1. Introduction
We are exposed to a diversity of pathogens throughout our lives. Our immune system, both its innate and adaptive arms, has developed molecular and cellular mechanisms to sense, prevent and respond to such infections. Cells in the first line of protection, the innate immune system, are equipped with pattern-recognition receptors (PRRs) that sense pathogen-associated molecular patterns (PAMPs), such as microbial products (e.g., viral RNA) [
1]. Activation of PRRs in infected cells leads to secretion of type I interferon (IFN), the main antiviral cytokine [
2,
3,
4,
5]. Binding of type I IFN to its receptor, in turn, induces the transcription of a family of interferon-stimulated genes (ISGs), whose protein products have both antiviral activity and immuno-modulatory effects [
2,
3,
6].
The survival of a viral population in a host depends on viruses replicating and avoiding intra-cellular defences. Many viruses have developed strategies to evade immune detection [
7], and thus, antagonise these defences [
7]. There exist a great diversity of such viral strategies and here we will only consider those mechanisms which interfere with intra-cellular pathways to either regulate type I IFN secretion or type I IFN signalling. For instance, influenza, hepatitis C virus, vaccinia or herpes simplex virus [
3,
8] all inhibit type I IFN responses.
Interferons are a family of cytokines which not only regulate cellular growth but also have antiviral properties and immuno-modulatory activity [
1]. They elicit an antiviral state defined by intra-cellular antimicrobial programmes, which require the induction of IFN stimulated genes. Interferons can be broadly classified into type I and type II. Type I interferons (or viral IFNs), which are secreted by infected cells, include IFN-
, IFN-
, IFN-
and IFN-
. Most cell types can produce IFN-
, which is the best characterised member of the family. Hematopoietic cells produce IFN-
. IFN-
is a type II IFN and is only produced by certain immune (professional) cells, such as natural killer cells, CD4
(helper) and CD8
(cytotoxic) T cells [
7].
Filoviruses, such as Ebola virus (EBOV) and Marburg virus, encode viral proteins with the ability to counteract type I IFN responses in order to replicate in an efficient manner and minimise the therapeutic antiviral power of IFNs. These type I IFN antagonist proteins, or viral antagonistic proteins (VAPs), are essential to guarantee viral replication, prevent the type I IFN-induced antiviral state in infected and bystander cells, as well as impair the ability of antigen presenting cells to initiate adaptive immune responses [
9]. This ability of filoviruses to replicate ‘unchecked’ by the host innate antiviral response can partly account for their lethality of infection. Early innate immune evasion facilitates fast and excessive viral replication, which in turn, activates a damaging host immune response [
2]. Unfortunately, filoviruses are not the only viral family to actively avoid immune surveillance. Other examples include influenza A virus, hepatitis B virus, and Bunyaviruses, such as Crimean-Congo haemorrhagic fever (CCHFV) or Rift Valley fever viruses [
10].
It is important to note that there exists stark contrast, and even conflicting evidence, between responses to in vivo and in vitro models of infection. In EBOV infection, for example, type I IFN production is abrogated after three days post-infection in in vitro infection, yet for in vivo infection, type I IFN cytokines are secreted during the entire infective period [
11,
12,
13]. In this paper, we develop mathematical models of the intra-cellular molecular processes that are known to antagonyse type I IFN production by viral proteins [
2,
6]. Existing mathematical models of intra-cellular production of type I IFN have many parameters that are difficult to estimate, or do not account for viral protein antagonism of PRR pathways [
14,
15,
16]. There are also models that describe inter-cellular interactions via IFN
receptors [
17]. Our goal is to model mechanisms of upstream and downstream viral protein antagonism, and to provide a case study applied to Ebola virus. We calibrate our models with clinical data from in vivo Ebola infection of rhesus macaques [
12]. Model selection allows us to compare different biological hypotheses. Our approach could serve as an example to characterise and quantify inhibition of type I IFN secretion by other pathogens, such as SARS-COV-2 virus [
18,
19], dengue and West Nile viruses [
20], hantaviruses [
21], and bunyaviruses [
22].
4. Discussion
The ability of highly pathogenic viruses, such as Ebola virus, SARS-CoV-2 or CCHFV, to subvert innate immune responses results in severe infection and high fatality rates. The current pandemic has tragically illustrated the need to improve our understanding of the strategies viruses make use of to evade immune surveillance [
48]. Of special relevance to viral infection are a family of cytokines: type I IFNs. Type I IFN responses (their expression induced by viral infection or the signalling cascades induced by their binding to specific IFN receptors) are frequently antagonised by viruses due to their importance in the initiation of innate immune responses [
8]. Therefore, we believe it is timely and useful to develop quantitative approaches to characterise and quantify these evasion mechanisms, in a way that can be applied to different viruses.
In this manuscript we have focused on viral strategies to antagonise cytosolic type I IFN secretion pathways via PRRs [
3], and have proposed three potential mathematical models with both upstream and downstream type I IFN inhibition mechanisms. These models have been formulated based on our current biological understanding of the interactions between the intra-cellular proteins involved [
8,
26]. In particular, we have investigated the role of a family of intra-cellular receptors, RLRs, which detect viral RNAs and promote IFN responses. VAPs may perturb a given RLR signalling pathway in its ‘upstream’ portion, at the level of dsRNA recognition, by competing with RLRs for dsRNA binding, or by removing PAMP signatures recognised by RLRs. Viral proteins such as NS1 (Influenza A), VP35 (Ebola), and N (SARS-COV), all bind viral RNAs, thus inhibiting PRRs from binding and signalling [
8,
26,
27,
49,
50]. PAMPs such as NP (Lassa Fever) and NSP14 (SARS-COV) are removed, preventing RLR recognition. ‘Downstream’ effects include the inhibition, mediated by VAPs, of RLR-induced antiviral proteins [
3,
8]. For instance, VAPs may modify binding sites of proteins, inhibit formation of signalling complexes, or prevent translocation and phosphorylation [
8]. Specific examples include the SARS-COV M protein which binds TRAF3 to form a long-lived complex, impeding its association with TBK1 and IKK-
kinases to forge a functional signalling complex [
51]. In the case of EBOV, its protein VP35 acts as a competing molecule for TBK1/IKK-
with interferon regulatory factors IRF3 and IRF7, but also prevents their translocation to the cellular nucleus [
52].
Mathematical models have been previously proposed to model interferon inhibition by viruses [
14,
15,
16] or to describe inter-cellular interactions via IFN-
receptors [
17]. These models are either virus specific or require detailed knowledge of many protein–protein interactions along the signalling pathway under consideration. Our aim in this manuscript is to characterise key biological hypotheses in a quantitative fashion, while avoiding, in principle, unnecessary complexity. Since clinical data sets from early viral infections are typically limited, it is important to have mathematical models in place that can be parameterised given this severe restriction, while our proposed models do not include all possible mechanisms of viral protein antagonism and inhibition of signalling pathways which result in type I IFN induction, the three models presented are a good first approximation. Moreover these models can be generalised to account for other mechanisms, proteins, or additional signalling pathways.
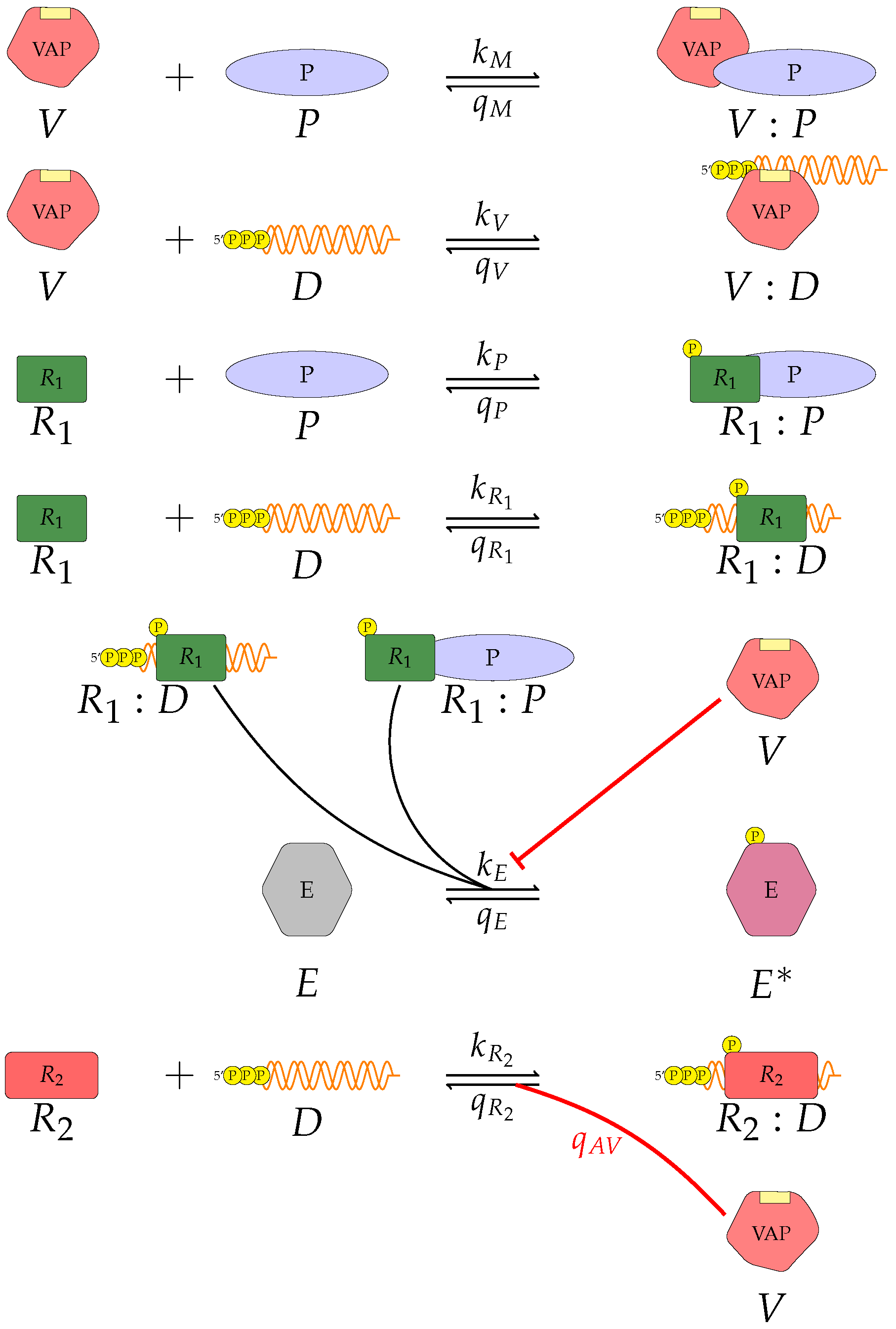
We proposed three separate models for the inhibition of type I interferon expression by VAPs. Each model considers a different biological mechanism or an alternate signalling pathway.
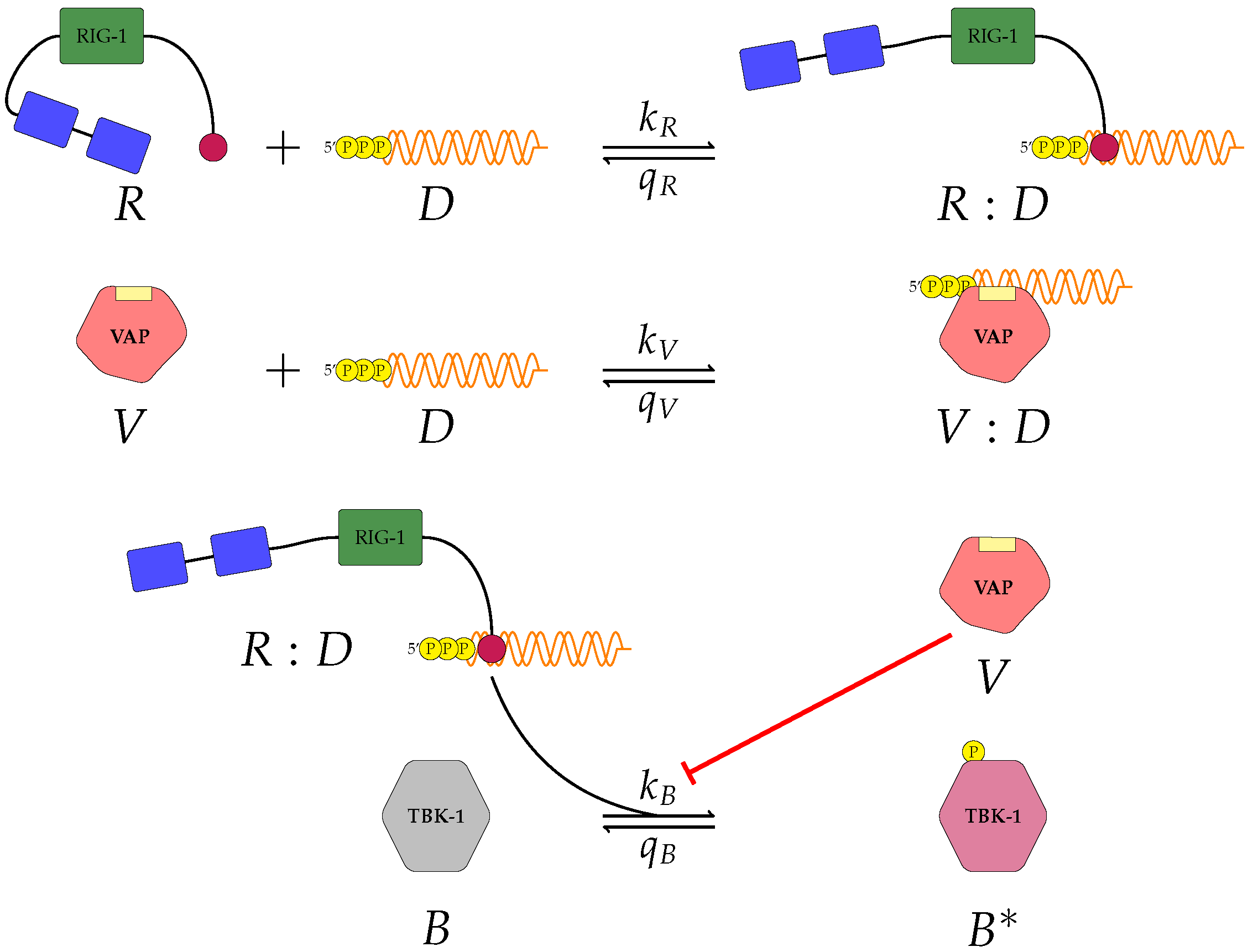
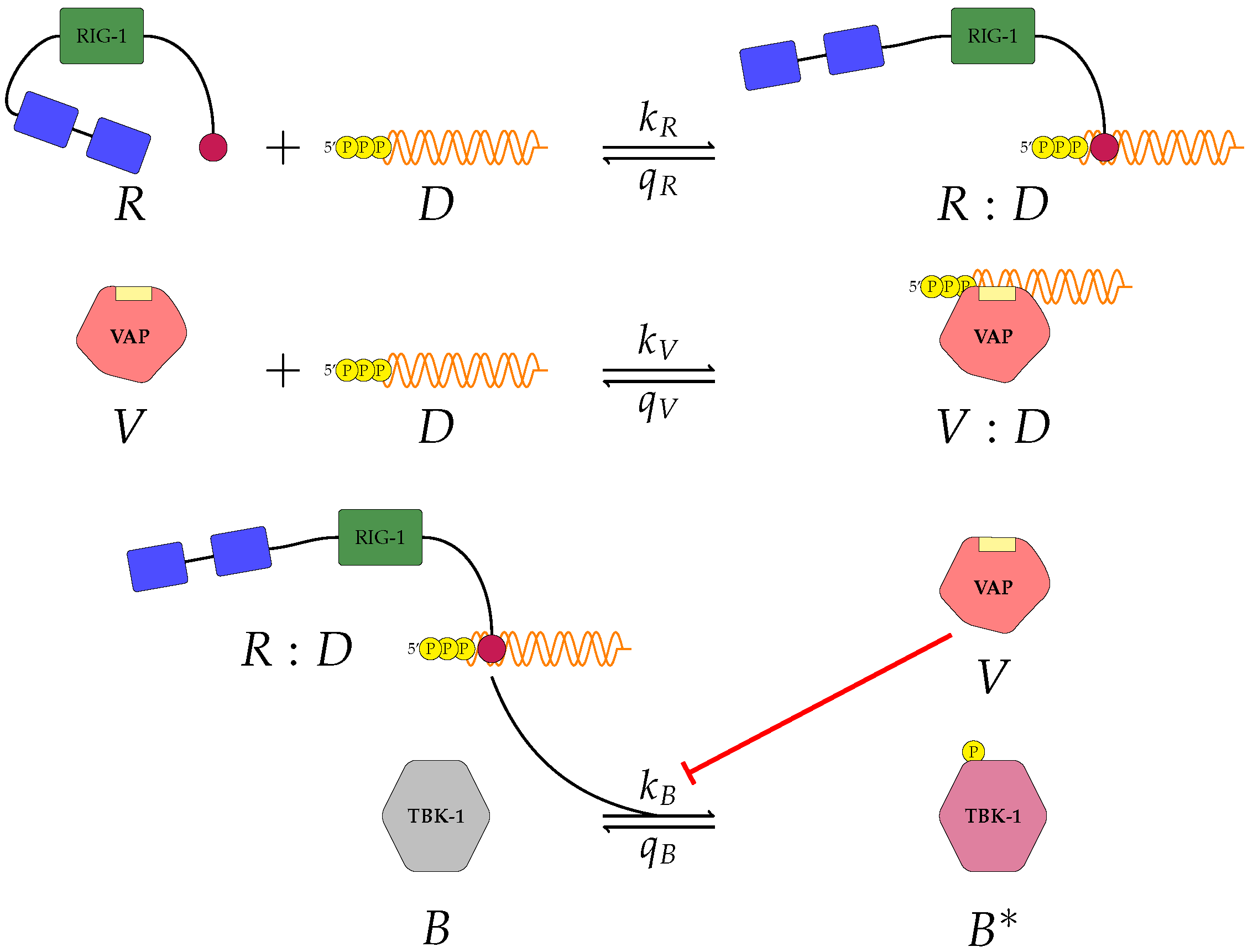
Figure 1,
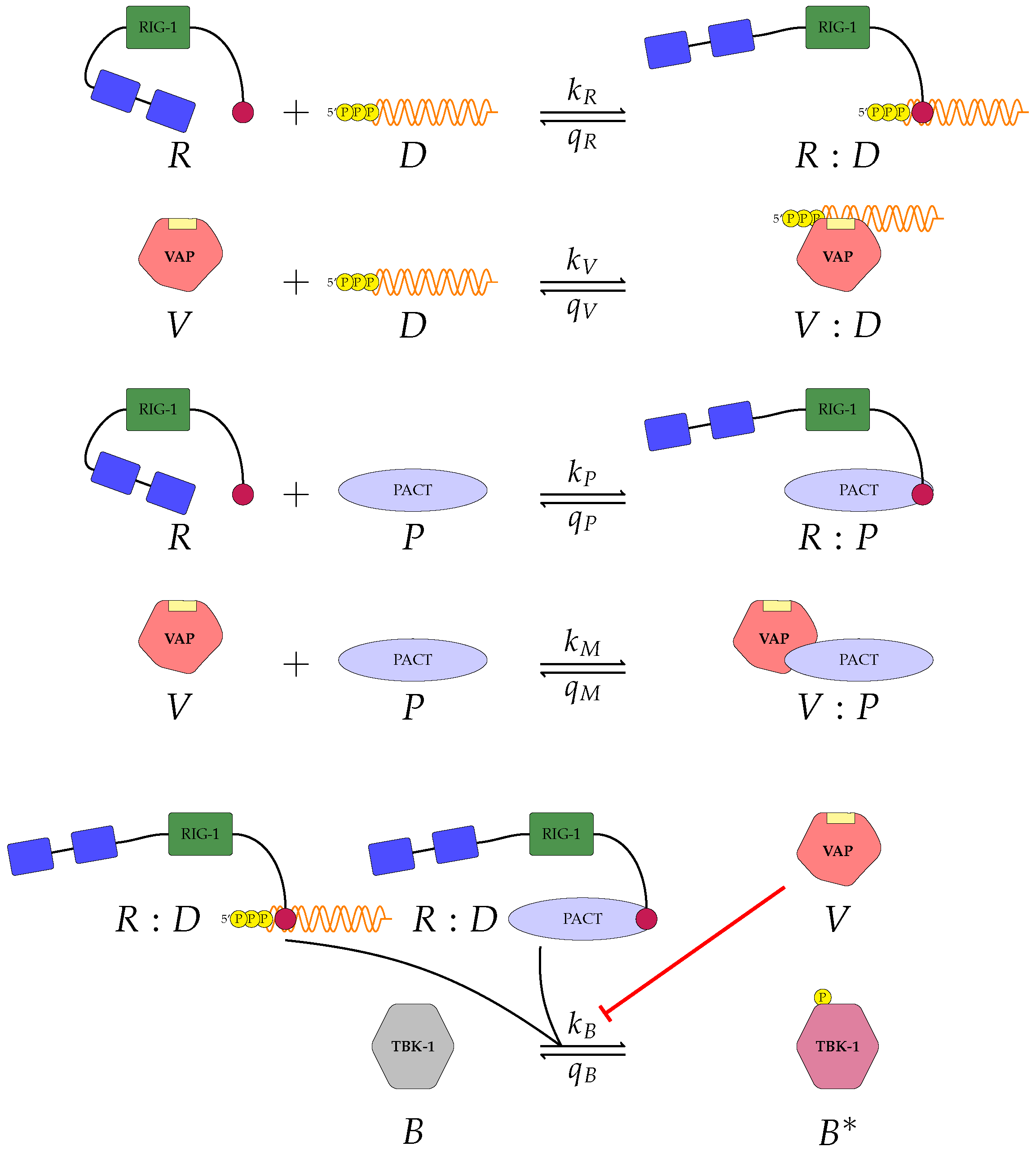
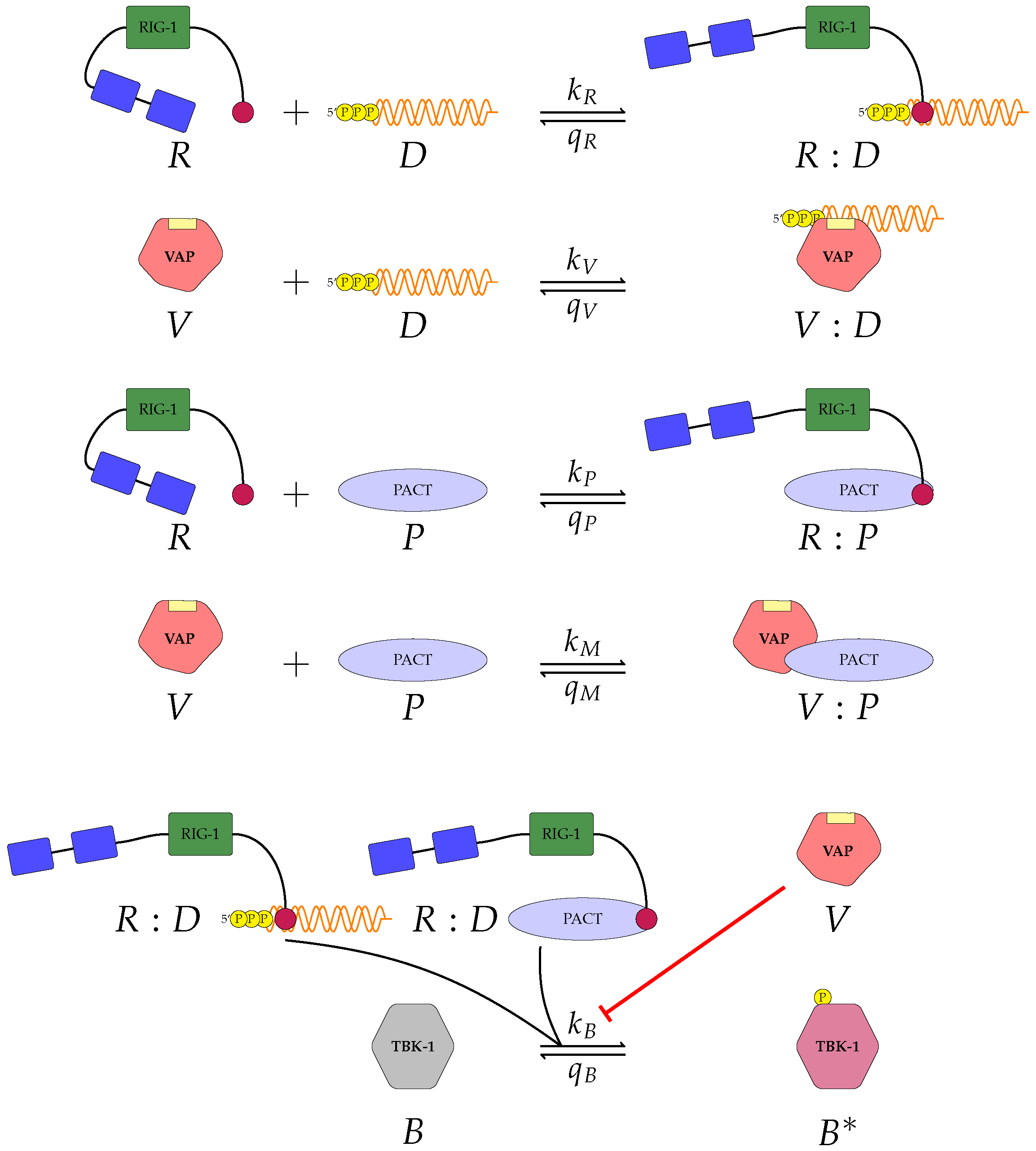
Figure 2 and
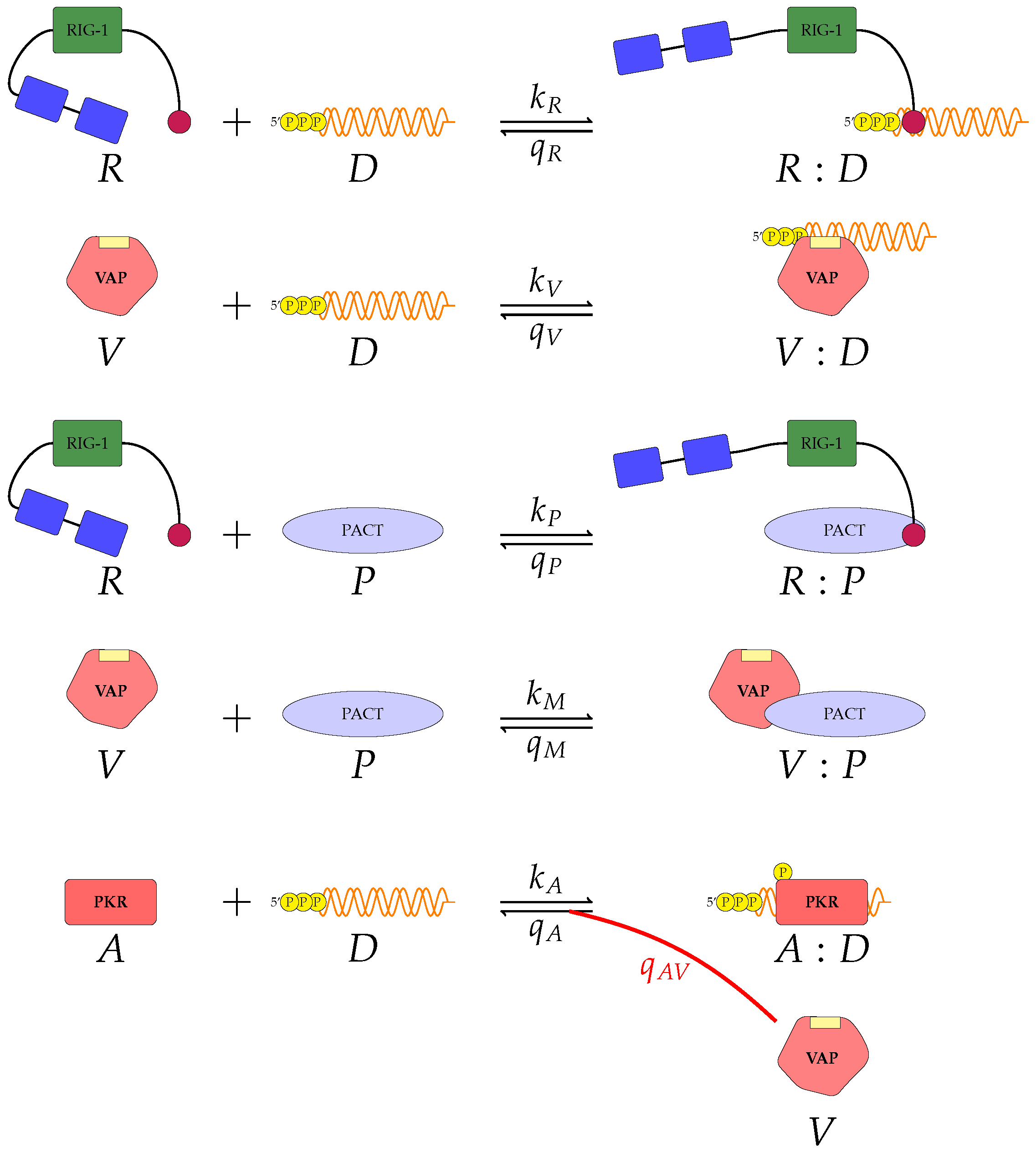
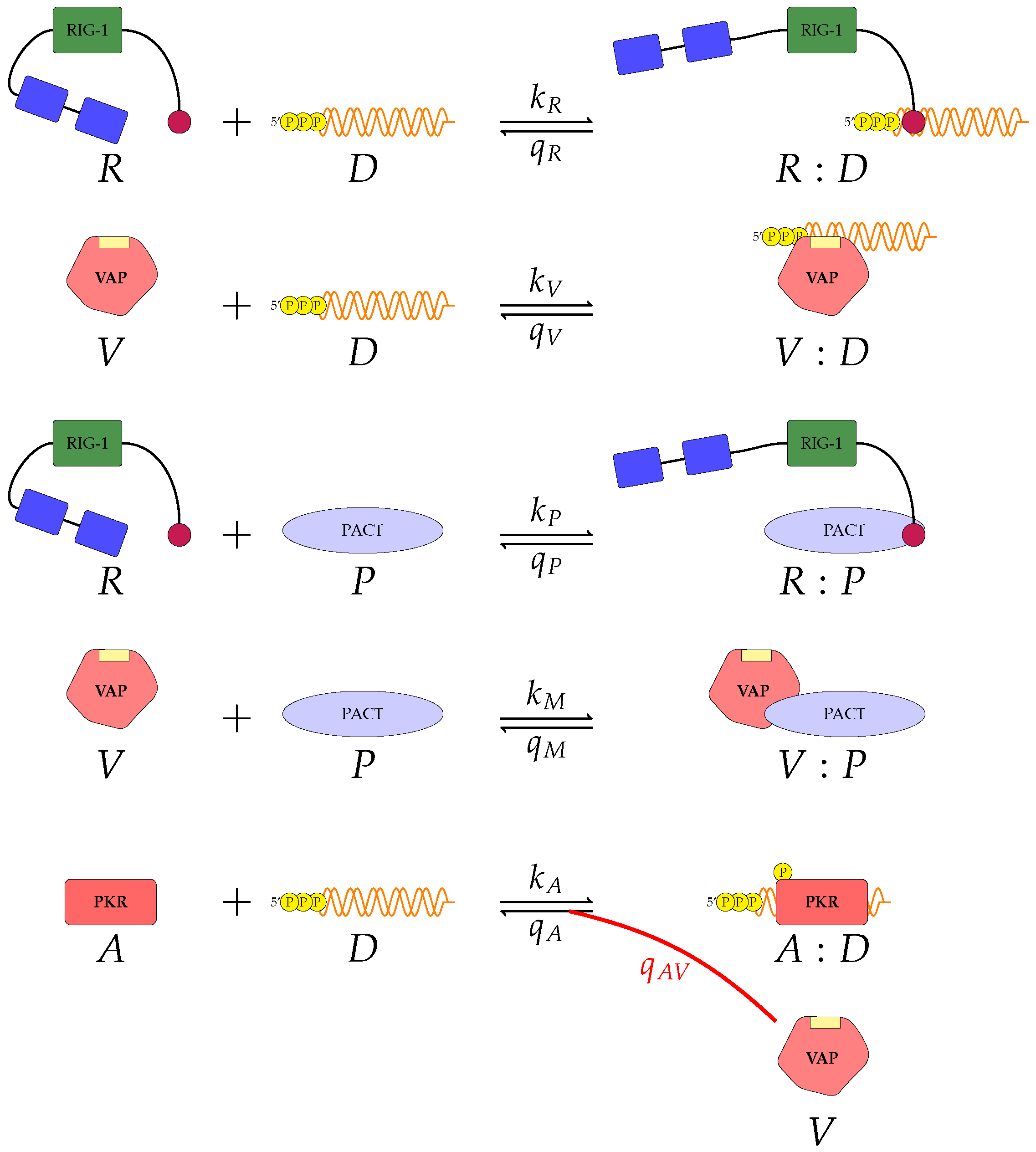
Figure 3 represent mechanisms which have been recently proposed in the literature [
3,
15,
27,
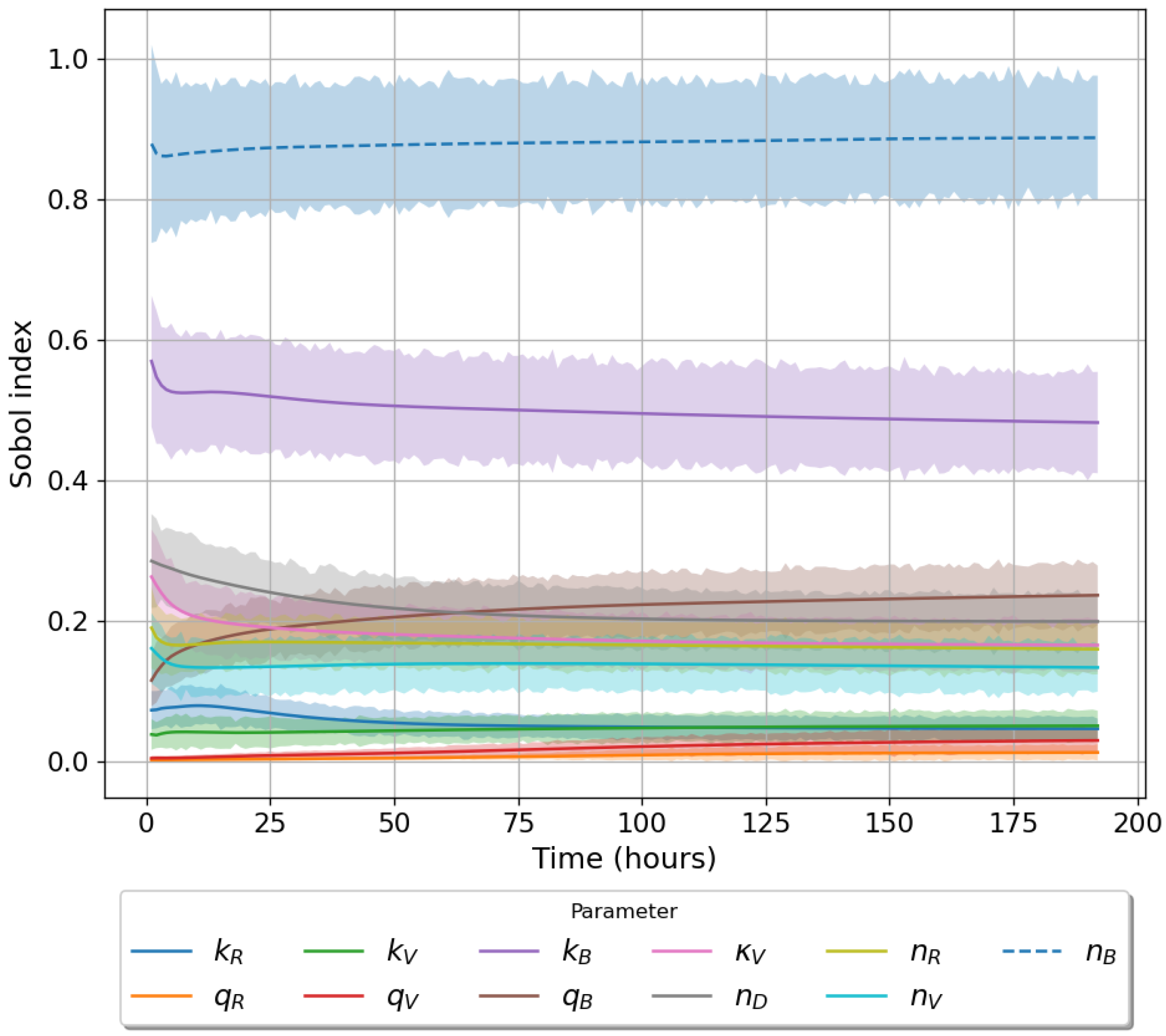
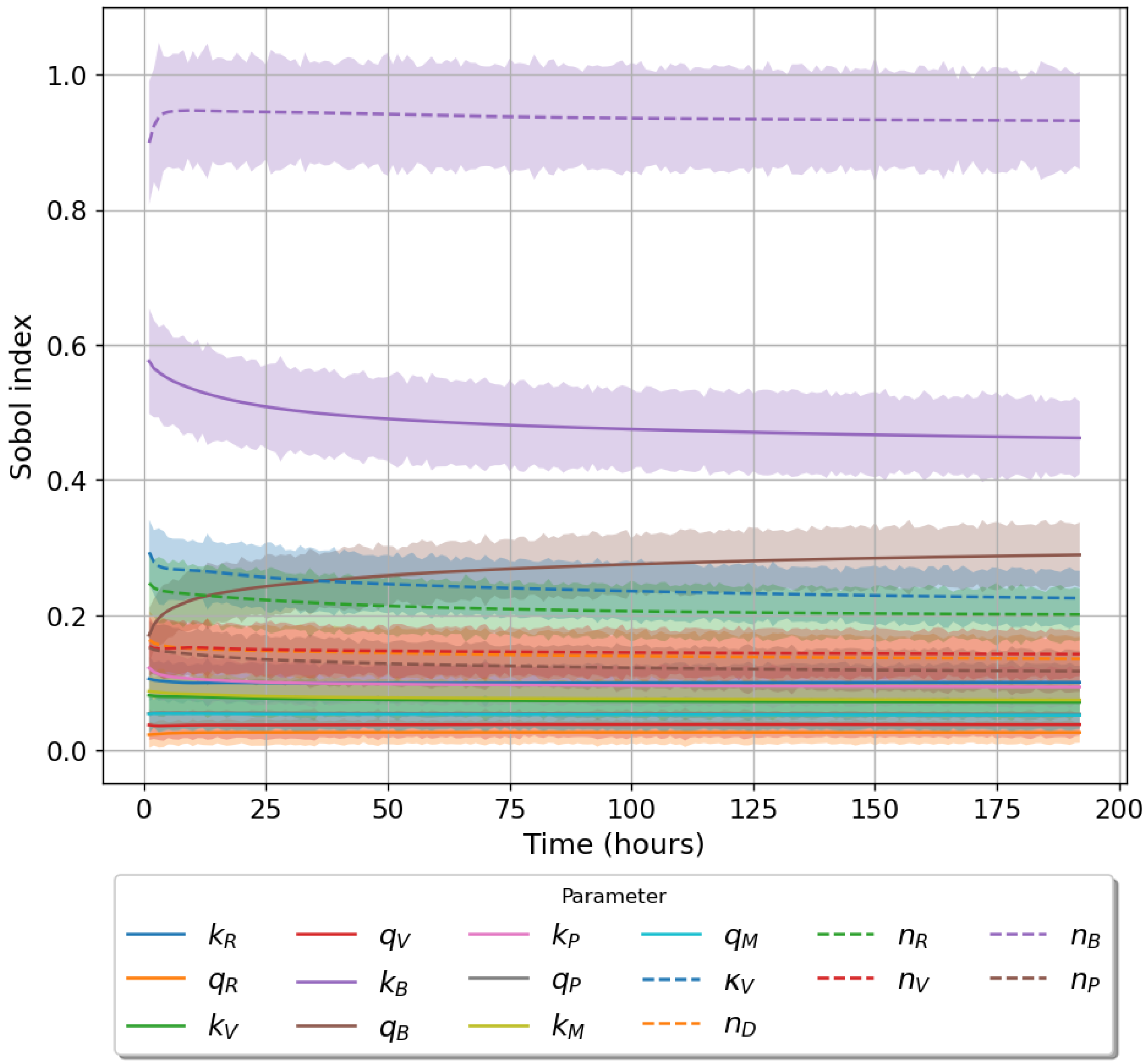
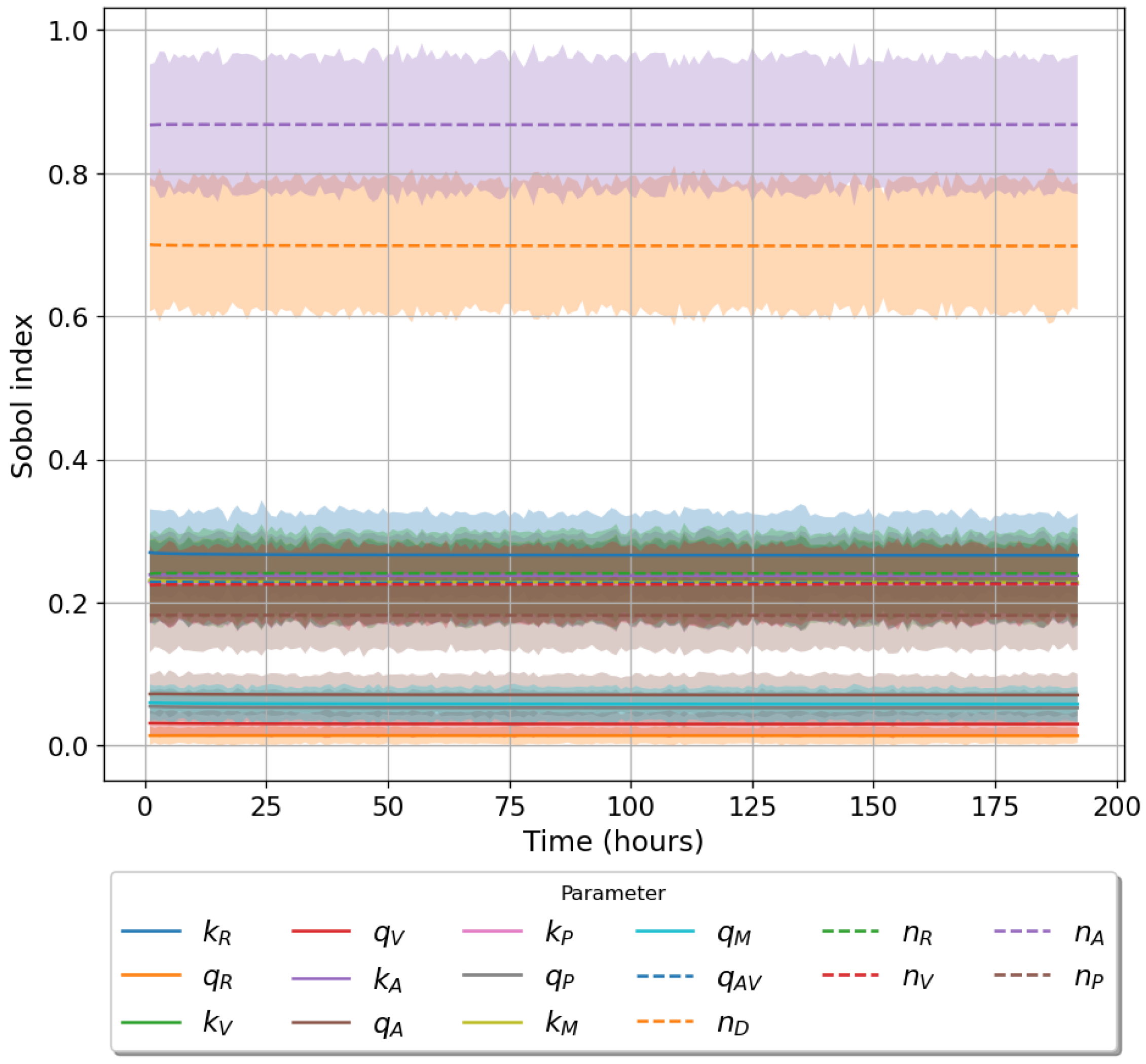
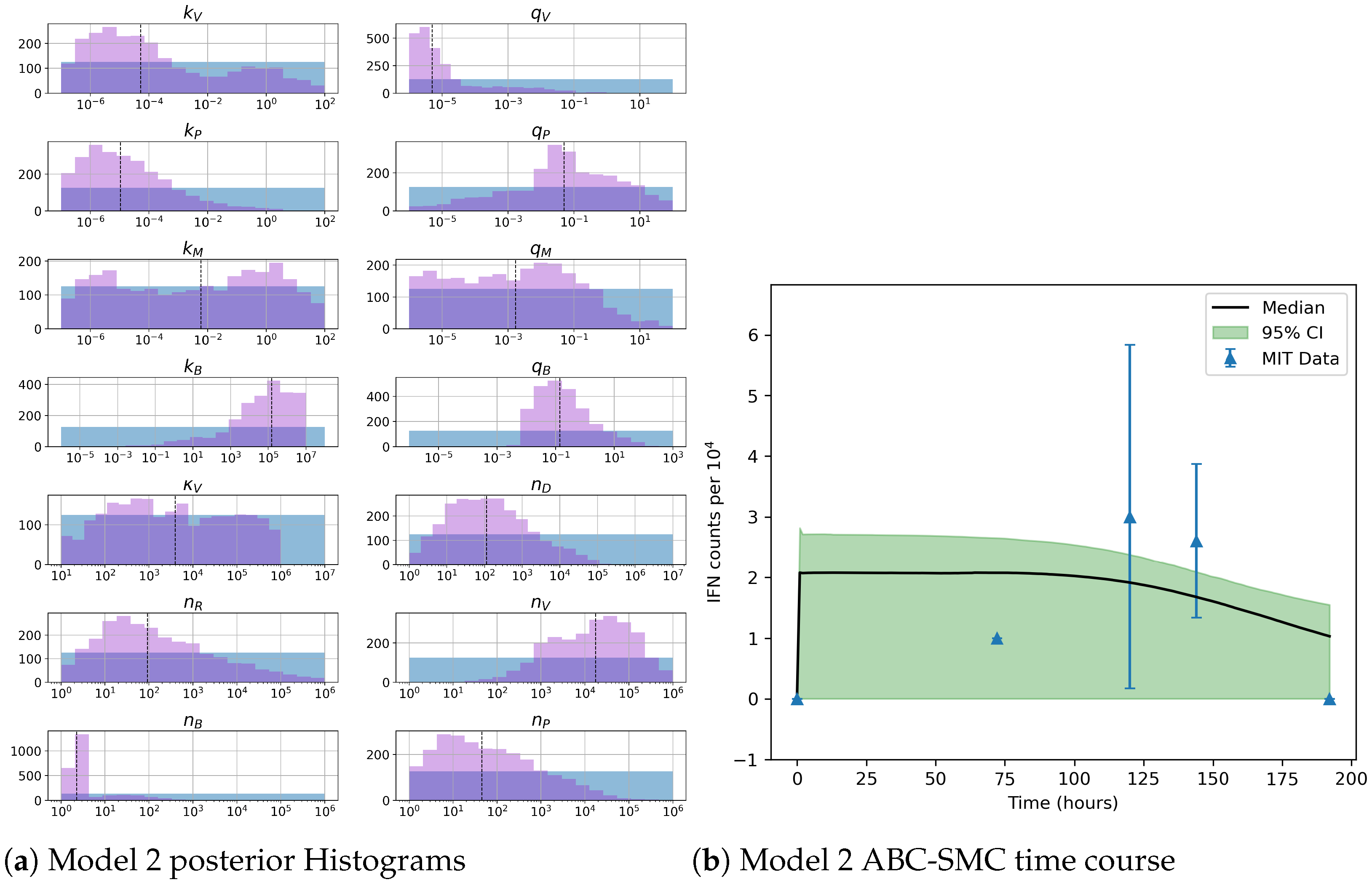
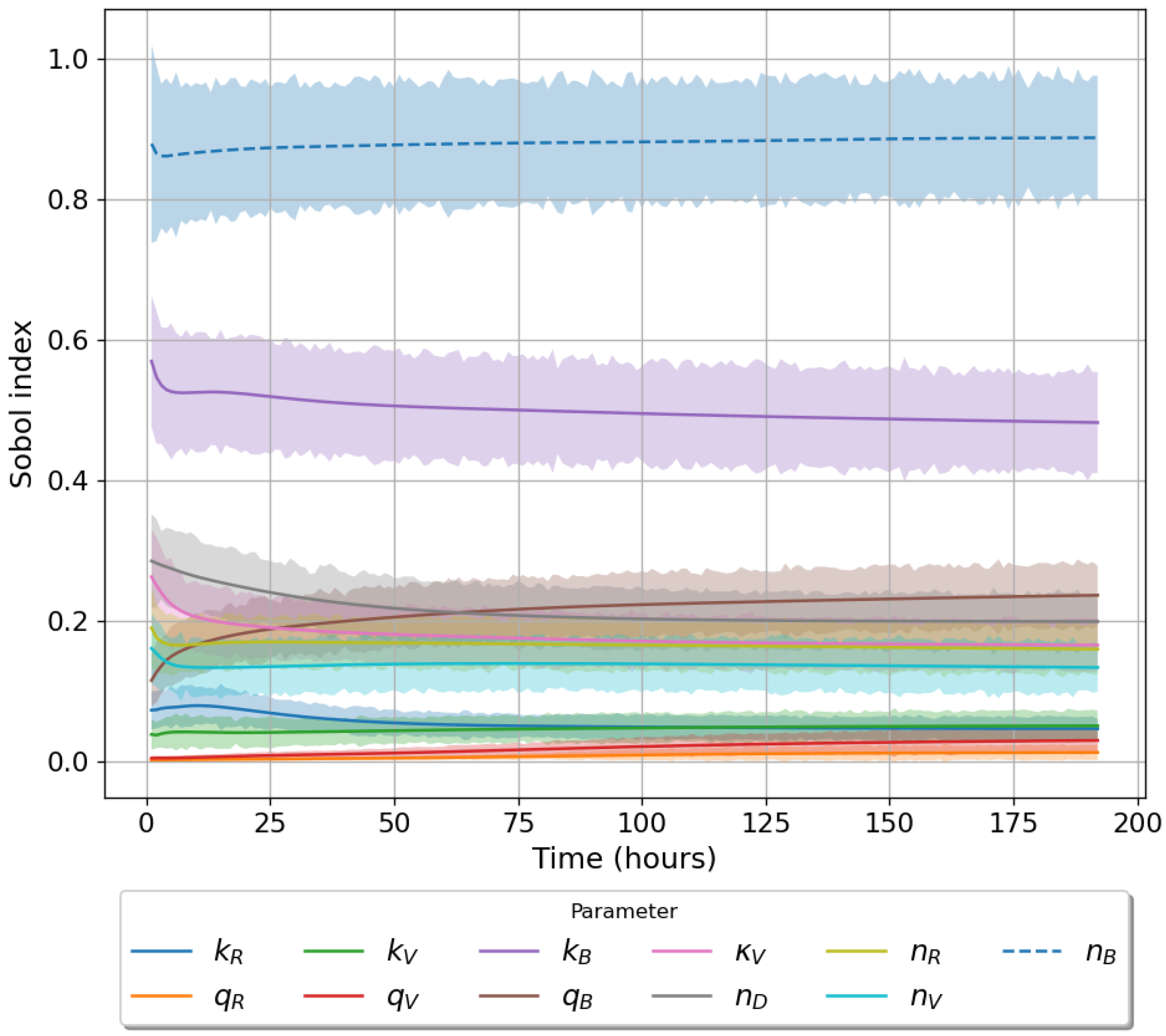
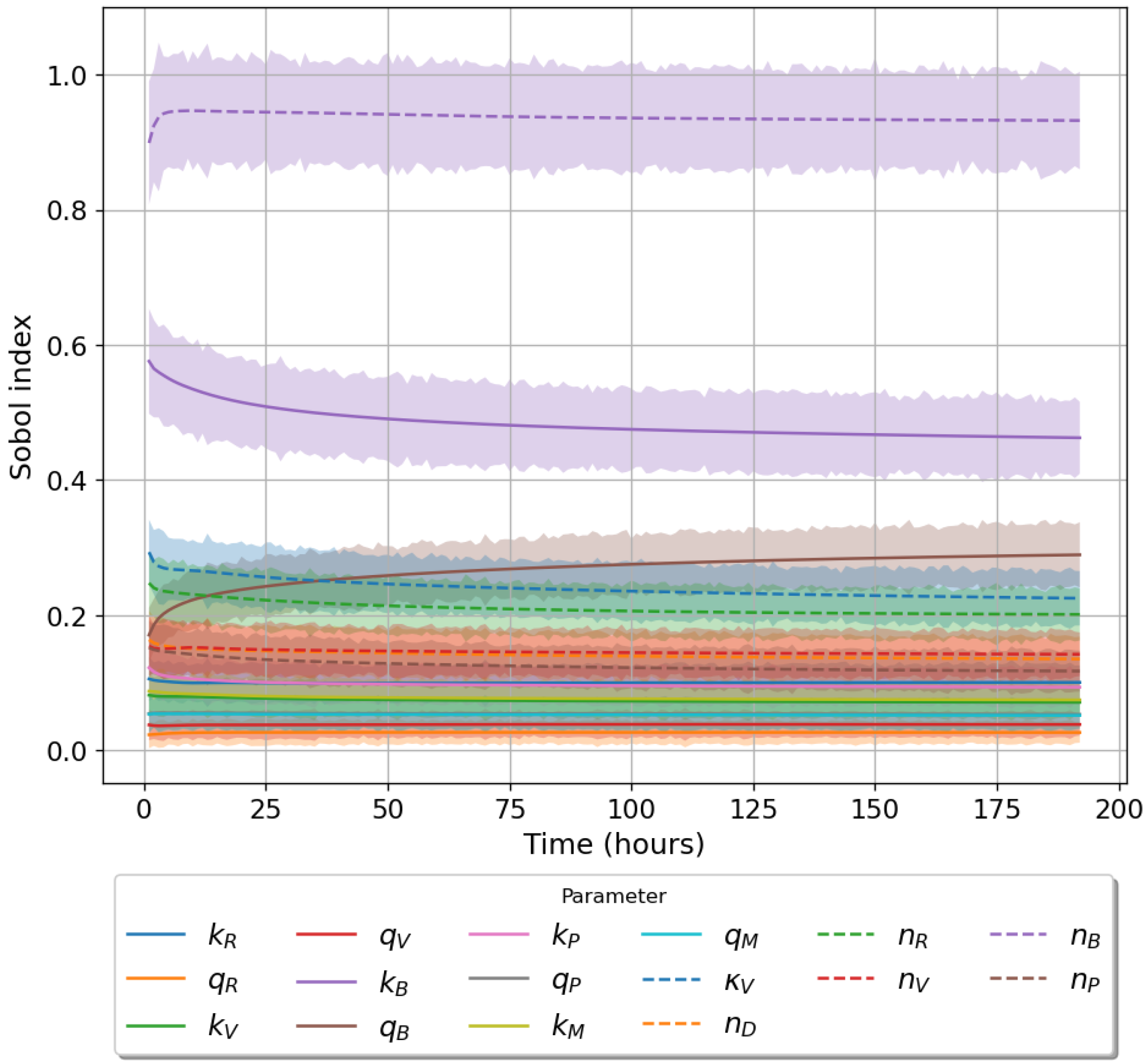
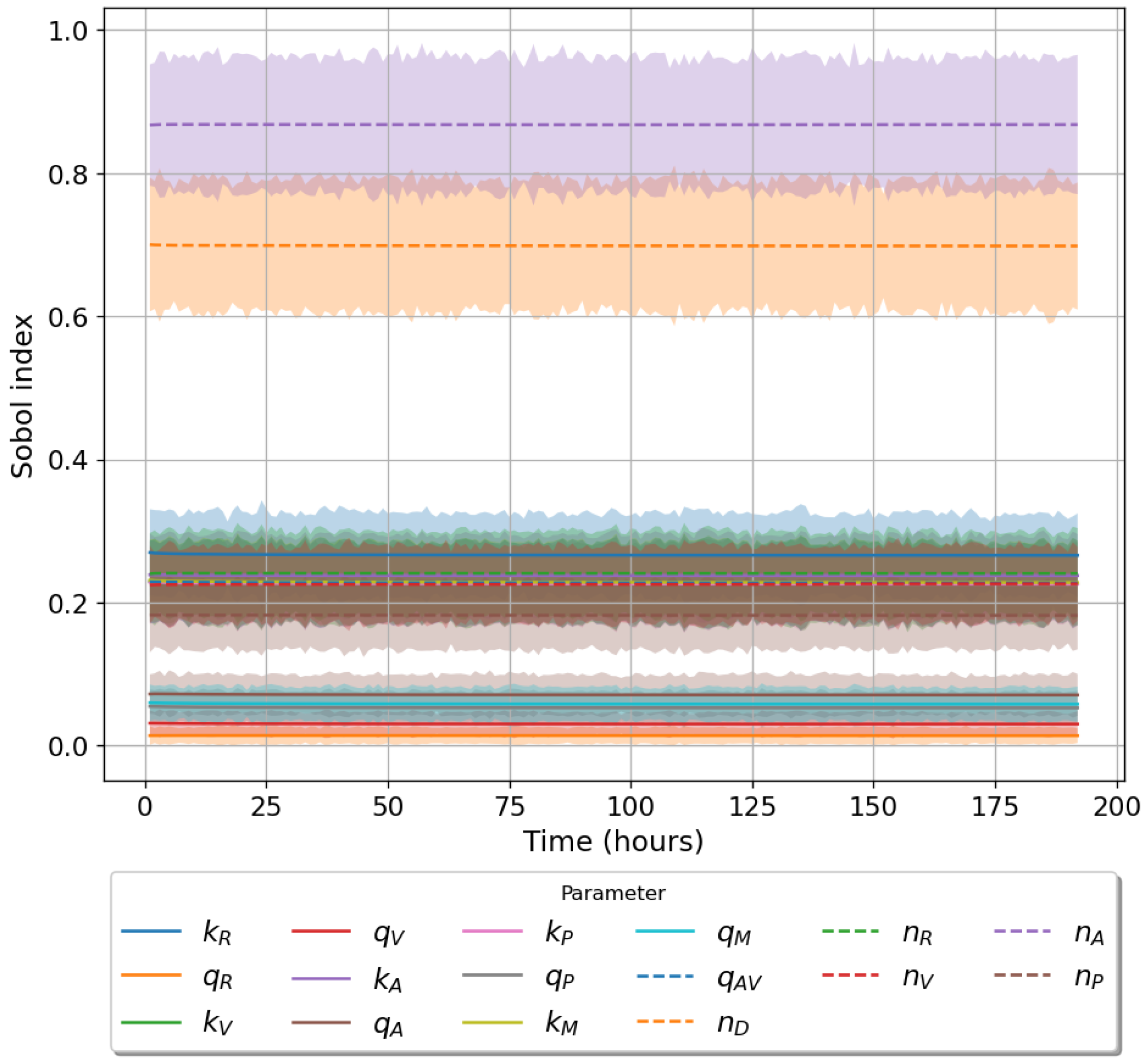
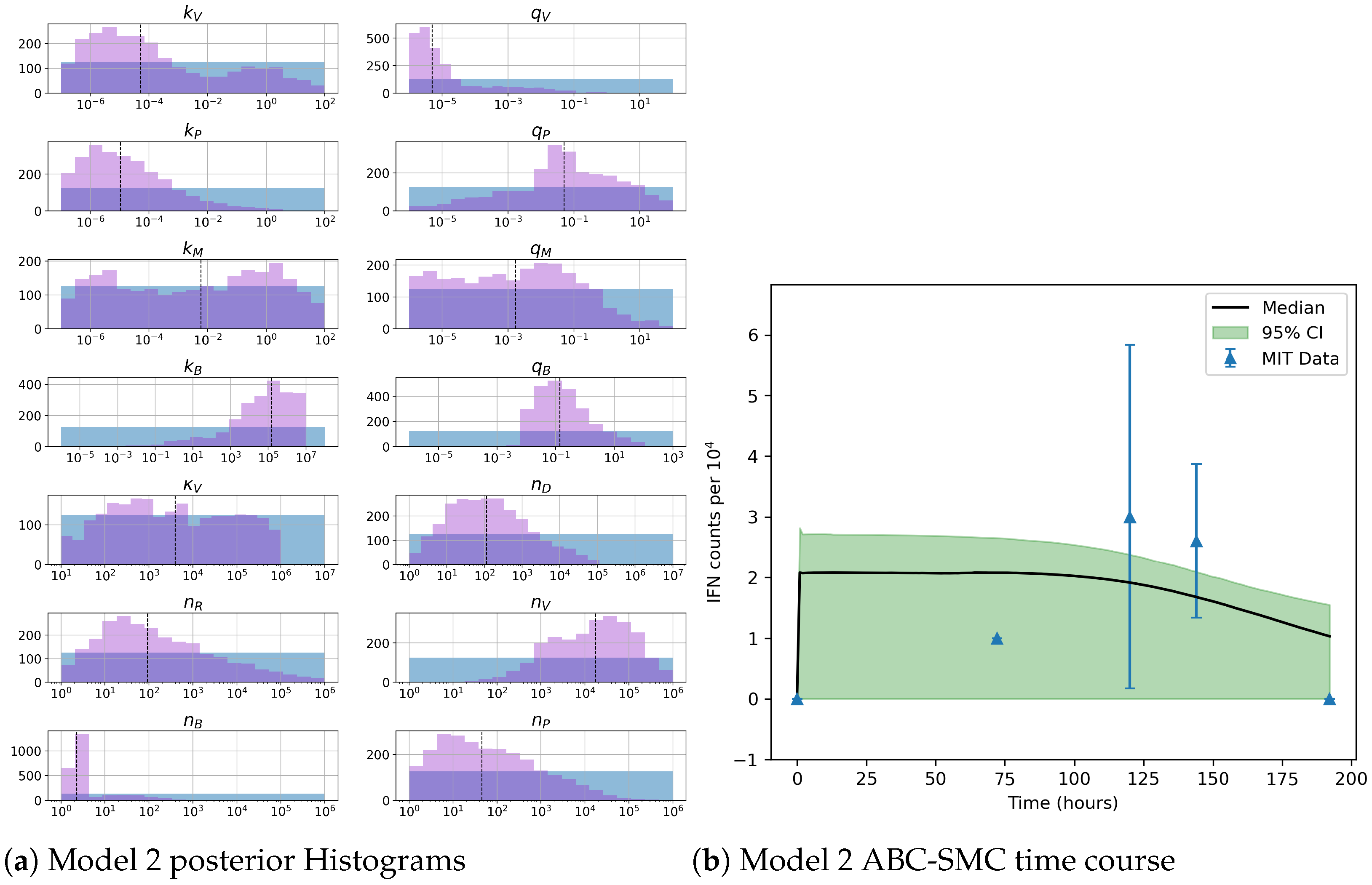
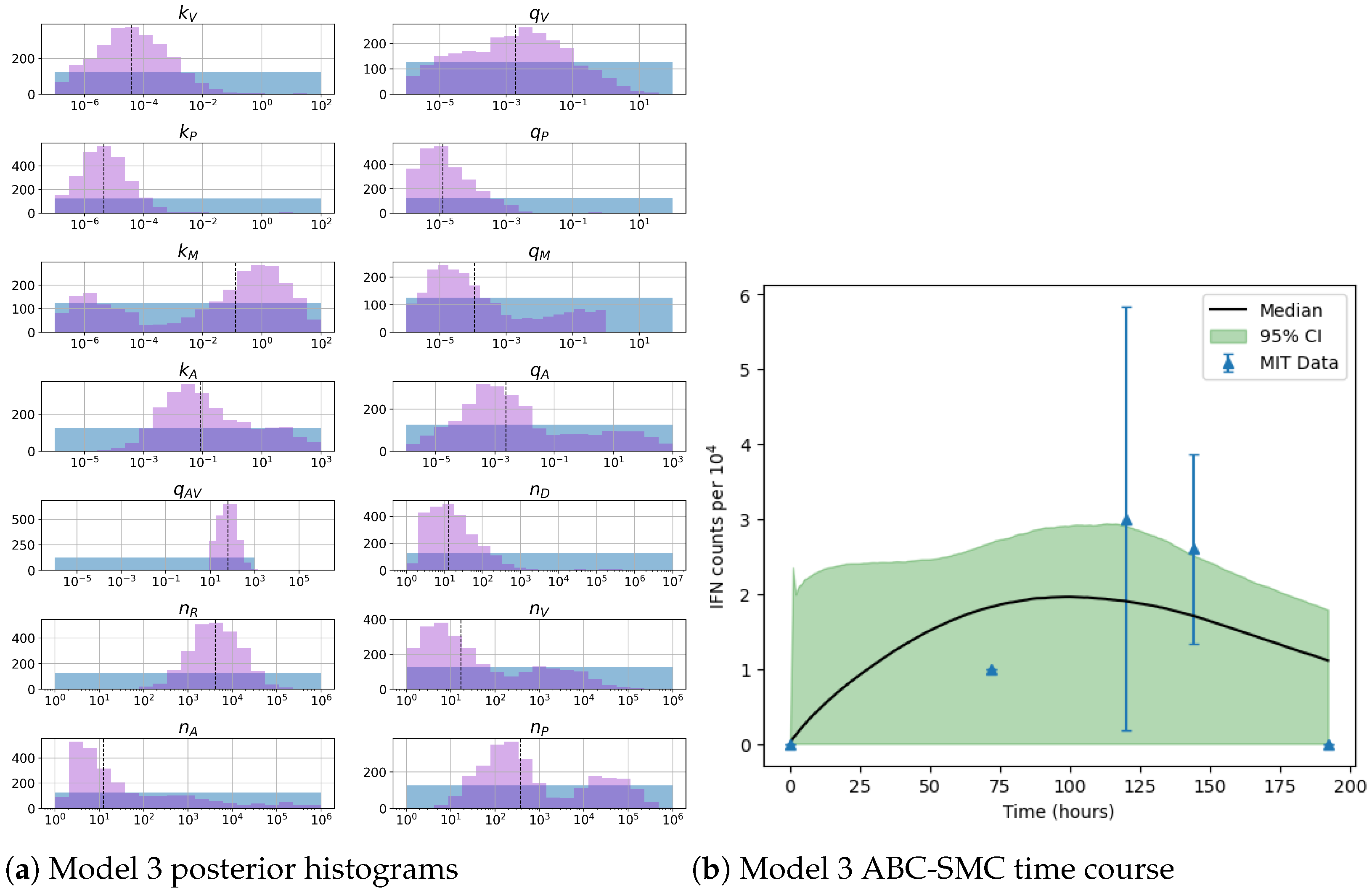
49]. For each model we have assessed its sensitivity and parameter identifiability, as well as carried out model selection and parameter calibration. In particular, we have made use of Sobol sensitivity analysis to identify, for each model and its output, which parameters would need to be closely controlled. We found that two parameters in each model need to be well characterised to avoid large variations in our model outputs. For models 1 and 2 these are the total number of TBK1 molecules,
, and its activation rate,
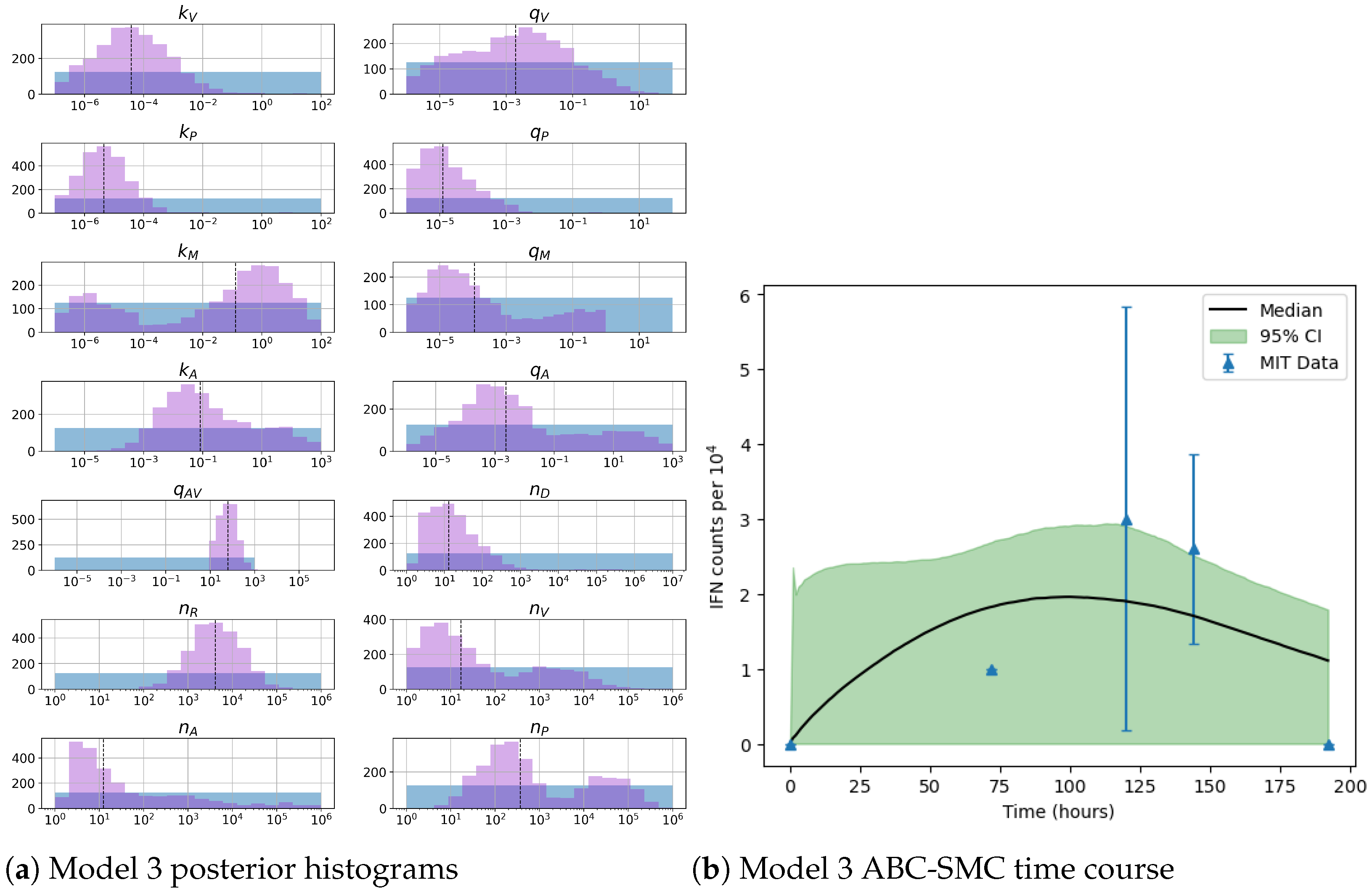
. In the case of model 3, the most important parameters are the total number of PKR molecules,
, and the total copy number of viral RNA molecules,
.
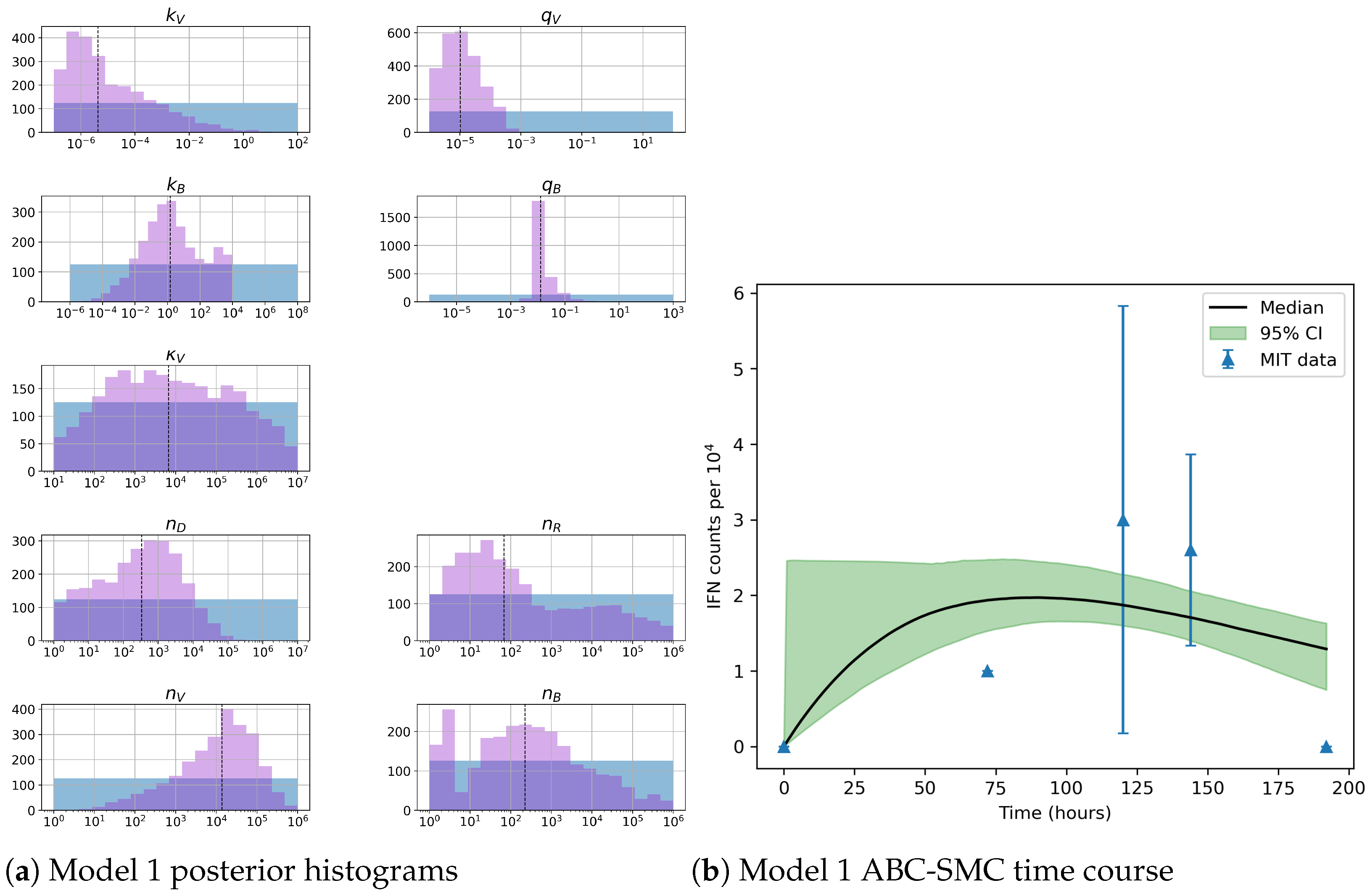
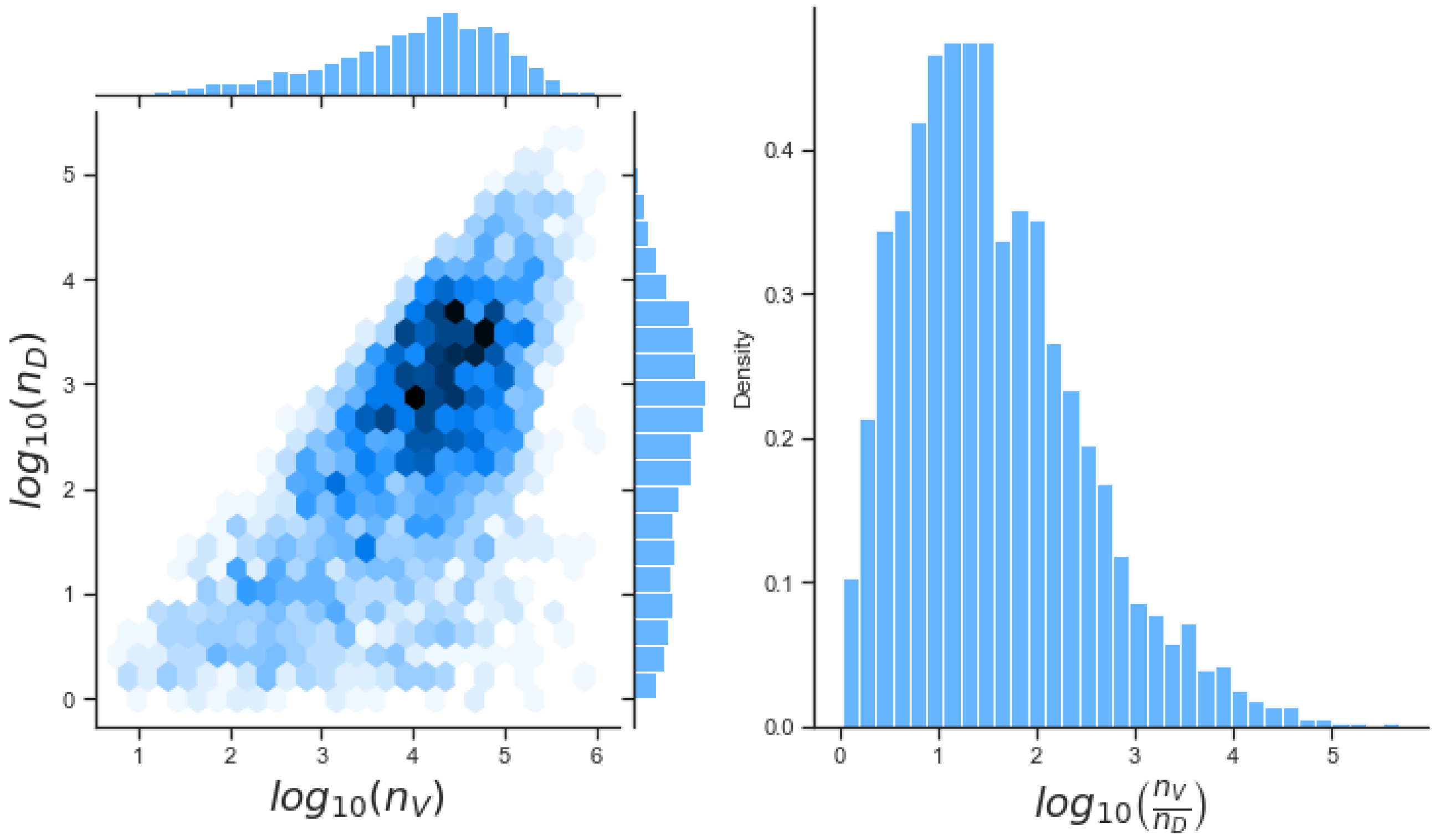
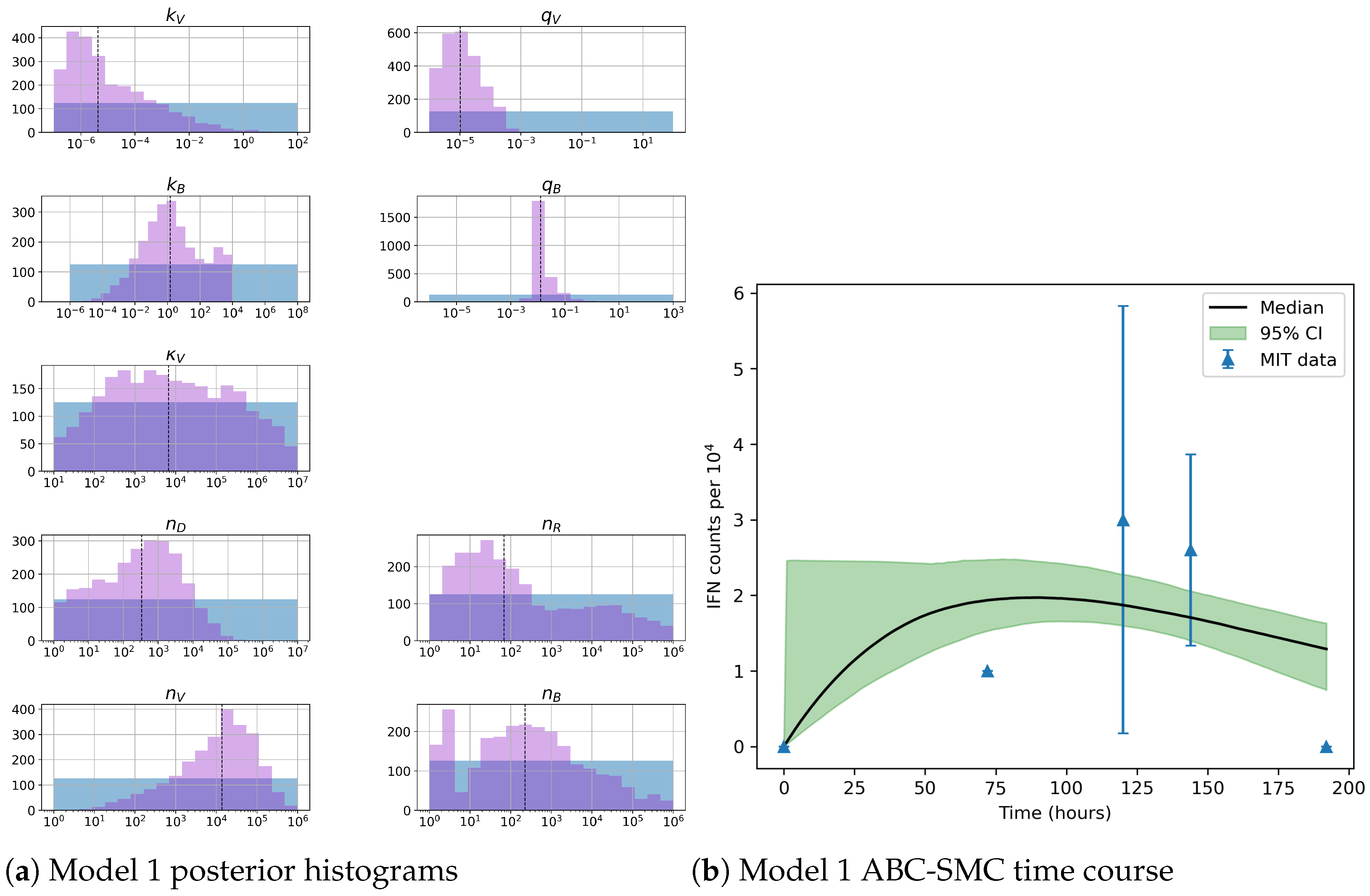
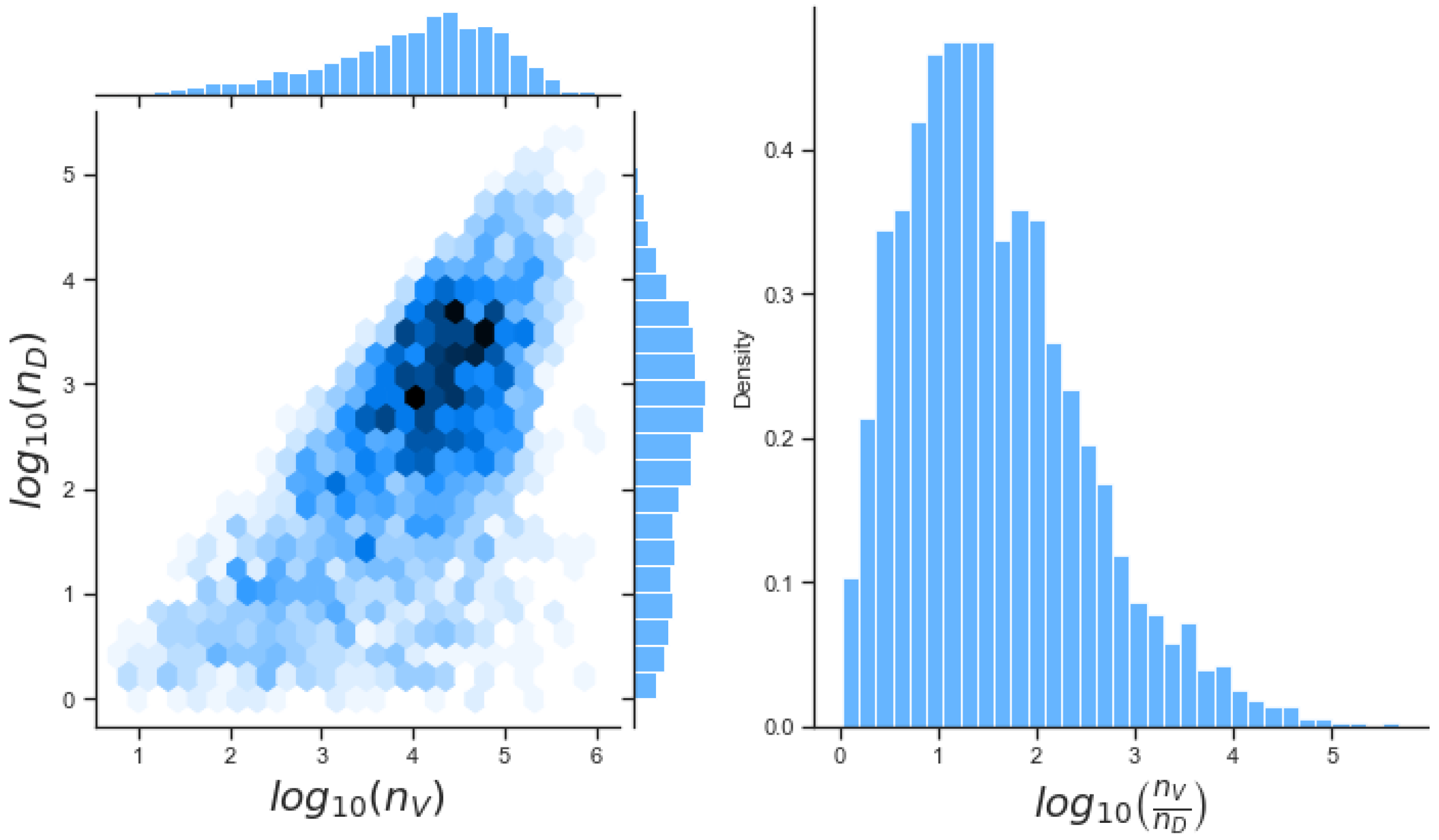
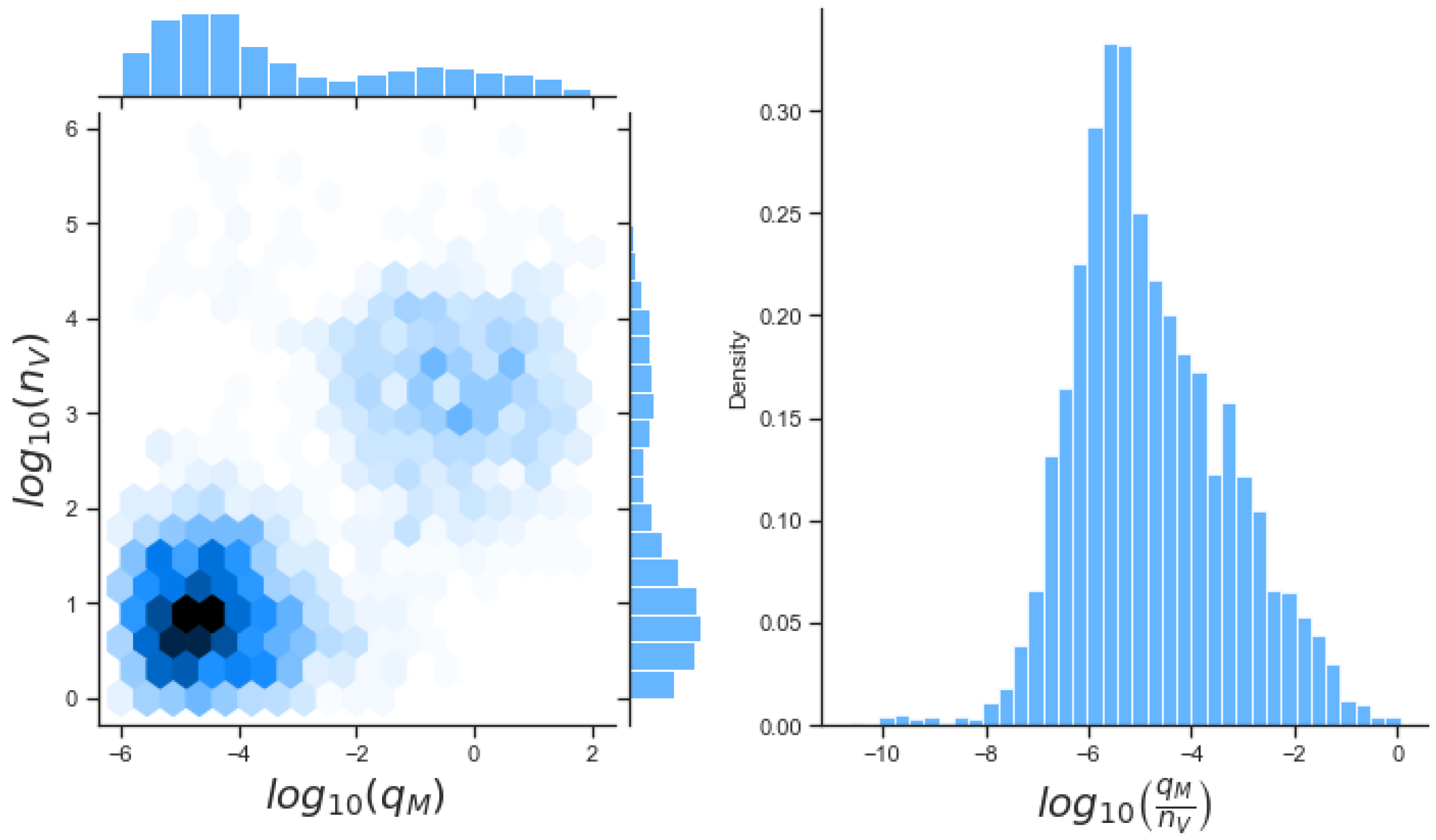
Unfortunately very little is known about the values for the parameters considered in our models. Thus, our aim was to carry out Bayesian inference to narrow down these values. To this end, it was also important to carry out a structural identifiability analysis. This analysis led to the following results: model 3 (considering the PKR pathway) is locally identifiable, but models 1 and 2 are not. We note that these results are in light of the limited data set we had at hand. Yet, this indicated that even with limited data, model 3 might be better, when compared to the other two models, at allowing us to infer parameter values. This was further supported by model selection and parameter inference [
37,
41]: the PKR signalling pathway has a higher percentage of acceptance as illustrated in
Table 7 and narrower posterior distributions for most parameter values (see
Figure 10). Overall model 2 was deemed the worst model of the three: many parameters were non-identifiable, it led to the worst percentage of parameter set acceptance from model selection, and wide posterior distributions for its parameters. Model 1 cannot be rejected since it had the lowest AIC coefficient. However as previously mentioned this model leads to poor learning for most parameter values and is structurally unidentifiable.
While the deterministic models we have presented can be generalised to be applicable to other viruses, it should be remembered that there exist additional signalling pathways and molecular mechanisms which have not been included in the models. Ebola virus infection, which we have examined as a case study, has a number of specific features not characterised by the mechanisms included in our models. Plasmacytoid dendritic cells (pDCs) have been shown to be refractory to EBOV infection, where as common dendritic cells are susceptible [
53]. This could be due to the fact that pDCs express basal (or constitutive) levels of IRF7 prior to infection [
54], and therefore, can be considered in an antiviral state. Thus, when considering the development of a mathematical model it is important to understand not only the virus but also the cellular tropism of the virus and the host (i.e., invertebrate or vertebrate); that is, which cells are the target cells of the virus [
55]. Here we have focused on type I IFNs as essential antiviral cytokines, yet immune responses require a complex and coordinated interaction of a large collection of cytokines and cells, which are out of the scope of this paper. We argue that a more comprehensive data set will be required to consider the development of mathematical models of such complexity [
56,
57].
A final point to consider is the difference between in vivo and in vitro infection. Our models have been parameterised with an in vivo data set. It has been recently highlighted that there exists a stark contrast in responses when comparing in vivo and in vitro infection; in particular, and for the in vitro case, type I IFN production is abrogated after three days post-infection, whereas in the case of in vivo infection type I IFN is present throughout the entire infective period [
11,
12]. Hence it is critical to keep this in mind when carrying out parameter calibration. Along this line of thought, it is also important to note that differences in in vivo experimental models can lead to rather different innate immune responses. For instance, bats are a proposed reservoir for Ebola virus but are known to be asymptomatic for disease. Experiments have indicated that bats have detectable viral RNA levels, but no detectable viremia [
58]. Yet, in the case of humans and non-human primates, the clinical presentation tends to be symptomatic and with measurable viremia [
59,
60]. Bat dendritic cells have shown an enhanced capacity to initiate IFN-dependent responses upon filovirus infection in comparison with, for example, human cells [
61]. Other studies have reported a difference in immune responses depending on the specific tissue analysed: Ebola viral RNA levels persist in male gonads even after a negative PCR test from blood samples [
62].
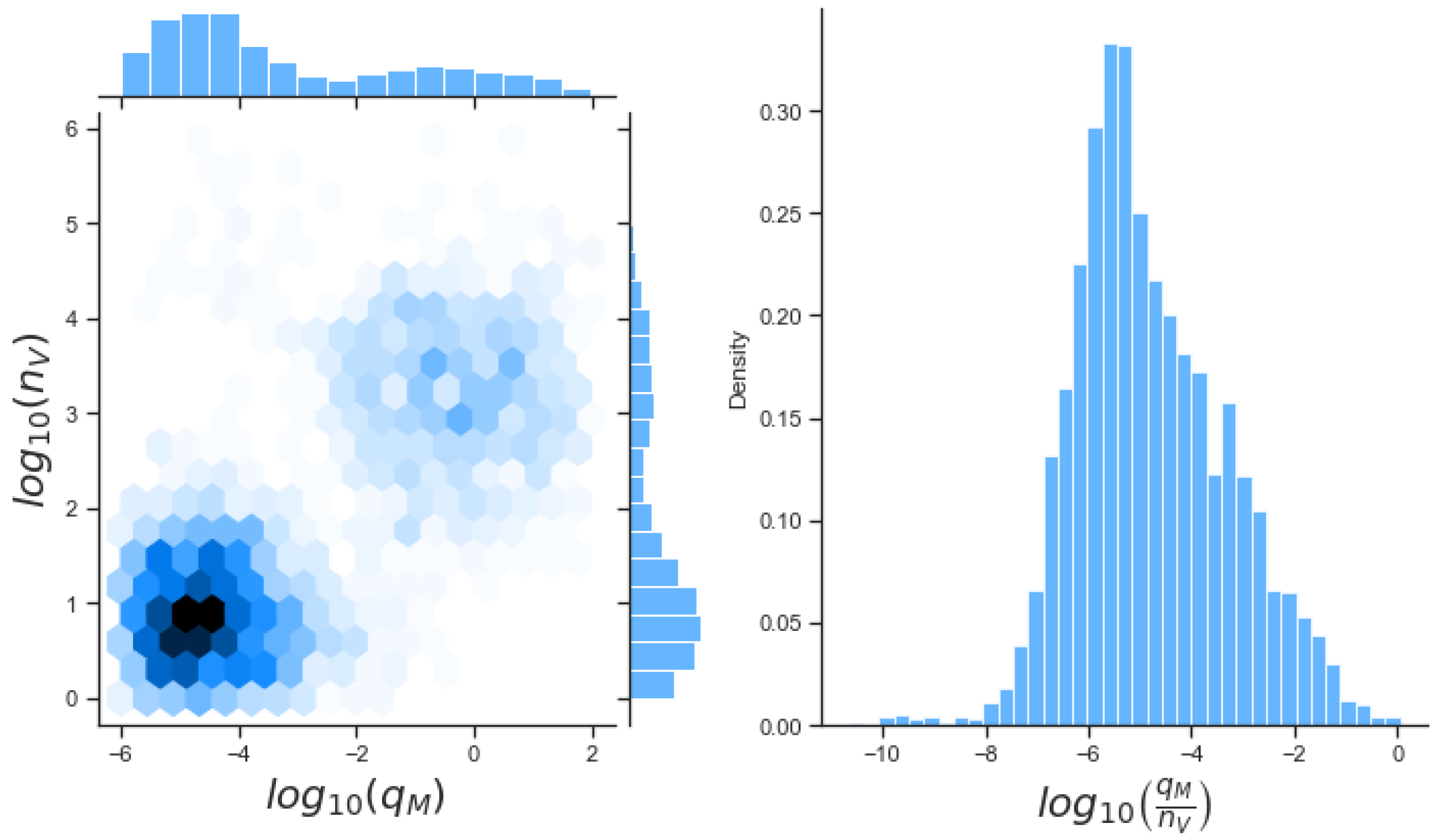
We believe the mathematical approaches presented in this manuscript have allowed us to explore different mechanisms of viral antagonism of type I IFN production. These models could be further expanded to incorporate other intra-cellular or viral mechanisms, or additional signalling pathways. Additional data sets, e.g., from quantitative proteomics, could be used to improve parameter inference, not only for Ebola virus but for other viruses which are of global concern and a public health threat (see
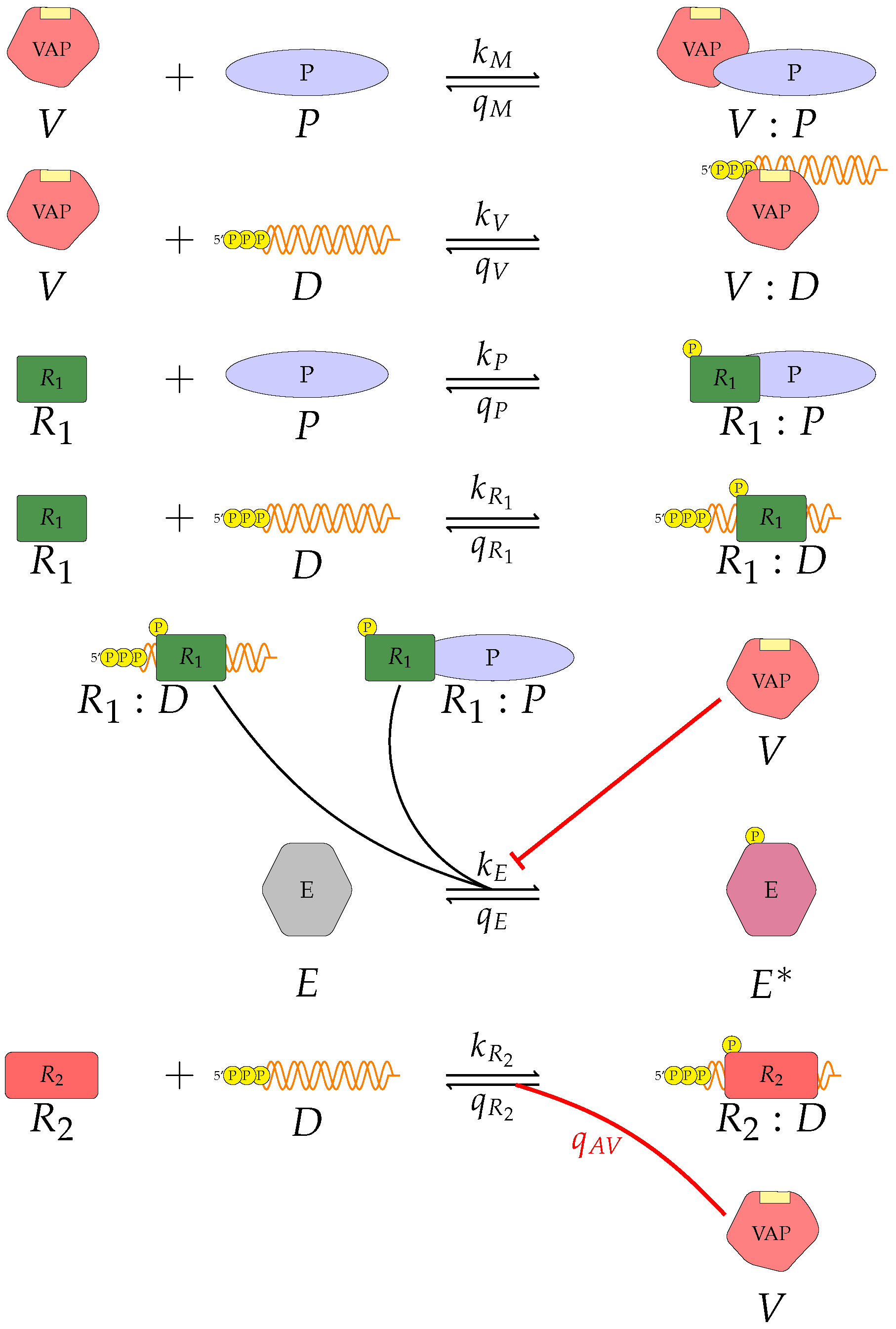
Figure 12). A general (or abstract) model of viral type I IFN inhibition should consider the following proteins and molecules:
V, the viral antagonistic protein for the virus under consideration;
D, the viral RNA;
, a PRR;
P, a dsRNA binding protein;
, an RNA-activated kinase; and
E, a downstream enzyme kinase. The model can be described by the following reactions, which assume mass action kinetics:
A pattern recognition receptor, binds to D to form , with forward rate and backward rate . This is the first step in the signalling pathway.
binds to P to form :P, with forward rate and backward rate .
V binds to D to form V:D, with forward rate and backward rate . This is an upstream inhibition mechanisms: V sequesters D from its potential binding to .
V binds to P to form V:P, with forward rate and backward rate . This is a second upstream inhibition mechanisms: V sequesters P from its potential binding to .
Downstream enzyme kinase, E, gets phosphorylated in the presence of complexes :P and :D. This phosphorylation event is inhibited by the presence of V. This encodes downstream inhibition by V. Phosphorylation and de-phosphorylation rates of E and are and , respectively.
A second intra-cellular signalling pathway can be considered, in addition to the one initiated by . To this end, we consider an RNA-activated kinase, , which can bind to D to form the signalling complex :D. A final mechanism of inhibition by V is included in this pathway. V enhances the disassociation of :D.
One last note to conclude. Our models have been restricted to be of a deterministic nature and not stochastic. We plan to generalise the models introduced here to birth and death Markov processes [
63] to include the effects that might arise due to a small number of proteins, as can be the case during the first stages of intra-cellular infection. Effects which have been completely ignored in our models, since we have assumed the initial number of molecules (
and
) is large to allow us for a deterministic approach. This might not be the case always, and thus, an stochastic perspective will be desirable and required.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}