
Novel Picornavirus Detected in Wild Deer: Identification, Genomic Characterisation, and Prevalence in Australia

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
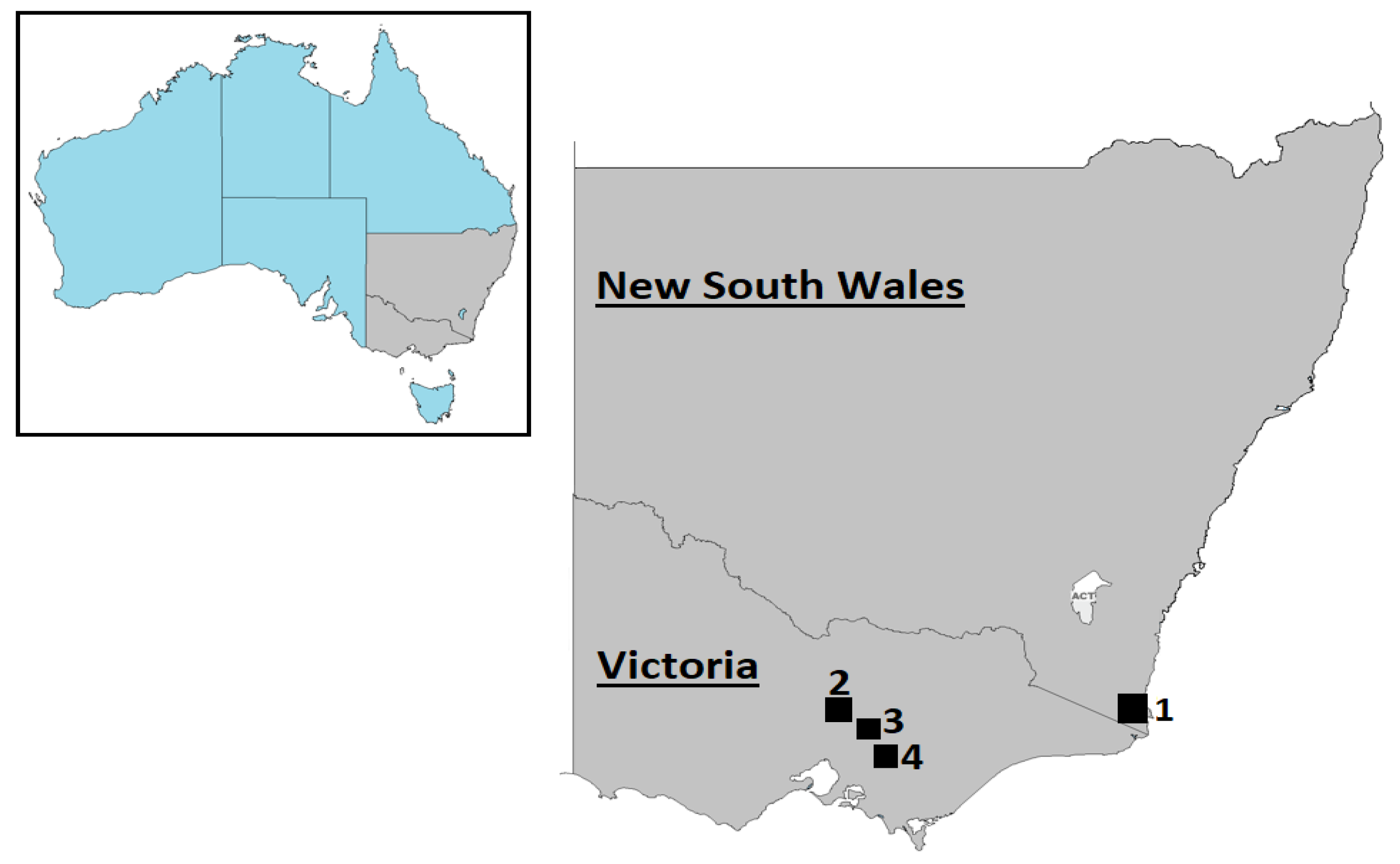
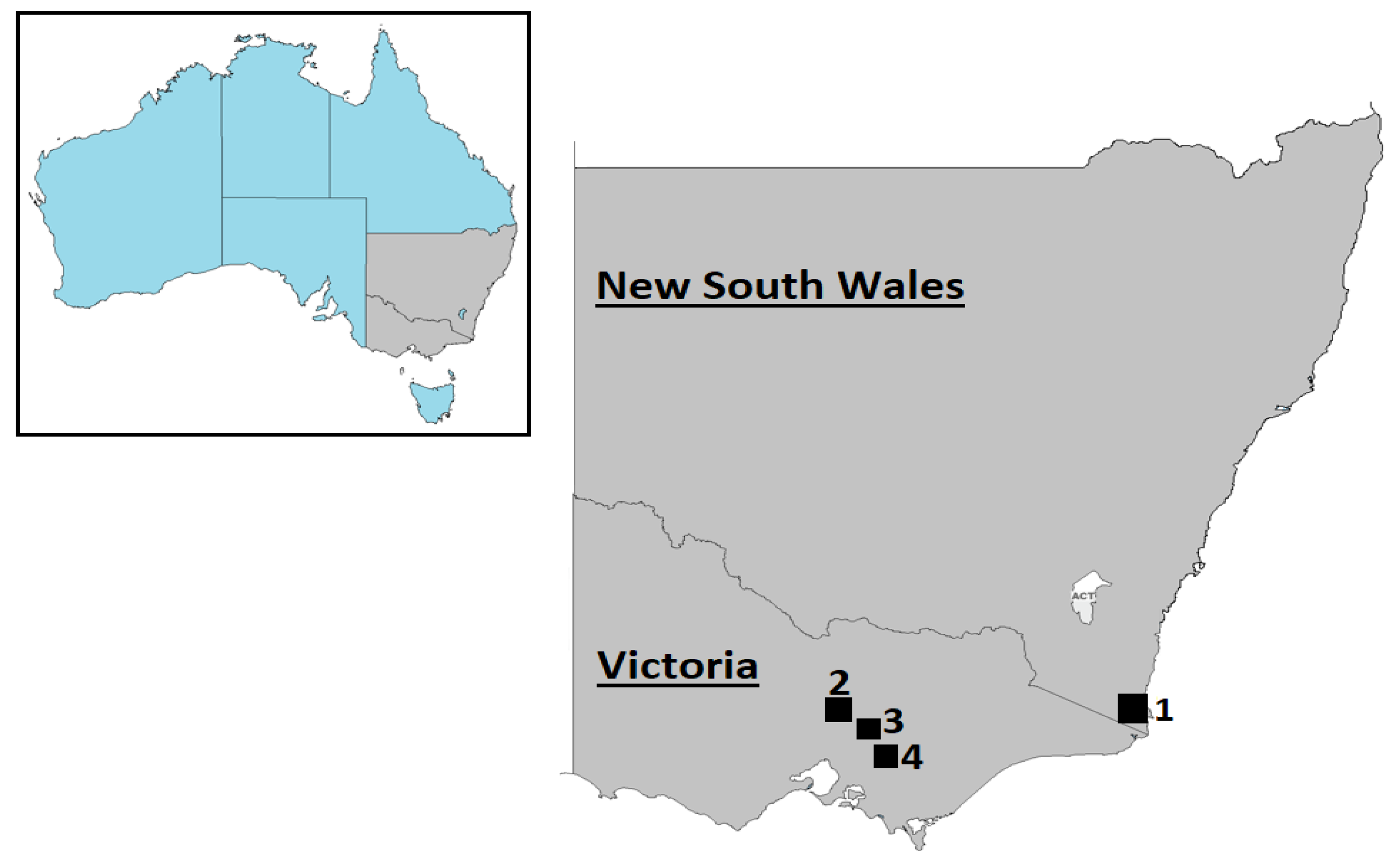
2.1. Sample Collection and Preparation
2.2. Viral Metagenomic Analysis
2.3. Bioinformatic Analysis
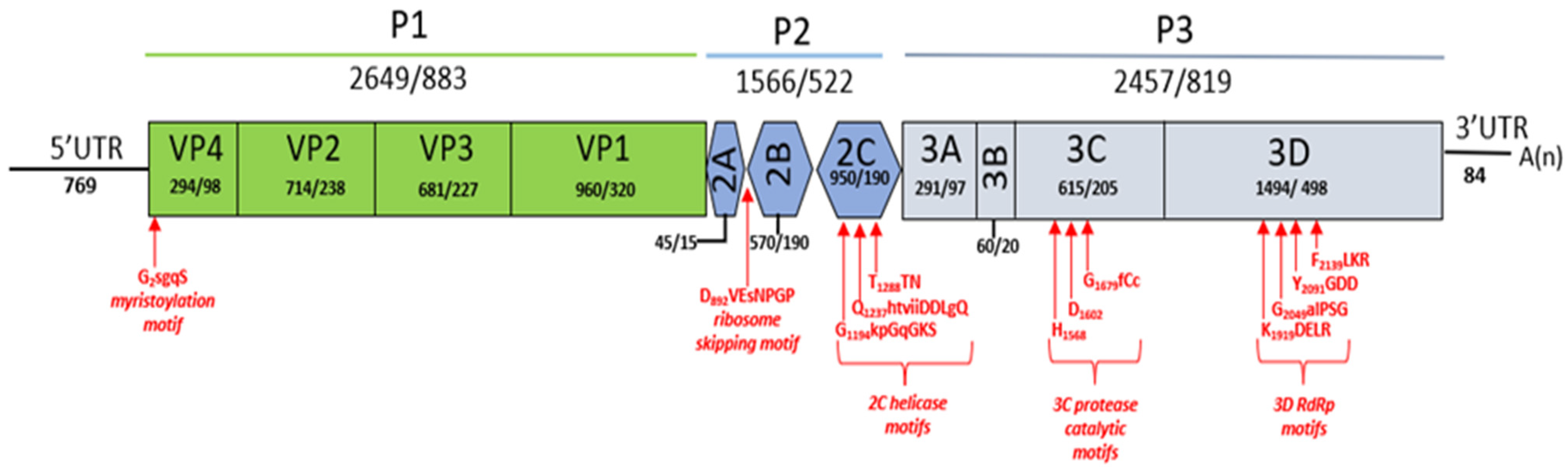
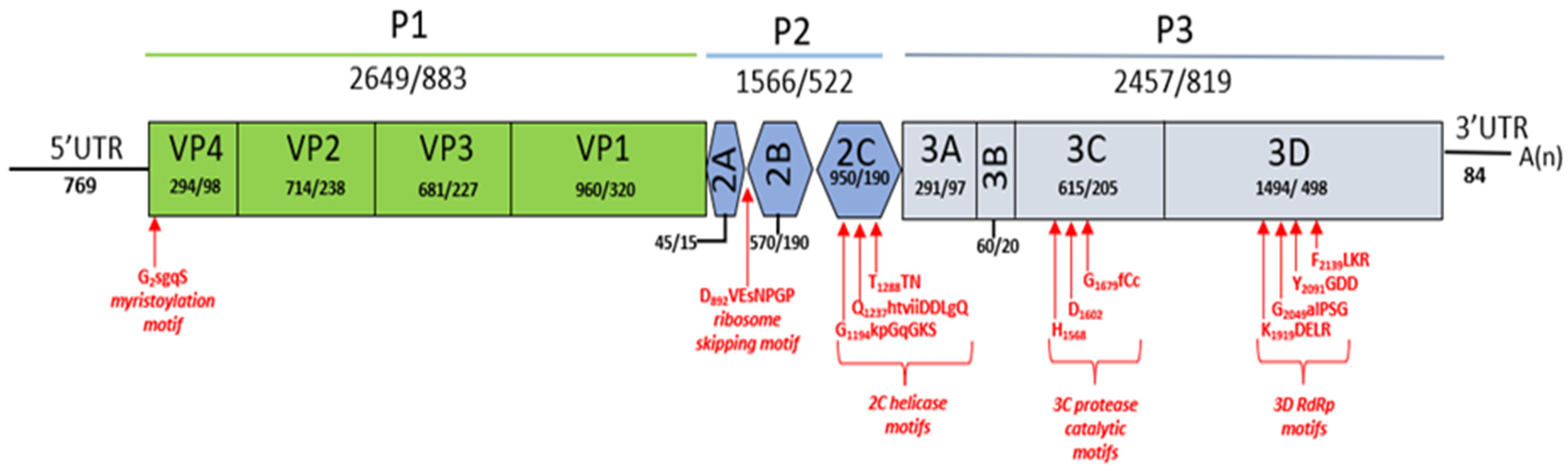
2.4. Genomic Characterisation
2.5. RT-PCR Detection of Deer/Bopivirus
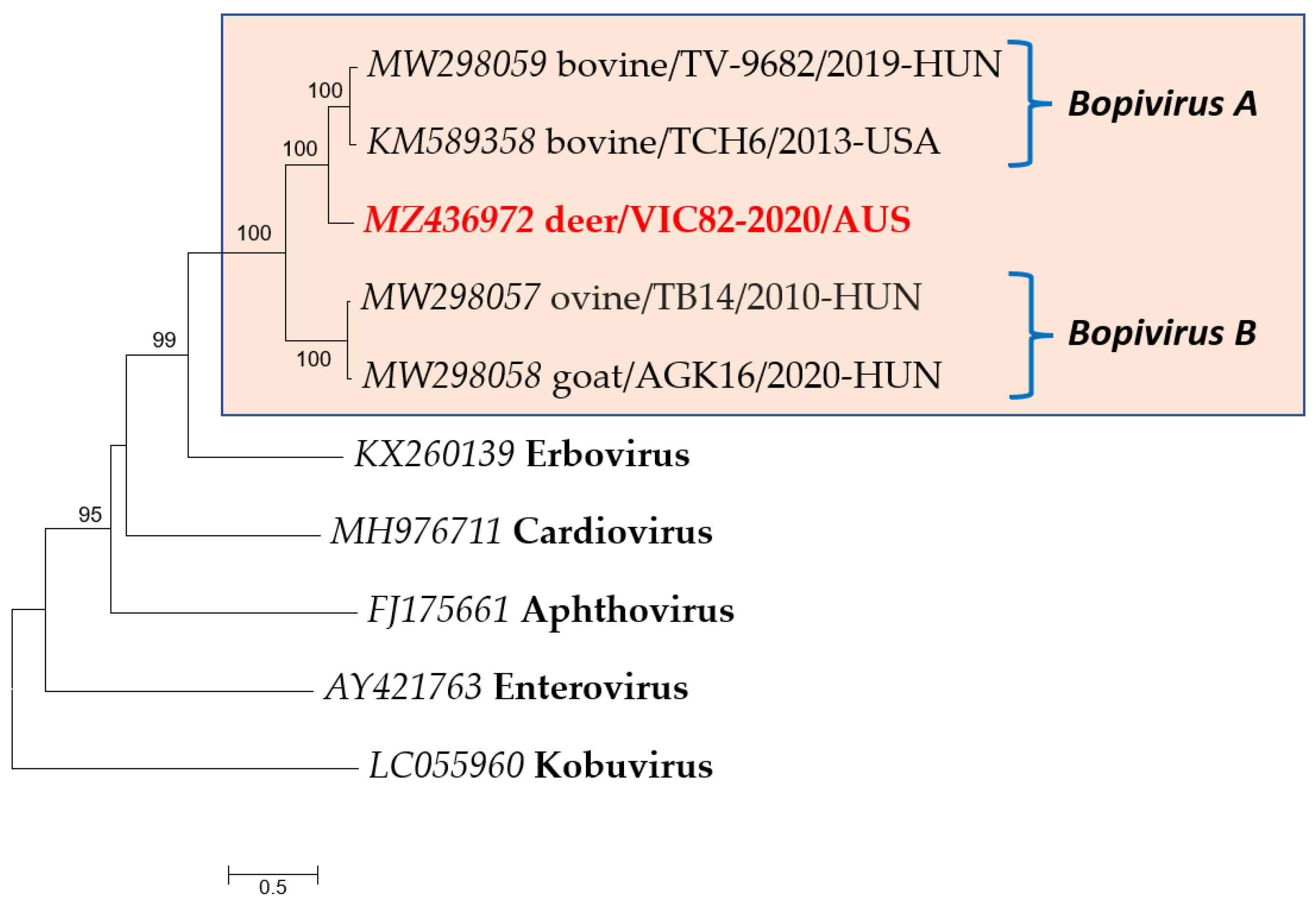
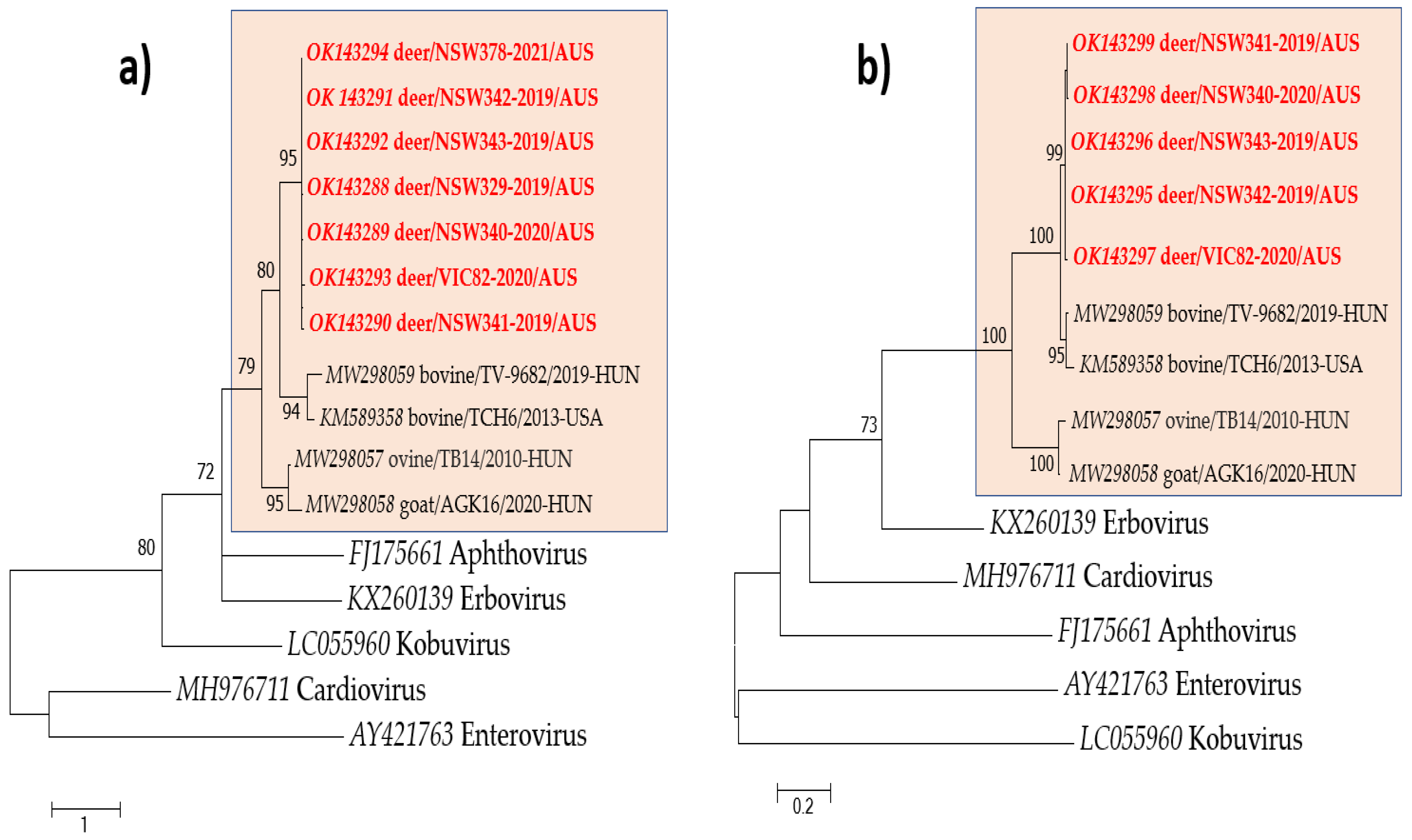
2.6. Phylogenetic Analysis
2.7. Virus Inoculation in Cell Cultures
2.8. Data Availability
3. Results
3.1. High-Throughput Sequencing and Assembly
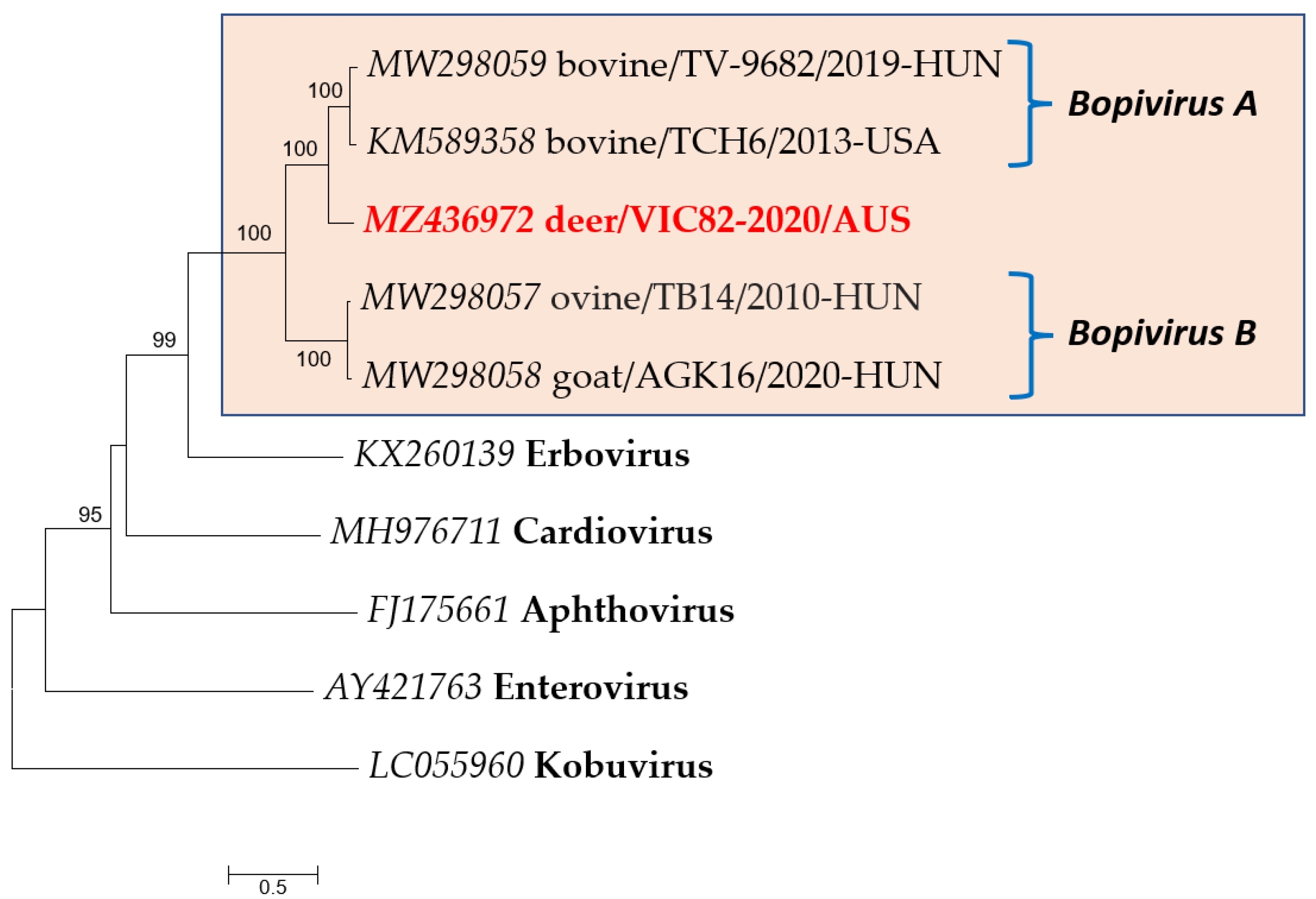
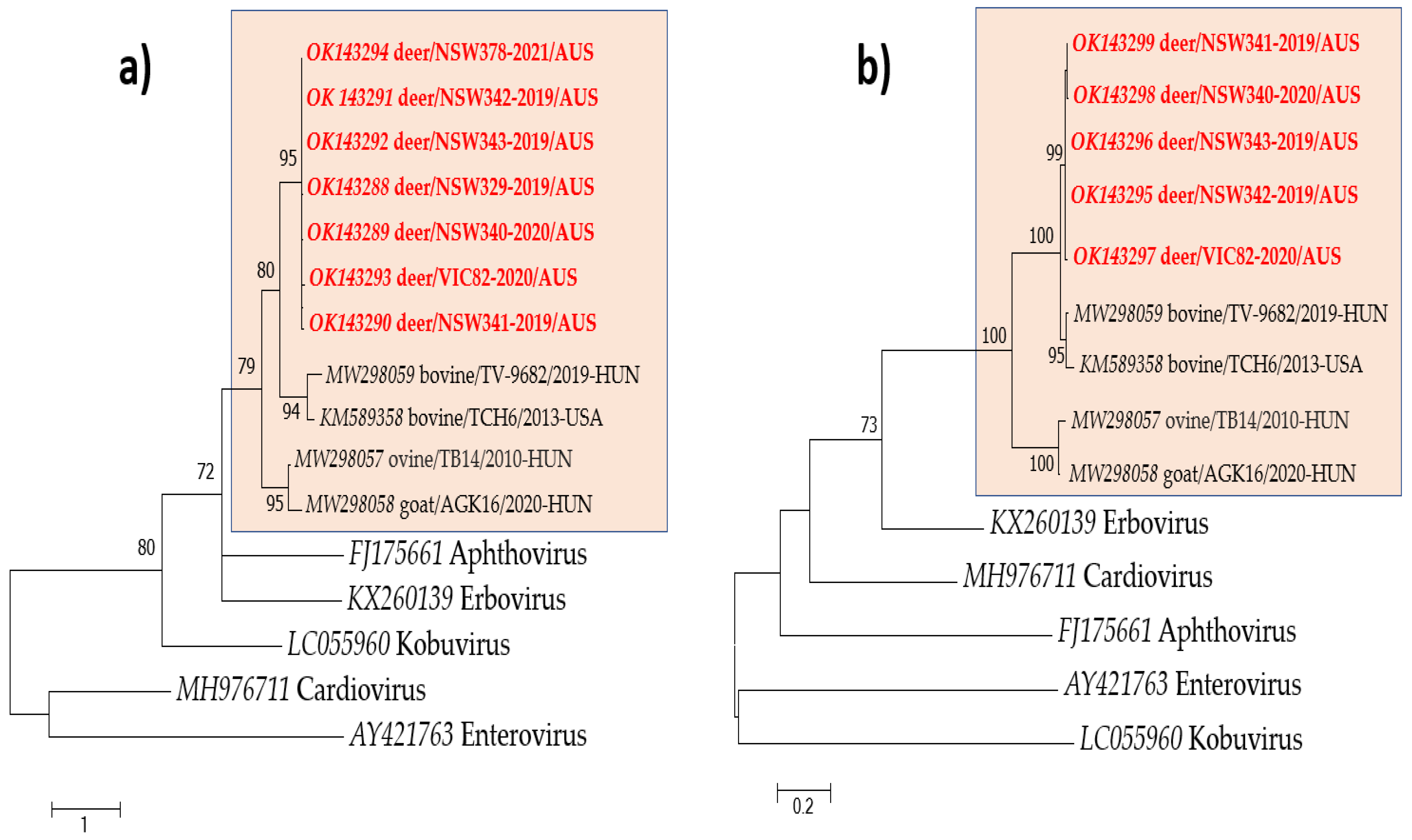
3.2. Genomic and Phylogenetic Analysis of Bopivirus-Like Sequence
3.3. Prevalence of Deer/Bopivirus
3.4. Viral Culture
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woods, R.; Reiss, A.; Cox-Witton, K.; Grillo, T.; Peters, A. The importance of wildlife disease monitoring as part of global surveillance for zoonotic diseases: The role of Australia. Trop. Med. Infect. Dis. 2019, 4, 29. [Google Scholar] [CrossRef] [Green Version]
- Mokili, J.L.; Rohwer, F.; Dutilh, B.E. Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2012, 2, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Zell, R. Picornaviridae-the ever-growing virus family. Arch. Virol. 2018, 163, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef]
- Hayer, J.; Wille, M.; Font, A.; Gonzalez-Aravena, M.; Norder, H.; Malmberg, M. Four novel picornaviruses detected in Magellanic Penguins (Spheniscus magellanicus) in Chile. Virology 2021, 560, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Shan, T.; Chen, Z.; Zhou, J.; Li, H.; Tong, G.; Liu, G. Isolation and complete genome analysis of a novel duck picornavirus in China. Vet. Microbiol. 2021, 253, 108950. [Google Scholar] [CrossRef]
- Zhang, M.; Li, Q.; Wu, F.; Ou, Z.; Li, Y.; You, F.; Chen, Q. Epidemiology, genetic characterization, and evolution of Hunnivirus carried by Rattus norvegicus and Rattus tanezumi: The first epidemiological evidence from Southern China. Pathogens 2021, 10, 661. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Sibley, S.D.; Chapman, C.A.; Knowles, N.J.; Lauck, M.; Johnson, J.C.; Lawson, C.C.; Lackemeyer, M.G.; Valenta, K.; Omeja, P.; et al. Discovery of Lanama virus, a distinct member of species Kunsagivirus C (Picornavirales: Picornaviridae), in wild vervet monkeys (Chlorocebus pygerythrus). Viruses 2020, 12, 1436. [Google Scholar] [CrossRef]
- Laszlo, Z.; Pankovics, P.; Reuter, G.; Csagola, A.; Balint, A.; Albert, M.; Boros, A. Multiple types of novel enteric bopiviruses (Picornaviridae) with the possibility of interspecies transmission identified from cloven-hoofed domestic livestock (ovine, caprine and bovine) in Hungary. Viruses 2021, 13, 66. [Google Scholar] [CrossRef]
- Cripps, J.K.; Pacioni, C.; Scroggie, M.P.; Woolnough, A.P.; Ramsey, D.S.L. Introduced deer and their potential role in disease transmission to livestock in Australia. Mammal Rev. 2019, 49, 60–77. [Google Scholar] [CrossRef] [Green Version]
- Huaman, J.L.; Pacioni, C.; Forsyth, D.M.; Pople, A.; Hampton, J.O.; Carvalho, T.G.; Helbig, K.J. Serosurveillance and molecular investigation of wild deer in Australia reveals seroprevalence of Pestivirus infection. Viruses 2020, 12, 752. [Google Scholar] [CrossRef] [PubMed]
- Huaman, J.L.; Pacioni, C.; Forsyth, D.M.; Pople, A.; Hampton, J.O.; Helbig, K.J.; Carvalho, T.G. Evaluation of haemoparasite and Sarcocystis infections in Australian wild deer. Int. J. Parasitol. Parasites Wildl. 2021, 15, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Huaman, J.L.; Pacioni, C.; Sarker, S.; Doyle, M.; Forsyth, D.M.; Pople, A.; Hampton, J.O.; Carvalho, T.; Helbig, K.J. Molecular epidemiology and characterization of Picobirnavirus in wild deer and cattle from Australia: Evidence of genogroup I and II in the upper respiratory tract. Viruses 2021, 13, 1492. [Google Scholar] [CrossRef]
- Di Martino, B.; Di Profio, F.; Melegari, I.; Di Felice, E.; Robetto, S.; Guidetti, C.; Orusa, R.; Martella, V.; Marsilio, F. Molecular detection of kobuviruses in European roe deer (Capreolus capreolus) in Italy. Arch. Virol. 2015, 160, 2083–2086. [Google Scholar] [CrossRef]
- Ley, V.; Higgins, J.; Fayer, R. Bovine enteroviruses as indicators of fecal contamination. Appl. Environ. Microbiol. 2002, 68, 3455–3461. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Mistry, J.; Schuster-Bockler, B.; Griffiths-Jones, S.; Hollich, V.; Lassmann, T.; Moxon, S.; Marshall, M.; Khanna, A.; Durbin, R.; et al. Pfam: Clans, web tools and services. Nucleic Acids Res. 2006, 34, D247–D251. [Google Scholar] [CrossRef] [Green Version]
- Kolekar, P.; Pataskar, A.; Kulkarni-Kale, U.; Pal, J.; Kulkarni, A. IRESPred: Web Server for Prediction of Cellular and Viral Internal Ribosome Entry Site (IRES). Sci. Rep. 2016, 6, 27436. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021, 49, D192–D200. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Marc, D.; Drugeon, G.; Haenni, A.L.; Girard, M.; van der Werf, S. Role of myristoylation of poliovirus capsid protein VP4 as determined by site-directed mutagenesis of its N-terminal sequence. EMBO J. 1989, 8, 2661–2668. [Google Scholar] [CrossRef]
- Donnelly, M.L.L.; Luke, G.; Mehrotra, A.; Li, X.; Hughes, L.E.; Gani, D.; Ryan, M.D. Analysis of the aphthovirus 2A/2B polyprotein ‘cleavage’ mechanism indicates not a proteolytic reaction, but a novel translational effect: A putative ribosomal ’skip’. J. Gen. Virol. 2001, 82, 1013–1025. [Google Scholar] [CrossRef]
- Corbic Ramljak, I.; Stanger, J.; Real-Hohn, A.; Dreier, D.; Wimmer, L.; Redlberger-Fritz, M.; Fischl, W.; Klingel, K.; Mihovilovic, M.D.; Blaas, D.; et al. Cellular N-myristoyltransferases play a crucial picornavirus genus-specific role in viral assembly, virion maturation, and infectivity. PLoS Pathog. 2018, 14, e1007203. [Google Scholar] [CrossRef]
- Hughes, P.J.; Stanway, G. The 2A proteins of three diverse picornaviruses are related to each other and to the H-rev107 family of proteins involved in the control of cell proliferation. J. Gen. Virol. 2000, 81, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Pallansch, M.A. Molecular evolution of the human enteroviruses: Correlation of serotype with VP1 sequence and application to picornavirus classification. J. Virol. 1999, 73, 1941–1948. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Wang, F.; Ni, Z.; Yun, T.; Yu, B.; Huang, J.; Chen, J. Genetic diversity of the VP1 gene of duck hepatitis virus type I (DHV-I) isolates from southeast China is related to isolate attenuation. Virus Res. 2008, 137, 137–141. [Google Scholar] [CrossRef]
- Cook, S.; Chung, B.Y.; Bass, D.; Moureau, G.; Tang, S.; McAlister, E.; Culverwell, C.L.; Glucksman, E.; Wang, H.; Brown, T.D.; et al. Novel virus discovery and genome reconstruction from field RNA samples reveals highly divergent viruses in dipteran hosts. PLoS ONE 2013, 8, e80720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuter, G.; Boros, A.; Pankovics, P. Kobuviruses-a comprehensive review. Rev. Med. Virol. 2011, 21, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Khamrin, P.; Maneekarn, N.; Okitsu, S.; Ushijima, H. Epidemiology of human and animal kobuviruses. Virusdisease 2014, 25, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Melegari, I.; Di Profio, F.; Sarchese, V.; Martella, V.; Marsilio, F.; Di Martino, B. First molecular evidence of kobuviruses in goats in Italy. Arch. Virol. 2016, 161, 3245–3248. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Sampling Location | Deer Species | Contig Length (nt) | Subject Cover | Best Hit (AC Number) | Perc. Identity |
|---|---|---|---|---|---|---|
| NSW301 | Kiah-NSW | Fallow deer a | 332 | 95% | Bopivirus A isolate TCH6 (KM589358) | 86% |
| 95% | Bopivirus sp. strain bovine/TV-9682/2019-HUN(MW298059) | 82% | ||||
| NSW341 | Kiah-NSW | Fallow deer a | 644 | 63% | Bopivirus A isolate TCH6 (KM589358) | 86% |
| 63% | Bopivirus sp. strain bovine/TV-9682/2019-HUN(MW298059) | 86% | ||||
| VIC82 | Victoria | Fallow deer a | 7554 | 55% | Bopivirus A isolate TCH6 (KM589358) | 79% |
| 61% | Bopivirus sp. strain bovine/TV-9682/2019-HUN(MW298059) | 78% |
| Virus | Accession Number | Host | Genomic Features | Pairwise Amino Acid Identity (%) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Size (nt) | GC Content | Polyprotein | P1 | 2C | 3C | 3D | |||
| bovine/TCH6/2013-USA | KM589358 | Cattle | 7018 | 50.4% | 79 | 66.7 | 91.2 | 90.7 | 88.5 |
| bovine/TV-9682/2019-HUN | MW298059 | Cattle | 7571 | 50.2% | 78.7 | 66.1 | 91.2 | 90.2 | 89.7 |
| ovine/TB14/2010-HUN | MW298057 | Sheep | 7385 | 54.9% | 58.7 | 57.2 | 62.5 | 66.8 | 66.9 |
| goat/AGK16/2020-HUN | MW298058 | Goat | 7426 | 55.1% | 58.2 | 55.7 | 62.1 | 66.8 | 67.3 |
| Location | Fallow Deer | Sambar Deer | Red Deer | Cattle | ||||
|---|---|---|---|---|---|---|---|---|
| N | n (%, CI) | N | n (%, CI) | N | n (%, CI) | N | n (%, CI) | |
| New South Wales | 59 | 5 (8.5, 3.7–18.4) | 3 | 0 (0–56.2) | 6 | 1 (16.7, 3.0–56.4) | 8 | 0 (0–32.4) |
| Victoria | 12 | 1 (8.3, 1.5–35.4) | 11 | 0 (0–25.9) | 0 | 0 | 15 | 0 (0–20.4) |
| Total | 71 | 6 (8.5, 3.9–17.2) | 14 | 0 (0–21.5) | 6 | 1 (16.7, 3.0–56.4) | 23 | 0 (0–14.3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huaman, J.L.; Pacioni, C.; Sarker, S.; Doyle, M.; Forsyth, D.M.; Pople, A.; Carvalho, T.G.; Helbig, K.J. Novel Picornavirus Detected in Wild Deer: Identification, Genomic Characterisation, and Prevalence in Australia. Viruses 2021, 13, 2412. https://doi.org/10.3390/v13122412
Huaman JL, Pacioni C, Sarker S, Doyle M, Forsyth DM, Pople A, Carvalho TG, Helbig KJ. Novel Picornavirus Detected in Wild Deer: Identification, Genomic Characterisation, and Prevalence in Australia. Viruses. 2021; 13(12):2412. https://doi.org/10.3390/v13122412
Chicago/Turabian StyleHuaman, Jose L., Carlo Pacioni, Subir Sarker, Mark Doyle, David M. Forsyth, Anthony Pople, Teresa G. Carvalho, and Karla J. Helbig. 2021. "Novel Picornavirus Detected in Wild Deer: Identification, Genomic Characterisation, and Prevalence in Australia" Viruses 13, no. 12: 2412. https://doi.org/10.3390/v13122412
APA StyleHuaman, J. L., Pacioni, C., Sarker, S., Doyle, M., Forsyth, D. M., Pople, A., Carvalho, T. G., & Helbig, K. J. (2021). Novel Picornavirus Detected in Wild Deer: Identification, Genomic Characterisation, and Prevalence in Australia. Viruses, 13(12), 2412. https://doi.org/10.3390/v13122412