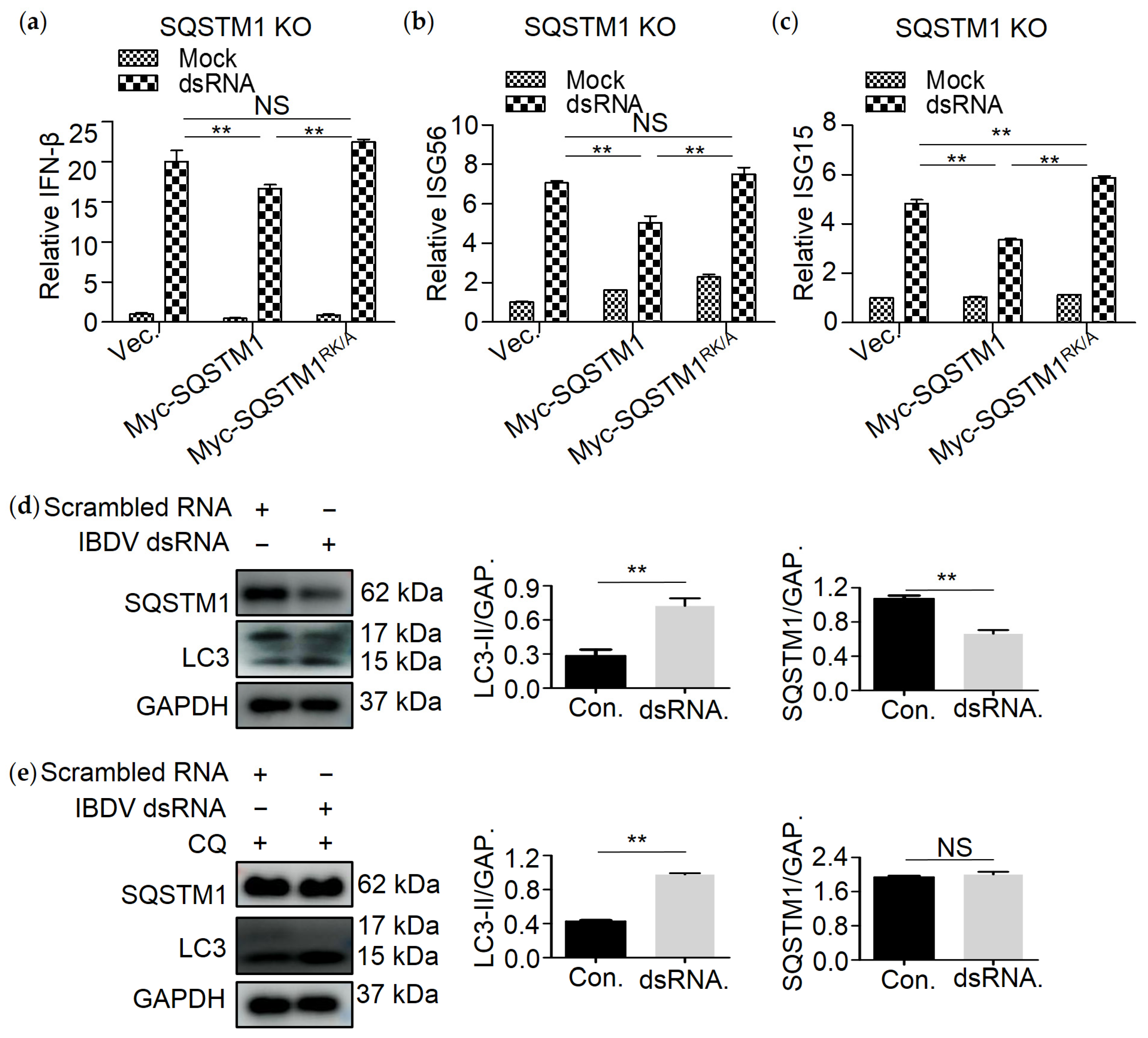
3.1. Autophagy Mediates the Degradation of IBDV dsRNA
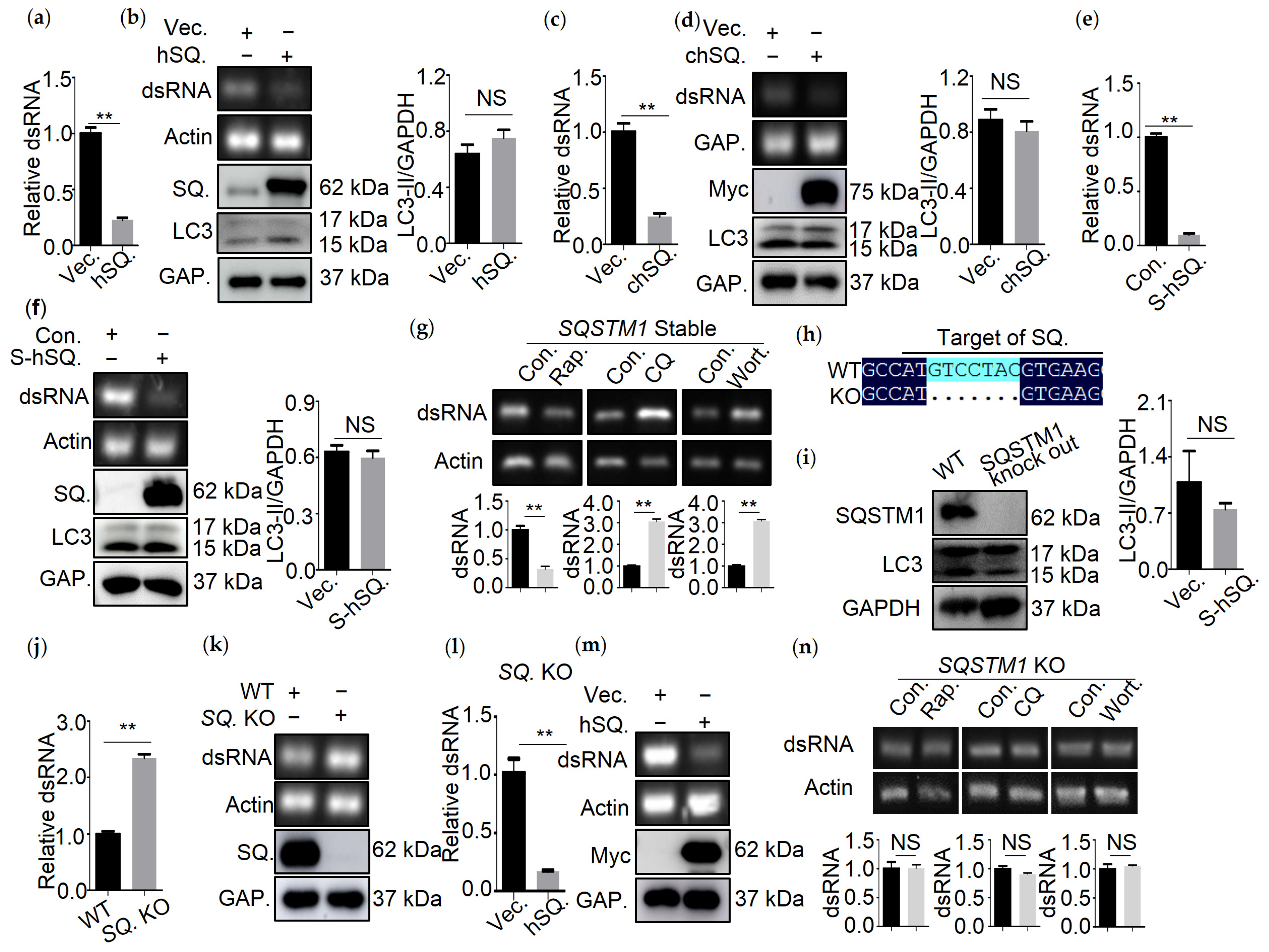
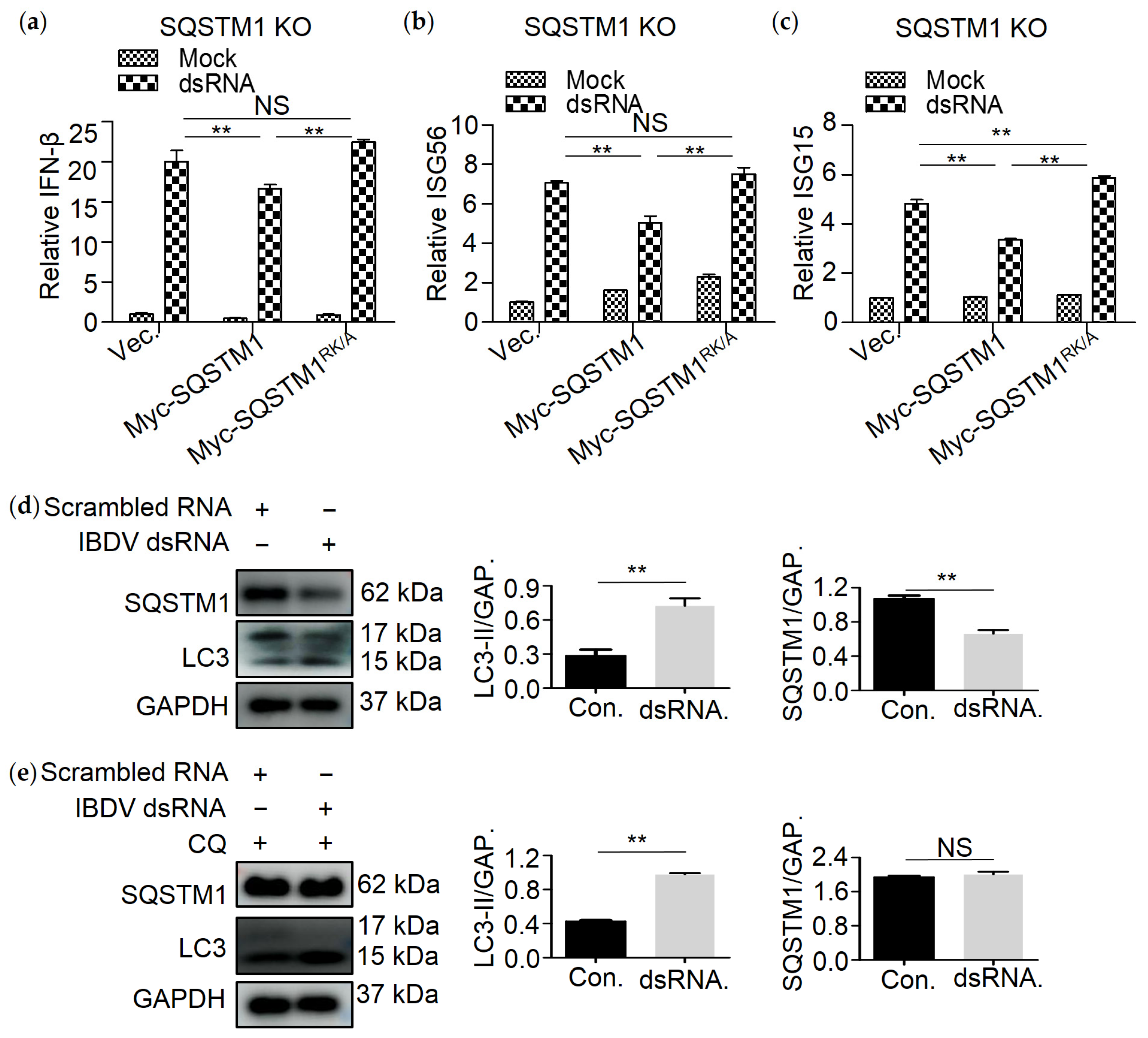
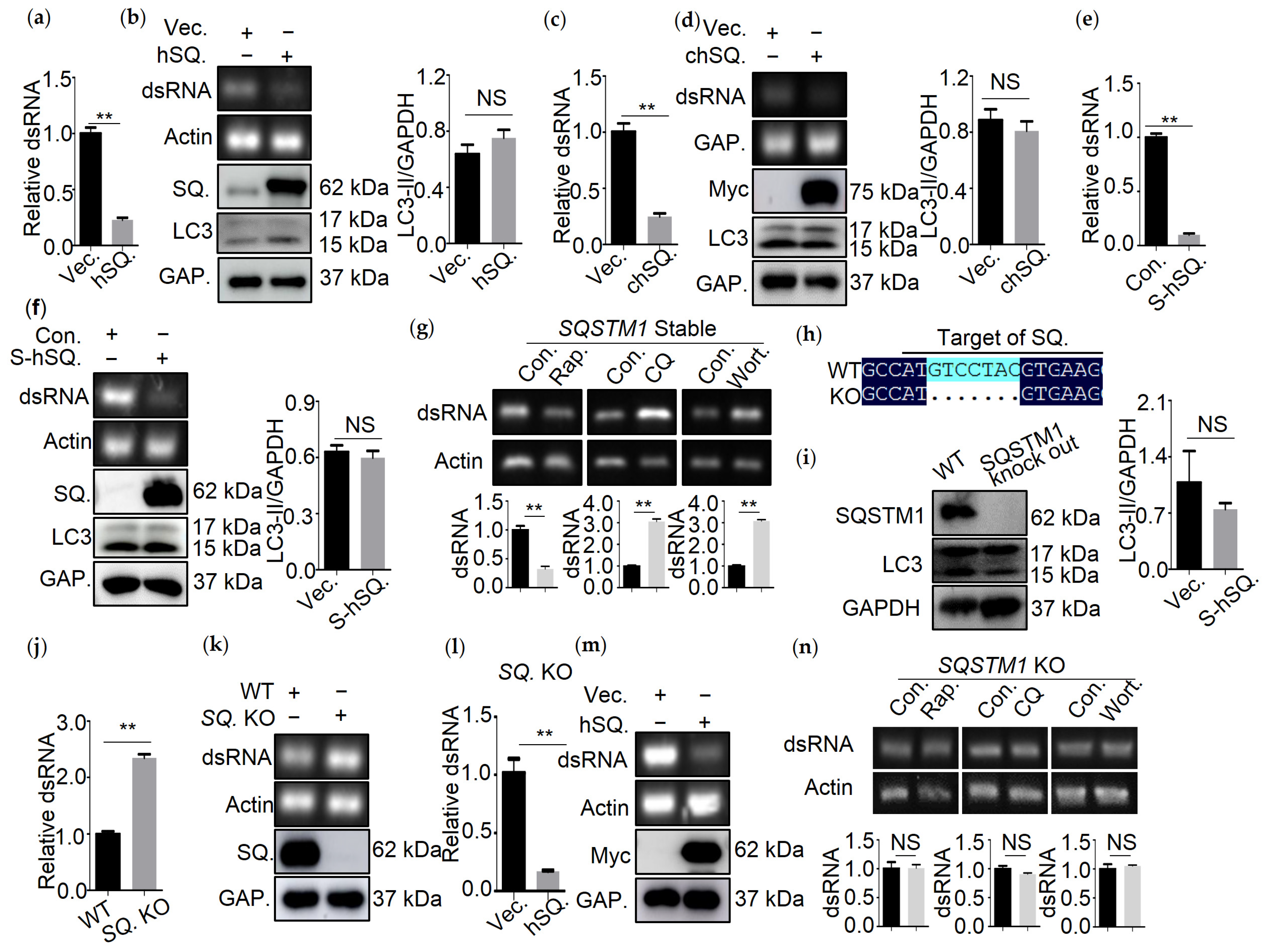
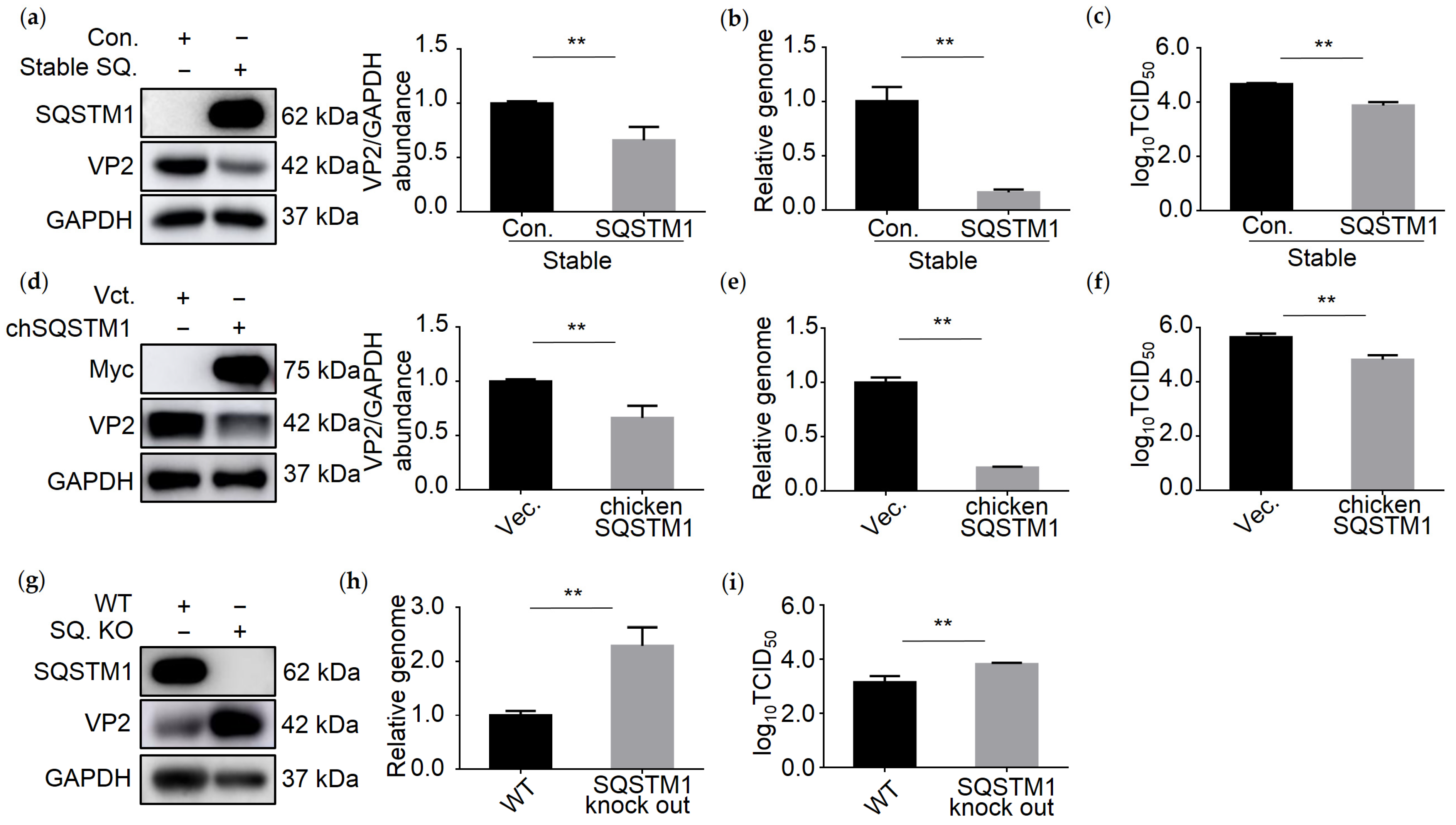
3.2. SQSTM1 Is Responsible for the Degradation of dsRNA
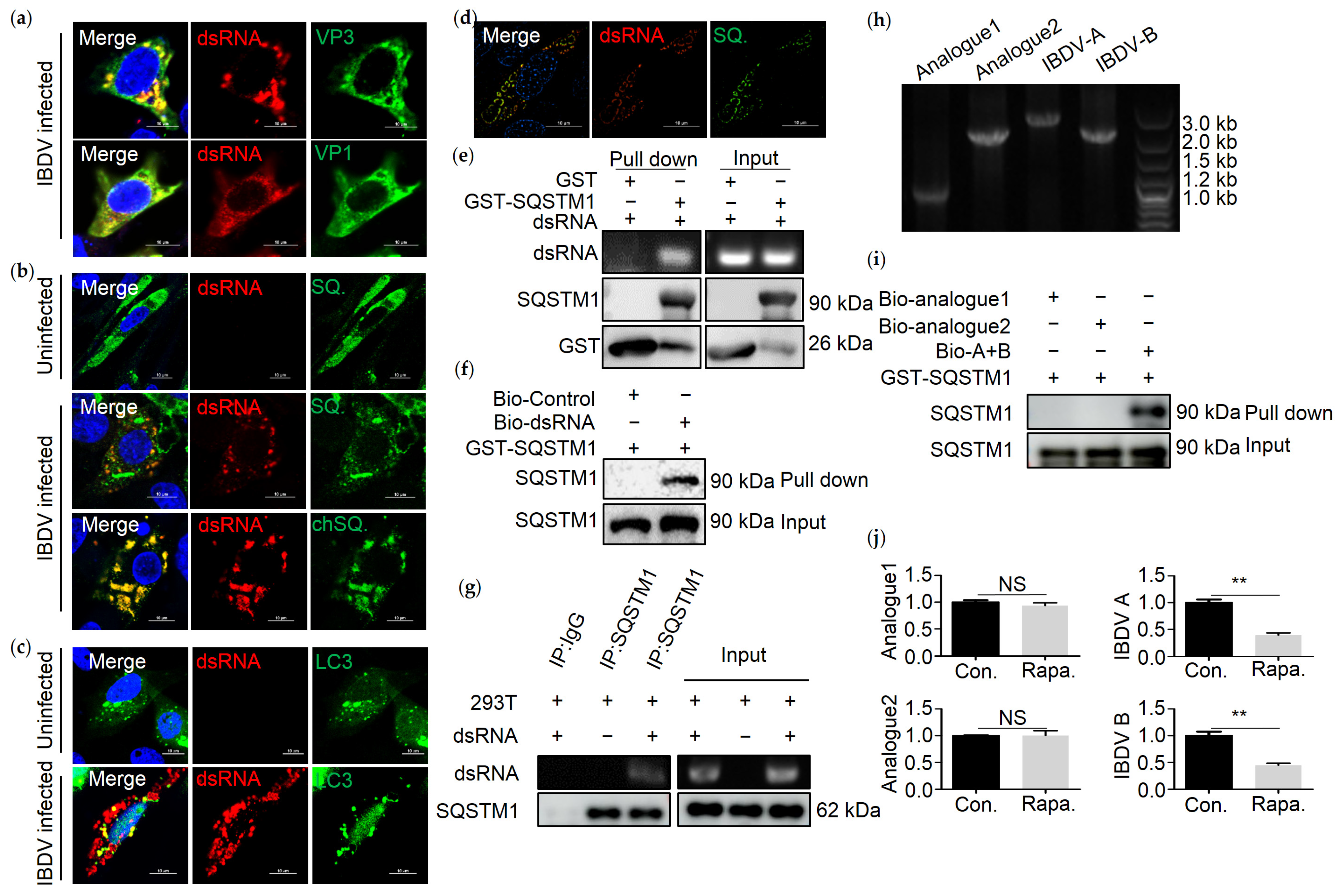
3.3. SQSTM1 Binds to dsRNA Directly
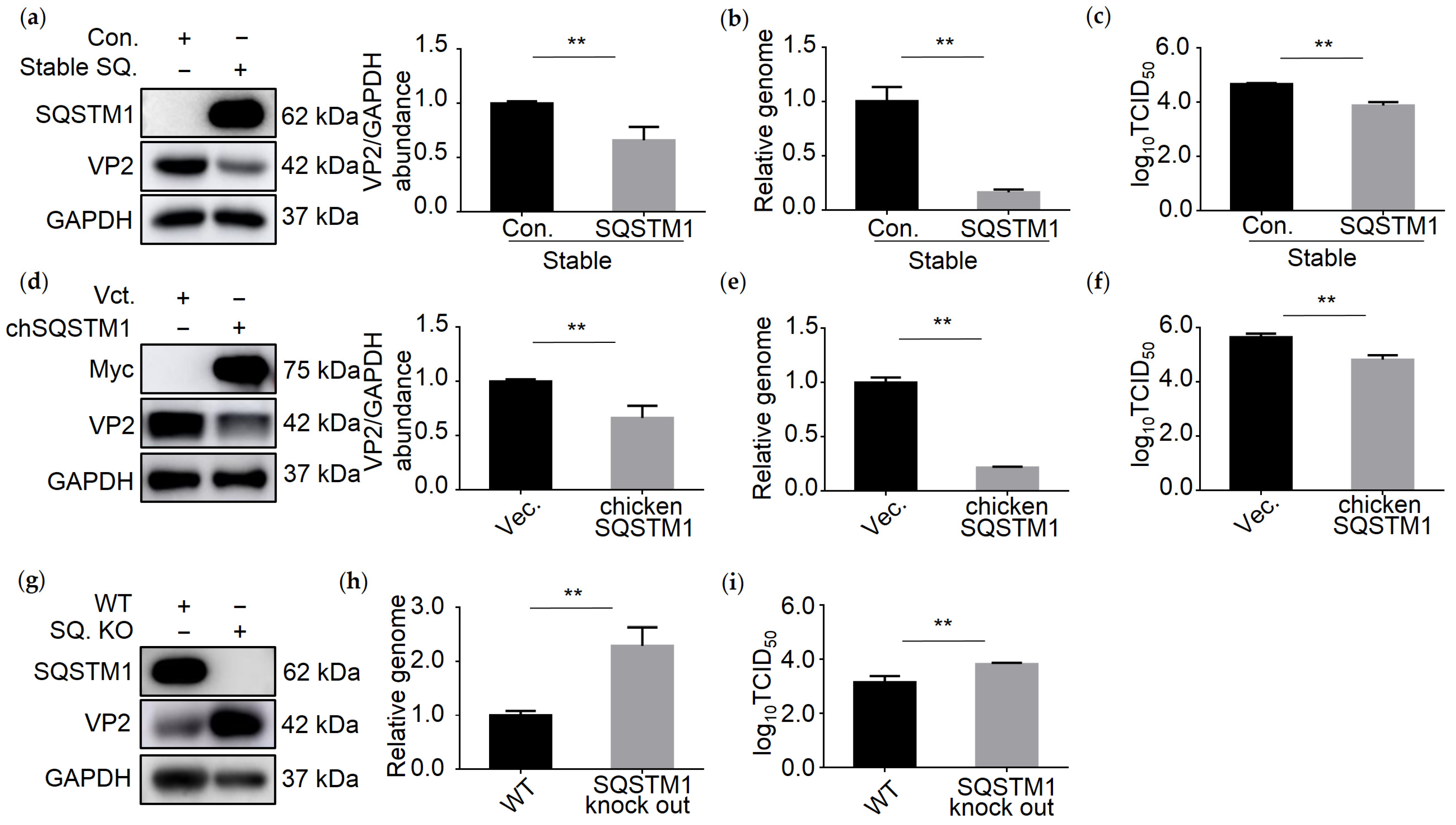
3.4. SQSTM1 Inhibits IBDV Replication
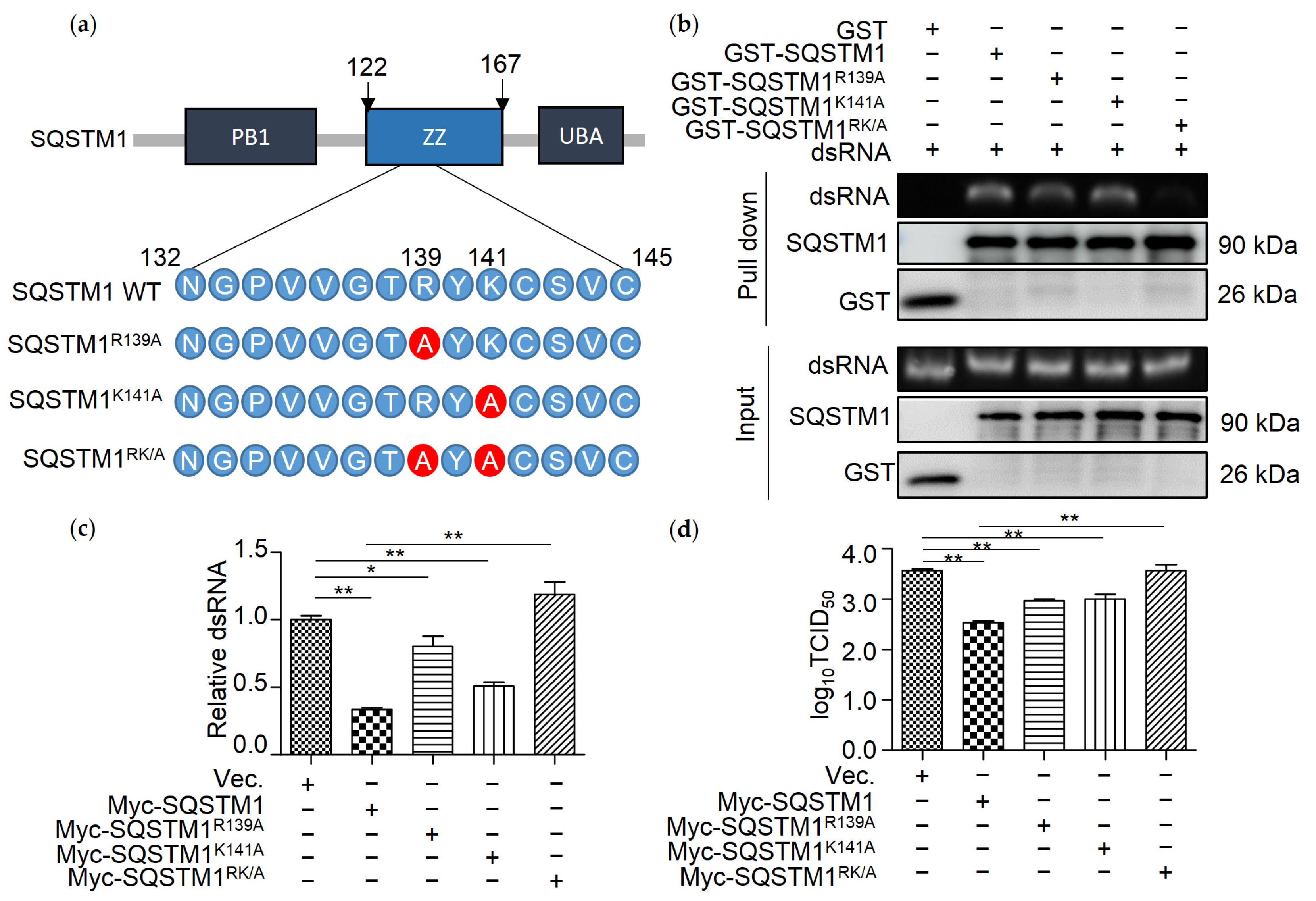
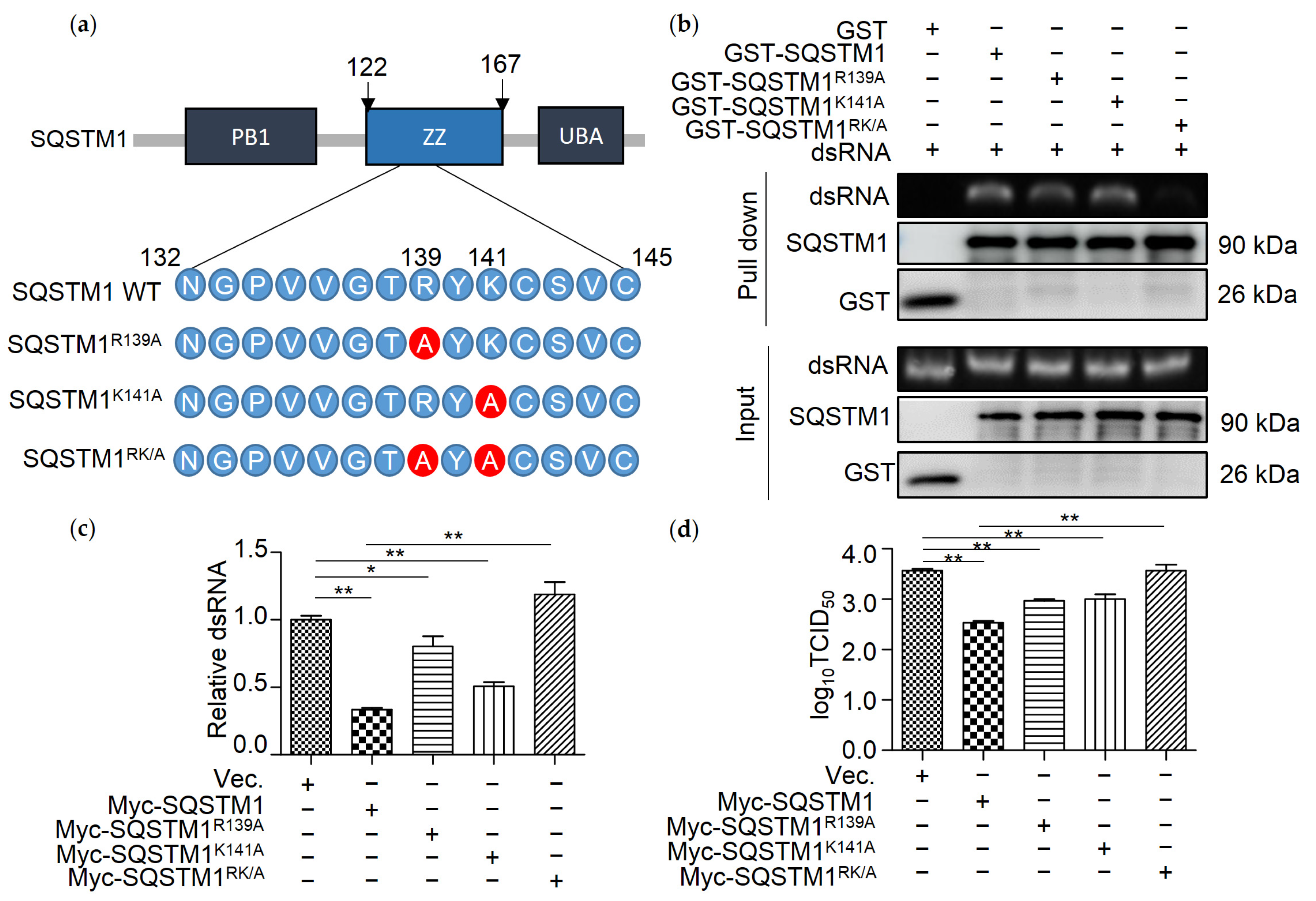
3.5. R139 and K141 Are Necessary for SQSTM1-Mediated Degradation of dsRNA
3.6. SQSTM1 Is Critical for dsRNA-Induced Production of Interferon β
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef]
- Rogov, V.; Dotsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaminets, A.; Beh, C.; Dikic, I. Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Horos, R.; Buscher, M.; Kleinendorst, R.; Alleaume, A.M.; Tarafder, A.K.; Schwarzl, T.; Dziuba, D.; Tischer, C.; Zielonka, E.M.; Adak, A.; et al. The Small Non-coding Vault RNA1-1 Acts as a Riboregulator of Autophagy. Cell 2019, 176, 1054–1067. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, S.F.; Parker, R. Principles and properties of eukaryotic mRNPs. Mol. Cell 2014, 54, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Doma, M.K.; Parker, R. RNA quality control in eukaryotes. Cell 2007, 131, 660–668. [Google Scholar] [CrossRef] [Green Version]
- Frankel, L.B.; Lubas, M.; Lund, A.H. Emerging connections between RNA and autophagy. Autophagy 2017, 13, 3–23. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Chitiprolu, M.; Gagnon, D.; Meng, L.; Perez-Iratxeta, C.; Lagace, D.; Gibbings, D. Autophagy supports genomic stability by degrading retrotransposon RNA. Nat. Commun. 2014, 5, 5276. [Google Scholar] [CrossRef]
- Shelly, S.; Lukinova, N.; Bambina, S.; Berman, A.; Cherry, S. Autophagy Is an Essential Component of Drosophila Immunity against Vesicular Stomatitis Virus. Immunity 2009, 30, 588–598. [Google Scholar] [CrossRef] [Green Version]
- Orvedahl, A.; MacPherson, S.; Sumpter, R., Jr.; Talloczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Hong, L.; Shi, L.; Guo, J.; Sun, Z.; Zhou, J. Proteomics Analysis of Host Cells Infected with Infectious Bursal Disease Virus. Mol. Cell. Proteom. 2008, 7, 612–625. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.; Jia, L.; Sun, Y.; Hu, B.; Wang, L.; Lu, X.; Zhou, J. Inhibition of antiviral innate immunity by birnavirus VP3 protein via blockage of viral double-stranded RNA binding to the host cytoplasmic RNA detector MDA5. J. Virol. 2014, 88, 11154–11165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Li, H.; Ma, G.; Zhou, J.; Hong, L.; Zheng, X.; Wu, Y.; Wang, Y.; Yan, Y. Competitive replication of different genotypes of infectious bursal disease virus on chicken embryo fibroblasts. Virus Genes 2009, 39, 46–52. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Zhang, Y.; Liu, F.; Hu, B.; Zhou, J. Construction of VPS34-Knockout A549/293T Cell Line by CRISPR/Cas9 System. Chin. J. Cell Biol. 2018, 40, 1719–1726. [Google Scholar]
- Wu, Y.; Peng, C.; Xu, L.; Zheng, X.; Liao, M.; Yan, Y.; Jin, Y.; Zhou, J. Proteome dynamics in primary target organ of infectious bursal disease virus. Proteomics 2012, 12, 1844–1859. [Google Scholar] [CrossRef]
- Zheng, X.; Hong, L.; Li, Y.; Guo, J.; Zhang, G.; Zhou, J. In vitro expression and monoclonal antibody of RNA-dependent RNA polymerase for infectious bursal disease virus. DNA Cell Biol. 2006, 25, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.C.; Sarin, L.P.; Bahar, M.W.; Myers, R.A.; Stuart, D.I.; Bamford, D.H.; Grimes, J.M. The N-Terminus of the RNA Polymerase from Infectious Pancreatic Necrosis Virus Is the Determinant of Genome Attachment. PLoS Pathog. 2011, 7, e1002085. [Google Scholar] [CrossRef]
- Kutateladze, T.G. Phosphatidylinositol 3-phosphate recognition and membrane docking by the FYVE domain. Biochim. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2006, 1761, 868–877. [Google Scholar] [CrossRef] [Green Version]
- Sameshima, M.; Liebhaber, S.A.; Schlessinger, D. Dual pathways for ribonucleic acid turnover in WI-38 but not in I-cell human diploid fibroblasts. Mol. Cell. Biol. 1981, 1, 75–81. [Google Scholar]
- Lardeux, B.R.; Mortimore, G.E. Amino acid and hormonal control of macromolecular turnover in perfused rat liver. Evidence for selective autophagy. J. Biol. Chem. 1987, 262, 14514–14519. [Google Scholar] [CrossRef]
- Mortimore, G.E.; Lardeux, B.R.; Heydrick, S.J. Mechanism and control of protein and RNA degradation in the rat hepatocyte: Two modes of autophagic sequestration. Revis. Sobre Biol. Cel. RBC 1989, 20, 79–96. [Google Scholar]
- Balavoine, S.; Feldmann, G.; Lardeux, B. Rates of RNA degradation in isolated rat hepatocytes. Effects of amino acids and inhibitors of lysosomal function. Eur. J. Biochem. 1990, 189, 617–623. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Furuta, A.; Kikuchi, H.; Aizawa, S.; Hatanaka, Y.; Konya, C.; Uchida, K.; Yoshimura, A.; Tamai, Y.; Wada, K.; et al. Discovery of a novel type of autophagy targeting RNA. Autophagy 2014, 9, 403–409. [Google Scholar] [CrossRef] [Green Version]
- Belancio, V.P.; Roy-Engel, A.M.; Deininger, P.L. All y’all need to know ‘bout retroelements in cancer. Semin. Cancer Biol. 2010, 20, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Elbarbary, R.A.; Lucas, B.A.; Maquat, L.E. Retrotransposons as regulators of gene expression. Science 2016, 351, aac7247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraft, C.; Deplazes, A.; Sohrmann, M.; Peter, M. Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat. Cell Biol. 2008, 10, 602–610. [Google Scholar] [CrossRef]
- Krisenko, M.O.; Higgins, R.L.; Ghosh, S.; Zhou, Q.; Trybula, J.S.; Wang, W.H.; Geahlen, R.L. Syk Is Recruited to Stress Granules and Promotes Their Clearance through Autophagy. J. Biol. Chem. 2015, 290, 27803–27815. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, S.; Cherkasova, V.; Bankhead, P.; Bukau, B.; Stoecklin, G. Translation suppression promotes stress granule formation and cell survival in response to cold shock. Mol. Biol. Cell 2012, 23, 3786–3800. [Google Scholar] [CrossRef]
- Hu, B.; Zhang, Y.; Jia, L.; Wu, H.; Fan, C.; Sun, Y.; Ye, C.; Liao, M.; Zhou, J. Binding of the pathogen receptor HSP90AA1 to avibirnavirus VP2 induces autophagy by inactivating the AKT-MTOR pathway. Autophagy 2015, 11, 503–515. [Google Scholar] [CrossRef] [Green Version]
- Watson, R.O.; Manzanillo, P.S.; Cox, J.S. Extracellular, M. Tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 2012, 150, 803–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, C.; Li, T.; Lei, J.; Zhang, Y.; Zhou, J.; Hu, B. The Autophagy Cargo Receptor SQSTM1 Inhibits Infectious Bursal Disease Virus Infection through Selective Autophagic Degradation of Double-Stranded Viral RNA. Viruses 2021, 13, 2494. https://doi.org/10.3390/v13122494
Xu C, Li T, Lei J, Zhang Y, Zhou J, Hu B. The Autophagy Cargo Receptor SQSTM1 Inhibits Infectious Bursal Disease Virus Infection through Selective Autophagic Degradation of Double-Stranded Viral RNA. Viruses. 2021; 13(12):2494. https://doi.org/10.3390/v13122494
Chicago/Turabian StyleXu, Chenyang, Tongtong Li, Jing Lei, Yina Zhang, Jiyong Zhou, and Boli Hu. 2021. "The Autophagy Cargo Receptor SQSTM1 Inhibits Infectious Bursal Disease Virus Infection through Selective Autophagic Degradation of Double-Stranded Viral RNA" Viruses 13, no. 12: 2494. https://doi.org/10.3390/v13122494
APA StyleXu, C., Li, T., Lei, J., Zhang, Y., Zhou, J., & Hu, B. (2021). The Autophagy Cargo Receptor SQSTM1 Inhibits Infectious Bursal Disease Virus Infection through Selective Autophagic Degradation of Double-Stranded Viral RNA. Viruses, 13(12), 2494. https://doi.org/10.3390/v13122494