
Innate Immune Signaling and Role of Glial Cells in Herpes Simplex Virus- and Rabies Virus-Induced Encephalitis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Viral Infection Routes to Enter the Central Nervous System
1.1. General Considerations
1.2. Which Factors Support RABV and HSV Replication in the Nervous System?
2. The Role of Glial Cells in Maintaining CNS Homeostasis
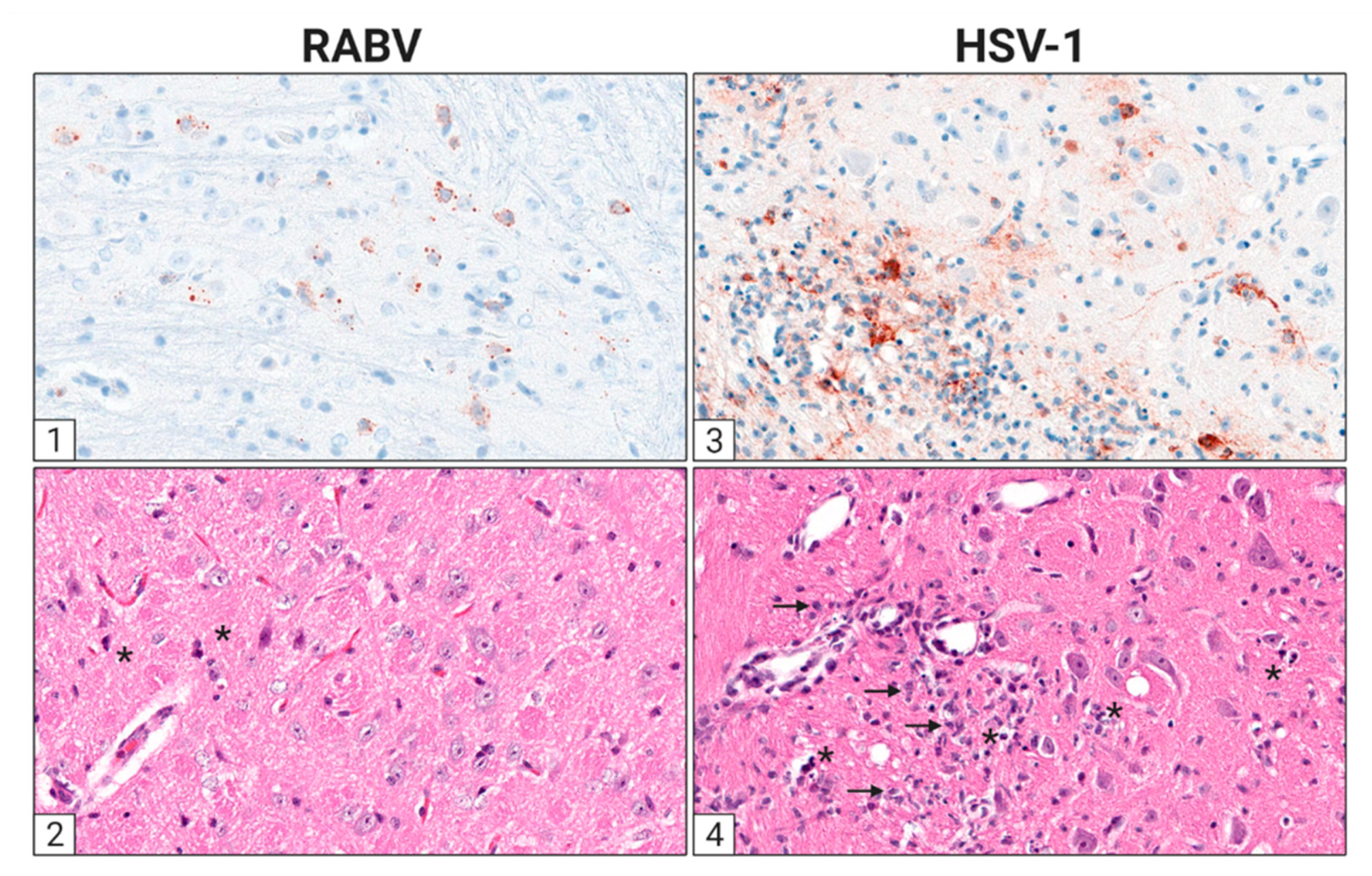
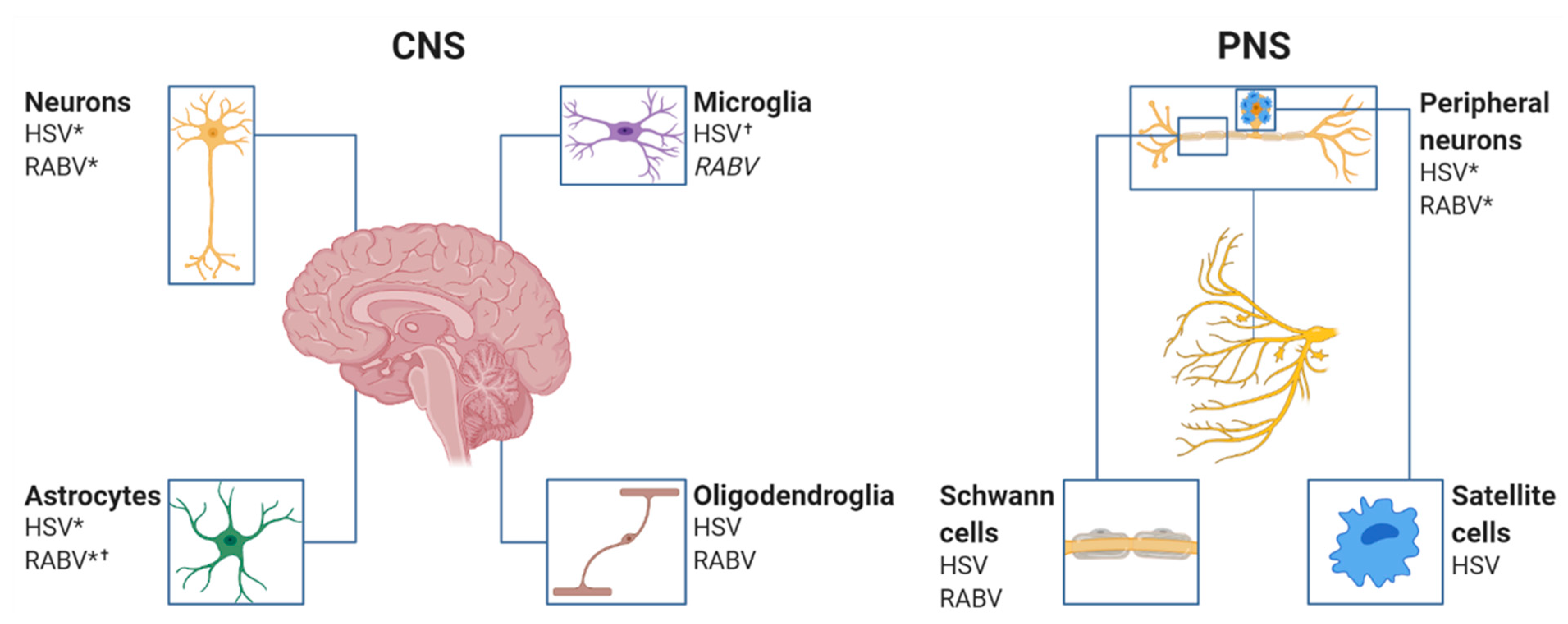
3. The Cellular Infection Pattern of RABV and HSV in the Nervous System
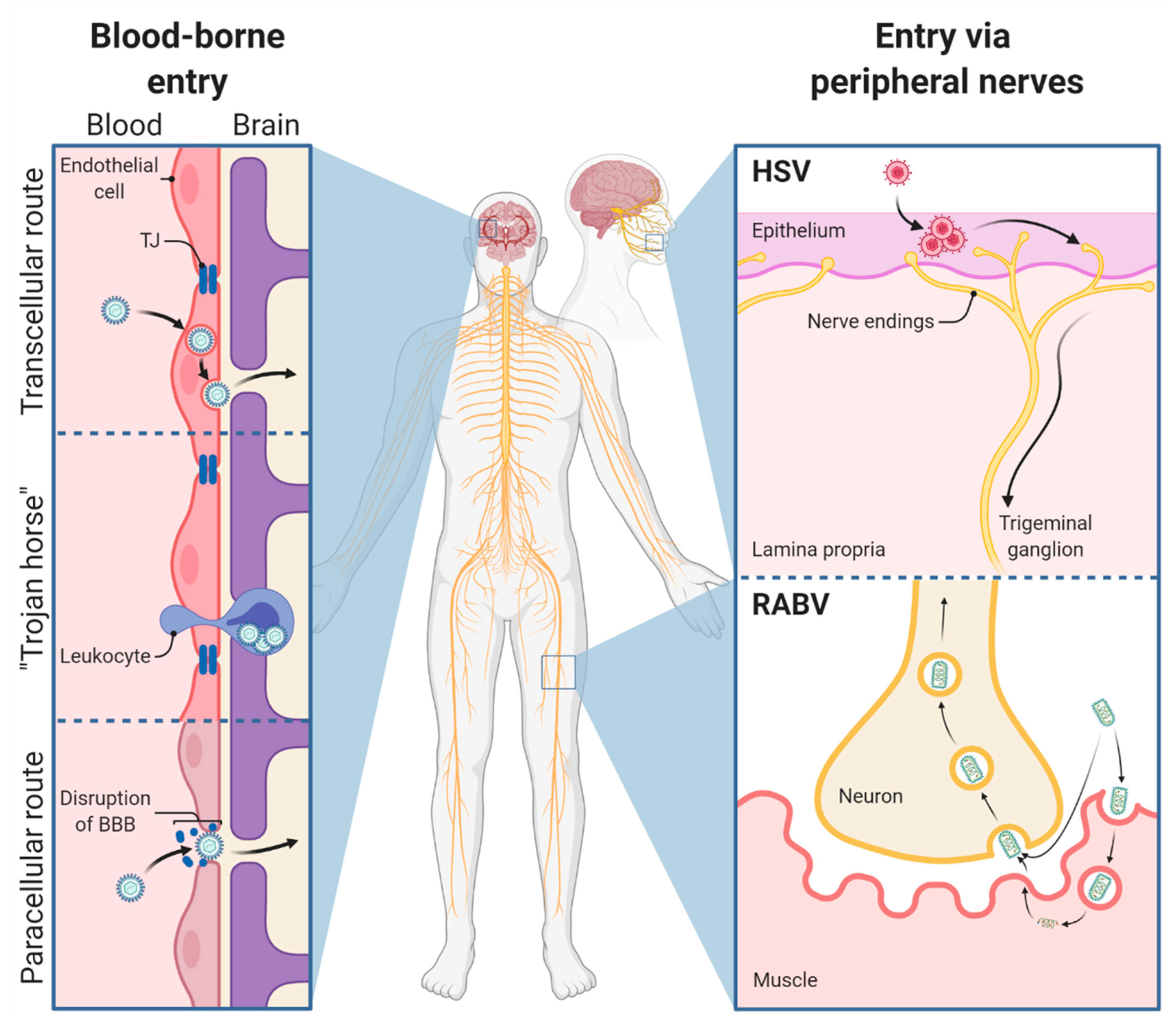
3.1. Viral Entry
3.2. RABV Tropism
3.2.1. Astrocytes
3.2.2. Microglia
3.2.3. Oligodendrocytes
3.2.4. Schwann Cells
3.3. HSV Tropism
3.3.1. Astrocytes
3.3.2. Microglia
3.3.3. Oligodendrocytes
3.3.4. Schwann Cells
4. The Role of the Blood–Brain Barrier in HSV and RABV Infections
4.1. Rabies Virus
4.2. Herpes Simplex Virus
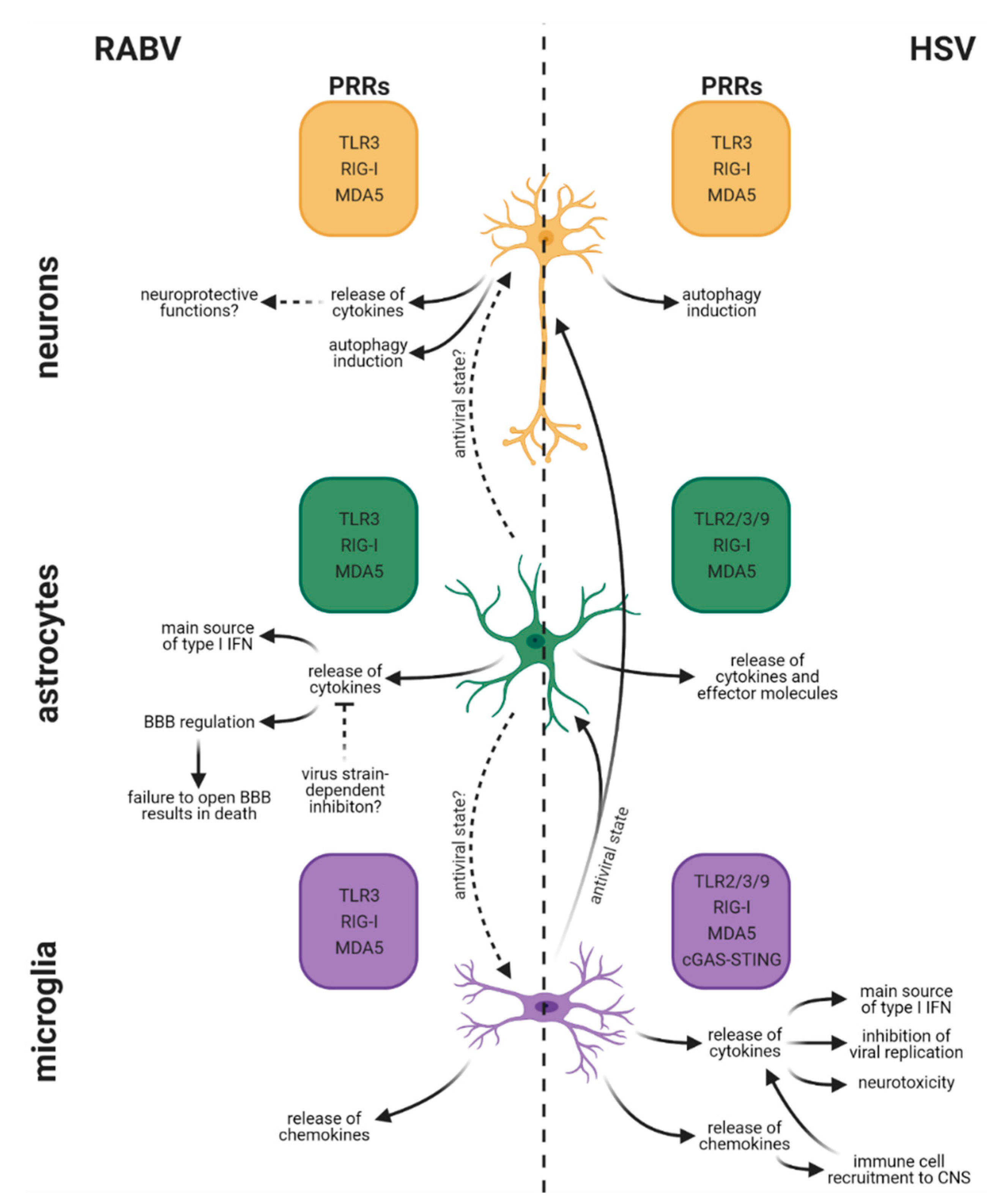
5. Differences in Cell-Type-Specific Innate Immune Responses between RABV and HSV Infection in the CNS
5.1. Rabies Virus
5.1.1. Neurons
5.1.2. Astrocytes
5.1.3. Microglia
5.2. Herpes Simplex Virus
5.2.1. Neurons
5.2.2. Astrocytes
5.2.3. Microglia
6. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Forrester, J.V.; McMenamin, P.G.; Dando, S.J. CNS infection and immune privilege. Nat. Rev. Neurosci. 2018, 19, 655–671. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood–brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef]
- Spindler, K.R.; Hsu, T.-H. Viral disruption of the blood-brain barrier. Trends Microbiol. 2012, 20, 282–290. [Google Scholar] [CrossRef]
- De Bock, M.; Van Haver, V.; Vandenbroucke, R.E.; Decrock, E.; Wang, N.; Leybaert, L. Into rather unexplored terrain-transcellular transport across the blood-brain barrier. Glia 2016, 64, 1097–1123. [Google Scholar] [CrossRef]
- Fooks, A.R.; Cliquet, F.; Finke, S.; Freuling, C.; Hemachudha, T.; Mani, R.S.; Müller, T.; Nadin-Davis, S.; Picard-Meyer, E.; Wilde, H.; et al. Rabies. Nat. Rev. Dis. Prim. 2017, 3, 17091. [Google Scholar] [CrossRef]
- Smith, G. Herpesvirus transport to the nervous system and back again. Annu. Rev. Microbiol. 2012, 66, 153–176. [Google Scholar] [CrossRef]
- Baer, G.M. The Natural History of Rabies, 2nd ed.; Academic Press: New York, NY, USA, 1975; Volume 2, ISBN 0120724014. [Google Scholar]
- Fooks, A.R.; Banyard, A.C.; Horton, D.L.; Johnson, N.; Mcelhinney, L.M.; Jackson, A.C. Current status of rabies and prospects for elimination. Lancet 2014, 384, 1389–1399. [Google Scholar] [CrossRef]
- Hampson, K.; Coudeville, L.; Lembo, T.; Sambo, M.; Kieffer, A.; Attlan, M.; Barrat, J.; Blanton, J.D.; Briggs, D.J.; Cleaveland, S.; et al. Estimating the Global Burden of Endemic Canine Rabies. PLoS Negl. Trop. Dis. 2015, 9, e0003709. [Google Scholar] [CrossRef]
- Watson, H.D.; Tignor, G.H.; Smith, A.L. Entry of rabies virus into the peripheral nerves of mice. J. Gen. Virol. 1981, 56, 372–382. [Google Scholar] [CrossRef]
- Murphy, F.A.; Bauer, S.P. Early street rabies virus infection in striated muscle and later progression to the central nervous system. Intervirology 1974, 3, 256–268. [Google Scholar] [CrossRef]
- Morimoto, K.; Patel, M.; Corisdeo, S.; Hooper, D.C.; Fu, Z.F.; Rupprecht, C.E.; Koprowski, H.; Dietzschold, B. Characterization of a unique variant of bat rabies virus responsible for newly emerging human cases in North America. Proc. Natl. Acad. Sci. USA 1996, 93, 5653–5658. [Google Scholar] [CrossRef]
- Ugolini, G. Rabies virus as a transneuronal tracer of neuronal connections. Adv. Virus Res. 2011, 79, 165–202. [Google Scholar] [CrossRef]
- Begeman, L.; GeurtsvanKessel, C.; Finke, S.; Freuling, C.M.; Koopmans, M.; Müller, T.; Ruigrok, T.J.H.; Kuiken, T. Comparative pathogenesis of rabies in bats and carnivores, and implications for spillover to humans. Lancet Infect. Dis. 2017, 18, e147–e159. [Google Scholar] [CrossRef]
- Velandia-Romero, M.L.; Castellanos, J.E.; Martínez-Gutiérrez, M. In Vivo differential susceptibility of sensory neurons to rabies virus infection. J. Neurovirol. 2013, 19, 367–375. [Google Scholar] [CrossRef]
- Jackson, A.C.; Fu, Z.F. Pathogenesis, 3rd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; ISBN 978-0-12-396547-9. [Google Scholar]
- Tang, Y.; Rampin, O.; Giuliano, F.; Ugolini, G. Spinal and brain circuits to motoneurons of the bulbospongiosus muscle: Retrograde transneuronal tracing with rabies virus. J. Comp. Neurol. 1999, 414, 167–192. [Google Scholar] [CrossRef]
- Graf, W.; Gerrits, N.; Yatim-Dhiba, N.; Ugolini, G. Mapping the oculomotor system: The power of transneuronal labelling with rabies virus. Eur. J. Neurosci. 2002, 15, 1557–1562. [Google Scholar] [CrossRef]
- Morcuende, S.; Delgado-García, J.M.; Ugolini, G.; Delgado-Garcia, J.-M.; Ugolini, G. Neuronal premotor networks involved in eyelid responses: Retrograde transneuronal tracing with rabies virus from the orbicularis oculi muscle in the rat. J. Neurosci. 2002, 22, 8808–8818. [Google Scholar] [CrossRef]
- Ugolini, G.; Klam, F.; Dans, M.D.; Dubayle, D.; Brandi, A.M.; Büttner-Ennever, J.; Graf, W. Horizontal eye movement networks in primates as revealed by retrograde transneuronal transfer of rabies virus: Differences in monosynaptic input to “slow” and “fast” abducens motoneurons. J. Comp. Neurol. 2006, 498, 762–785. [Google Scholar] [CrossRef]
- Ugolini, G. Advances in viral transneuronal tracing. J. Neurosci. Methods 2010, 194, 2–20. [Google Scholar] [CrossRef]
- Dhingra, V.; Li, X.; Liu, Y.; Fu, Z.F. Proteomic profiling reveals that rabies virus infection results in differential expression of host proteins involved in ion homeostasis and synaptic physiology in the central nervous system. J. Neurovirol. 2007, 13, 107–117. [Google Scholar] [CrossRef]
- Fu, Z.F.; Jackson, A.C. Neuronal dysfunction and death in rabies virus infection. J. Neurovirol. 2005, 11, 101–106. [Google Scholar] [CrossRef]
- Hemachudha, T. Human rabies: Clinical aspects, pathogenesis, and potential therapy. Curr. Top. Microbiol. Immunol. 1994, 187, 121–143. [Google Scholar] [CrossRef]
- Hemachudha, T.; Ugolini, G.; Wacharapluesadee, S.; Sungkarat, W.; Shuangshoti, S.; Laothamatas, J. Human rabies: Neuropathogenesis, diagnosis, and management. Lancet Neurol. 2013, 12, 498–513. [Google Scholar] [CrossRef]
- Almeida, M.F.; Martorelli, L.F.A.; Aires, C.C.; Sallum, P.C.; Durigon, E.L.; Massad, E. Experimental rabies infection in haematophagous bats Desmodus rotundus. Epidemiol. Infect. 2005, 133, 523–527. [Google Scholar] [CrossRef]
- Obregón-Morales, C.; Aguilar-Setién, Á.; Perea Martínez, L.; Galvez-Romero, G.; Martínez-Martínez, F.O.; Aréchiga-Ceballos, N. Experimental infection of Artibeus intermedius with a vampire bat rabies virus. Comp. Immunol. Microbiol. Infect. Dis. 2017, 52, 43–47. [Google Scholar] [CrossRef]
- Benavides, J.A.; Velasco-Villa, A.; Godino, L.C.; Satheshkumar, P.S.; Nino, R.; Rojas-Paniagua, E.; Shiva, C.; Falcon, N.; Streicker, D.G. Abortive vampire bat rabies infections in Peruvian peridomestic livestock. PLoS Negl. Trop. Dis. 2020, 14, e0008194. [Google Scholar] [CrossRef]
- Turmelle, A.S.; Jackson, F.R.; Green, D.; McCracken, G.F.; Rupprecht, C.E. Host immunity to repeated rabies virus infection in big brown bats. J. Gen. Virol. 2010, 91, 2360–2366. [Google Scholar] [CrossRef]
- Jackson, F.R.; Turmelle, A.S.; Farino, D.M.; Franka, R.; McCracken, G.F.; Rupprecht, C.E. Experimental rabies virus infection of big brown bats (Eptesicus fuscus). J. Wildl. Dis. 2008, 44, 612–621. [Google Scholar] [CrossRef]
- Gnanadurai, C.W.; Zhou, M.; He, W.; Leyson, C.M.; Huang, C.-T.; Salyards, G.; Harvey, S.B.; Chen, Z.; He, B.; Yang, Y.; et al. Presence of virus neutralizing antibodies in cerebral spinal fluid correlates with non-lethal rabies in dogs. PLoS Negl. Trop. Dis. 2013, 7, e2375. [Google Scholar] [CrossRef]
- Manickama, R.; Basheer, M.D.; Jayakumar, R. Post-exposure prophylaxis (PEP) of rabies-infected Indian street dogs. Vaccine 2008, 26, 6564–6568. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.M.; de Oliveira, S.V.; Heinemann, M.B.; Gonçalves, V.S.P. Epidemiological Profile of Wild Rabies in Brazil (2002–2012). Transbound. Emerg. Dis. 2017, 64, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Kotait, I.; Oliveira, R.d.N.; Carrieri, M.L.; Castilho, J.G.; Macedo, C.I.; Pereira, P.M.C.; Boere, V.; Montebello, L.; Rupprecht, C.E. Non-human primates as a reservoir for rabies virus in Brazil. Zoonoses Public Health 2019, 66, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Machado, G.P.; de Paula Antunes, J.M.A.; Uieda, W.; Biondo, A.W.; de Andrade Cruvinel, T.M.; Kataoka, A.P.; Martorelli, L.F.A.; de Jong, D.; Amaral, J.M.G.; Hoppe, E.G.L.; et al. Exposure to rabies virus in a population of free-ranging capuchin monkeys (Cebus apella nigritus) in a fragmented, environmentally protected area in southeastern Brazil. Primates 2012, 53, 227–231. [Google Scholar] [CrossRef] [PubMed]
- WHO. Weekly Epidemiological Record, 2018, vol. 93, 16 [full issue]. Wkly. Epidemiol. Rec. 2018, 93, 201–220. [Google Scholar]
- O’Brien, K.L.; Nolan, T. The WHO position on rabies immunization—2018 updates. Vaccine 2019, 37, A85–A87. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, R.E.J.; Tieves, K.S.; Hoffman, G.M.; Ghanayem, N.S.; Amlie-Lefond, C.M.; Schwabe, M.J.; Chusid, M.J.; Rupprecht, C.E. Survival after treatment of rabies with induction of coma. N. Engl. J. Med. 2005, 352, 2508–2514. [Google Scholar] [CrossRef]
- Zeiler, F.A.; Jackson, A.C. Critical Appraisal of the Milwaukee Protocol for Rabies: This Failed Approach Should Be Abandoned. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 2016, 43, 44–51. [Google Scholar] [CrossRef]
- Whitley, R.; Kimberlin, D.W. Herpes simplex: Encephalitis children and adolescents. Semin. Pediatr. Infect. Dis. 2005, 16, 17–23. [Google Scholar] [CrossRef]
- Price, R.; Chernik, N.L.; Horta-Barbosa, L.; Posner, J.B. Herpes simplex encephalitis in an anergic patient. Am. J. Med. 1973, 54, 222–228. [Google Scholar] [CrossRef]
- Whitley, R.; Lakeman, A.D.; Nahmias, A.; Roizman, B. DNA Restriction-Enzyme Analysis of Herpes Simplex Virus Isolates Obtained from Patients with Encephalitis. N. Engl. J. Med. 1982, 307, 1060–1062. [Google Scholar] [CrossRef] [PubMed]
- Gnann, J.; Whitley, R. Herpes Simplex Encephalitis: An Update. Curr. Infect. Dis. Rep. 2017, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.R.; Moran, C.J.; Cross, D.W.T.; Wippold, F.J.; Schlesinger, Y.; Storch, G.A. MR imaging of herpes simplex type I encephalitis in infants and young children: A separate pattern of findings. Am. J. Roentgenol. 2000, 174, 1651–1655. [Google Scholar] [CrossRef] [PubMed]
- Vossough, A.; Zimmerman, R.A.; Bilaniuk, L.T.; Schwartz, E.M. Imaging findings of neonatal herpes simplex virus type 2 encephalitis. Neuroradiology 2008, 50, 355–366. [Google Scholar] [CrossRef]
- Baskin, H.J.; Hedlund, G. Neuroimaging of herpesvirus infections in children. Pediatr. Radiol. 2007, 37, 949–963. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, M.J.; Venkatesan, A. Herpes Simplex Virus-1 Encephalitis in Adults: Pathophysiology, Diagnosis, and Management. Neurotherapeutics 2016, 13, 493–508. [Google Scholar] [CrossRef]
- Steiner, I.; Benninger, F. Manifestations of Herpes Virus Infections in the Nervous System. Neurol. Clin. 2018, 36, 725–738. [Google Scholar] [CrossRef]
- Whitley, R.J.; Gnann, J.W. Viral encephalitis: Familiar infections and emerging pathogens. Lancet 2002, 359, 507–513. [Google Scholar] [CrossRef]
- Stahl, J.P.; Mailles, A.; De Broucker, T. Herpes simplex encephalitis and management of acyclovir in encephalitis patients in France. Epidemiol. Infect. 2012, 140, 372–381. [Google Scholar] [CrossRef]
- Stahl, J.P.; Mailles, A. Herpes simplex virus encephalitis update. Curr. Opin. Infect. Dis. 2019, 32, 239–243. [Google Scholar] [CrossRef]
- Etessami, R.; Conzelmann, K.-K.; Fadai-Ghotbi, B.; Natelson, B.; Tsiang, H.; Ceccaldi, P.-E. Spread and pathogenic characteristics of a G-deficient rabies virus recombinant: An in vitro and in vivo study. J. Gen. Virol. 2000, 81, 2147–2153. [Google Scholar] [CrossRef] [PubMed]
- Le Blanc, I.; Luyet, P.-P.; Pons, V.; Ferguson, C.; Emans, N.; Petiot, A.; Mayran, N.; Demaurex, N.; Fauré, J.; Sadoul, R.; et al. Endosome-to-cytosol transport of viral nucleocapsids. Nat. Cell Biol. 2005, 7, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Piccinotti, S.; Kirchhausen, T.; Whelan, S.P.J. Uptake of Rabies Virus into Epithelial Cells by Clathrin-Mediated Endocytosis Depends upon Actin. J. Virol. 2013, 87, 11637–11647. [Google Scholar] [CrossRef] [PubMed]
- Lafon, M. Rabies virus receptors. J. Neurovirol. 2005, 11, 82–87. [Google Scholar] [CrossRef]
- Baquero, E.; Albertini, A.A.V.; Gaudin, Y. Recent mechanistic and structural insights on class III viral fusion glycoproteins. Curr. Opin. Struct. Biol. 2015, 33, 52–60. [Google Scholar] [CrossRef]
- Gaudin, Y.; Tuffereau, C.; Segretain, D.; Knossow, M.; Flamand, A. Reversible conformational changes and fusion activity of rabies virus glycoprotein. J. Virol. 1991, 65, 4853–4859. [Google Scholar] [CrossRef]
- Bauer, A.; Nolden, T.; Schröter, J.; Römer-Oberdörfer, A.; Gluska, S.; Perlson, E.; Finke, S. Anterograde glycoprotein-dependent transport of newly generated rabies virus in dorsal root ganglion neurons. J. Virol. 2014, 88, 14172–14183. [Google Scholar] [CrossRef]
- Gluska, S.; Zahavi, E.E.; Chein, M.; Gradus, T.; Bauer, A.; Finke, S.; Perlson, E. Rabies Virus Hijacks and Accelerates the p75NTR Retrograde Axonal Transport Machinery. PLoS Pathog. 2014, 10, e1004348. [Google Scholar] [CrossRef]
- Klingen, Y.; Conzelmann, K.-K.; Finke, S. Double-Labeled Rabies Virus: Live Tracking of Enveloped Virus Transport. J. Virol. 2008, 82, 237–245. [Google Scholar] [CrossRef]
- Raux, H.; Flamand, A.; Blondel, D. Interaction of the Rabies Virus P Protein with the LC8 Dynein Light Chain. J. Virol. 2000, 74, 10212–10216. [Google Scholar] [CrossRef]
- Jacob, Y.; Badrane, H.; Ceccaldi, P.E.; Tordo, N. Cytoplasmic dynein LC8 interacts with lyssavirus phosphoprotein. J. Virol. 2000, 74, 10217–10222. [Google Scholar] [CrossRef]
- Mebatsion, T. Extensive attenuation of rabies virus by simultaneously modifying the dynein light chain binding site in the P protein and replacing Arg333 in the G protein. J. Virol. 2001, 75, 11496–11502. [Google Scholar] [CrossRef]
- Bauer, A.; Nolden, T.; Nemitz, S.; Perlson, E.; Finke, S. A Dynein Light Chain 1 Binding Motif in Rabies Virus Polymerase L Protein Plays a Role in Microtubule Reorganization and Viral Primary Transcription. J. Virol. 2015, 89, 9591–9600. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Yang, J.; Wu, X.; Fu, Z.F. Interactions amongst rabies virus nucleoprotein, phosphoprotein and genomic RNA in virus-infected and transfected cells. J. Gen. Virol. 2004, 85, 3725–3734. [Google Scholar] [CrossRef]
- Finke, S.; Mueller-Waldeck, R.; Conzelmann, K.K. Rabies virus matrix protein regulates the balance of virus transcription and replication. J. Gen. Virol. 2003, 84, 1613–1621. [Google Scholar] [CrossRef]
- Abraham, G.; Banerjee, A.K. Sequential transcription of the genes of vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 1976, 73, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Okumura, A.; Harty, R.N. Rabies virus assembly and budding. Adv. Virus Res. 2011, 79, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Mebatsion, T.; Konig, M.; Conzelmann, K.K. Budding of rabies virus particles in the absence of the spike glycoprotein. Cell 1996, 84, 941–951. [Google Scholar] [CrossRef]
- Mebatsion, T.; Weiland, F.; Conzelmann, K.K. Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J. Virol. 1999, 73, 242–250. [Google Scholar] [CrossRef]
- Jackson, A.C.; Ye, H.; Phelan, C.C.; Ridaura-Sanz, C.; Zheng, Q.; Li, Z.; Wan, X.; Lopez-Corella, E. Extraneural organ involvement in human rabies. Lab. Investig. 1999, 79, 945–951. [Google Scholar]
- Jogai, S.; Radotra, B.D.; Banerjee, A.K. Rabies viral antigen in extracranial organs: A post-mortem study. Neuropathol. Appl. Neurobiol. 2002, 28, 334–338. [Google Scholar] [CrossRef]
- Huffmaster, N.J.; Sollars, P.J.; Richards, A.L.; Pickard, G.E.; Smith, G.A. Dynamic ubiquitination drives herpesvirus neuroinvasion. Proc. Natl. Acad. Sci. USA 2015, 112, 12818–12823. [Google Scholar] [CrossRef]
- Lee, J.I.; Sollars, P.J.; Baver, S.B.; Pickard, G.E.; Leelawong, M.; Smith, G.A. A herpesvirus encoded deubiquitinase is a novel neuroinvasive determinant. PLoS Pathog. 2009, 5, e1000387. [Google Scholar] [CrossRef]
- Aggarwal, A.; Miranda-Saksena, M.; Boadle, R.A.; Kelly, B.J.; Diefenbach, R.J.; Alam, W.; Cunningham, A.L. Ultrastructural Visualization of Individual Tegument Protein Dissociation during Entry of Herpes Simplex Virus 1 into Human and Rat Dorsal Root Ganglion Neurons. J. Virol. 2012, 86, 6123–6137. [Google Scholar] [CrossRef] [PubMed]
- Luxton, G.W.; Haverlock, S.; Coller, K.E.; Antinone, S.E.; Pincetic, A.; Smith, G.A. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 5832–5837. [Google Scholar] [CrossRef] [PubMed]
- Piccinotti, S.; Whelan, S.P.J. Rabies Internalizes into Primary Peripheral Neurons via Clathrin Coated Pits and Requires Fusion at the Cell Body. PLOS Pathog. 2016, 12, e1005753. [Google Scholar] [CrossRef]
- Antinone, S.E.; Smith, G.A. Retrograde axon transport of herpes simplex virus and pseudorabies virus: A live-cell comparative analysis. J. Virol. 2010, 84, 1504–1512. [Google Scholar] [CrossRef]
- Twelvetrees, A.E.; Pernigo, S.; Sanger, A.; Guedes-Dias, P.; Schiavo, G.; Steiner, R.A.; Dodding, M.P.; Holzbaur, E.L. The Dynamic Localization of Cytoplasmic Dynein in Neurons Is Driven by Kinesin-1. Neuron 2016, 90, 1000–1015. [Google Scholar] [CrossRef]
- Wolfstein, A.; Nagel, C.H.; Radtke, K.; Dohner, K.; Allan, V.J.; Sodeik, B. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 2006, 7, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, O.O.; MacGibeny, M.A.; Enquist, L.W. Latent versus productive infection: The alpha herpesvirus switch. Future Virol. 2018, 13, 431–443. [Google Scholar] [CrossRef]
- Nicoll, M.P.; Proenca, J.T.; Efstathiou, S. The molecular basis of herpes simplex virus latency. FEMS Microbiol. Rev. 2012, 36, 684–705. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of neurons. J. Clin. Investig. 2017, 127, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, S.; Gritti, L.; Crooks, D.; Dombrowski, Y. Oligodendrocytes in Development, Myelin Generation and Beyond. Cells 2019, 8, 1424. [Google Scholar] [CrossRef] [PubMed]
- George, D.; Ahrens, P.; Lambert, S. Satellite glial cells represent a population of developmentally arrested Schwann cells. Glia 2018, 66, 1496–1506. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, P.; Poole, D.P.; Veldhuis, N.A. Chapter Four—Role of Nonneuronal TRPV4 Signaling in Inflammatory Processes. In Advances in Pharmacology; Geraghty, D.P., Rash, L.D., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 79, pp. 117–139. ISBN 1054-3589. [Google Scholar]
- Soung, A.; Klein, R.S. Viral Encephalitis and Neurologic Diseases: Focus on Astrocytes. Trends Mol. Med. 2018, 24, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, D.; Li, G. The role of microglia in viral encephalitis: A review. J. Neuroinflamm. 2019, 16, 76. [Google Scholar] [CrossRef]
- Metchnikoff, E.; Mesnil, F.; Weinberg, M. Etudes biologiques sur la vieillesse: II. Recherches sur la vieillesse des perroquets. Ann. Inst. Pasteur 1902, 16, 912–917. [Google Scholar]
- Carty, M.; Reinert, L.; Paludan, S.R.; Bowie, A.G. Innate antiviral signalling in the central nervous system. Trends Immunol. 2014, 35, 79–87. [Google Scholar] [CrossRef]
- Broderick, C.; Hoek, R.M.; Forrester, J.V.; Liversidge, J.; Sedgwick, J.D.; Dick, A.D. Constitutive retinal CD200 expression regulates resident microglia and activation state of inflammatory cells during experimental autoimmune uveoretinitis. Am. J. Pathol. 2002, 161, 1669–1677. [Google Scholar] [CrossRef]
- Hoek, R.M.; Ruuls, S.R.; Murphy, C.A.; Wright, G.J.; Goddard, R.; Zurawski, S.M.; Blom, B.; Homola, M.E.; Streit, W.J.; Brown, M.H.; et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 2000, 290, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.H.; Zhang, E.; Kang, J.W.; Shin, Y.N.; Byun, J.Y.; Oh, S.H.; Seo, J.H.; Lee, Y.H.; Kim, D.W. Expression of CD200 in alternative activation of microglia following an excitotoxic lesion in the mouse hippocampus. Brain Res. 2012, 1481, 90–96. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, X.J.; Tian, L.P.; Pan, J.; Lu, G.Q.; Zhang, Y.J.; Ding, J.Q.; Chen, S. Di CD200-CD200R dysfunction exacerbates microglial activation and dopaminergic neurodegeneration in a rat model of Parkinson’s disease. J. Neuroinflamm. 2011, 8, 154. [Google Scholar] [CrossRef]
- Burmeister, A.R.; Marriott, I. The Interleukin-10 Family of Cytokines and Their Role in the CNS. Front. Cell. Neurosci. 2018, 12, 458. [Google Scholar] [CrossRef] [PubMed]
- Szelényi, J. Cytokines and the central nervous system. Brain Res. Bull. 2001, 54, 329–338. [Google Scholar] [CrossRef]
- Almolda, B.; González, B.; Castellano, B. Activated microglial cells acquire an immature dendritic cell phenotype and may terminate the immune response in an acute model of EAE. J. Neuroimmunol. 2010, 223, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Almolda, B.; Gonzalez, B.; Castellano, B. Antigen presentation in EAE: Role of microglia, macrophages and dendritic cells. Front. Biosci. (Landmark Ed.) 2011, 16, 1157–1171. [Google Scholar] [CrossRef]
- Griffin, D.E. Immune responses to RNA-virus infections of the CNS. Nat. Rev. Immunol. 2003, 3, 493–502. [Google Scholar] [CrossRef]
- Malone, K.E.; Stohlman, S.A.; Ramakrishna, C.; Macklin, W.; Bergmann, C.C. Induction of class I antigen processing components in oligodendroglia and microglia during viral encephalomyelitis. Glia 2008, 56, 426–435. [Google Scholar] [CrossRef]
- Hamo, L.; Stohlman, S.A.; Otto-Duessel, M.; Bergmann, C.C. Distinct regulation of MHC molecule expression on astrocytes and microglia during viral encephalomyelitis. Glia 2007, 55, 1169–1177. [Google Scholar] [CrossRef]
- Frank, E.; Pulver, M.; de Tribolet, N. Expression of class II major histocompatibility antigens on reactive astrocytes and endothelial cells within the gliosis surrounding metastases and abscesses. J. Neuroimmunol. 1986, 12, 29–36. [Google Scholar] [CrossRef]
- Rostami, J.; Fotaki, G.; Sirois, J.; Mzezewa, R.; Bergström, J.; Essand, M.; Healy, L.; Erlandsson, A. Astrocytes have the capacity to act as antigen-presenting cells in the Parkinson’s disease brain. J. Neuroinflamm. 2020, 17, 119. [Google Scholar] [CrossRef]
- Sriram, S. Role of glial cells in innate immunity and their role in CNS demyelination. J. Neuroimmunol. 2011, 239, 13–20. [Google Scholar] [CrossRef]
- Appolinário, C.M.; Allendorf, S.D.; Peres, M.G.; Ribeiro, B.D.; Fonseca, C.R.; Vicente, A.F.; De Paula Antunes, J.M.A.; Jane, M. Profile of cytokines and chemokines triggered by wild-type strains of rabies virus in mice. Am. J. Trop. Med. Hyg. 2016, 94, 378–383. [Google Scholar] [CrossRef]
- Reinert, L.S.; Lopušná, K.; Winther, H.; Sun, C.; Thomsen, M.K.; Nandakumar, R.; Mogensen, T.H.; Meyer, M.; Vægter, C.; Nyengaard, J.R.; et al. Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Pfefferkorn, C.; Kallfass, C.; Lienenklaus, S.; Spanier, J.; Kalinke, U.; Rieder, M.; Conzelmann, K.-K.; Michiels, T.; Staeheli, P. Abortively Infected Astrocytes Appear To Represent the Main Source of Interferon Beta in the Virus-Infected Brain. J. Virol. 2016, 90, 2031–2038. [Google Scholar] [CrossRef]
- Detje, C.N.; Lienenklaus, S.; Chhatbar, C.; Spanier, J.; Prajeeth, C.K.; Soldner, C.; Tovey, M.G.; Schlüter, D.; Weiss, S.; Stangel, M.; et al. Upon intranasal vesicular stomatitis virus infection, astrocytes in the olfactory bulb are important interferon Beta producers that protect from lethal encephalitis. J. Virol. 2015, 89, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Kallfass, C.; Ackerman, A.; Lienenklaus, S.; Weiss, S.; Heimrich, B.; Staeheli, P. Visualizing production of beta interferon by astrocytes and microglia in brain of La Crosse virus-infected mice. J. Virol. 2012, 86, 11223–11230. [Google Scholar] [CrossRef] [PubMed]
- Adle-Biassette, H.; Bourhy, H.; Gisselbrecht, M.; Chrétien, F.; Wingertsmann, L.; Baudrimont, M.; Rotivel, Y.; Godeau, B.; Gray, F. Rabies encephalitis in a patient with AIDS: A clinicopathological study. Acta Neuropathol. 1996, 92, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Miao, F.-M.; Zhang, S.-F.; Wang, S.-C.; Liu, Y.; Zhang, F.; Hu, R.-L. Comparison of immune responses to attenuated rabies virus and street virus in mouse brain. Arch. Virol. 2017, 162, 247–257. [Google Scholar] [CrossRef]
- Gnanadurai, C.W.; Yang, Y.; Huang, Y.; Li, Z.; Leyson, C.M.; Cooper, T.L.; Platt, S.R.; Harvey, S.B.; Hooper, D.C.; Faber, M.; et al. Differential host immune responses after infection with wild-type or lab-attenuated rabies viruses in dogs. PLoS Negl. Trop. Dis. 2015, 9, e0004023. [Google Scholar] [CrossRef]
- Brzózka, K.; Finke, S.; Brzo, K. Identification of the Rabies Virus Alpha/Beta Interferon Antagonist: Phosphoprotein P Interferes with Phosphorylation of Interferon Regulatory Factor 3 Identification of the Rabies Virus Alpha/Beta Interferon Antagonist: Phosphoprotein P Interferes. J. Virol. 2005, 79, 7673–7681. [Google Scholar] [CrossRef]
- Prehaud, C.; Lay, S.; Dietzschold, B.; Lafon, M. Glycoprotein of nonpathogenic rabies viruses is a key determinant of human cell apoptosis. J. Virol. 2003, 77, 10537–10547. [Google Scholar] [CrossRef] [PubMed]
- Baloul, L.; Lafon, M. Apoptosis and rabies virus neuroinvasion. Biochimie 2003, 85, 777–788. [Google Scholar] [CrossRef]
- Steiner, I.; Kennedy, P.G.; Pachner, A.R. The neurotropic herpes viruses: Herpes simplex and varicella-zoster. Lancet Neurol. 2007, 6, 1015–1028. [Google Scholar] [CrossRef]
- Ito, N.; Moseley, G.W.; Sugiyama, M. The importance of immune evasion in the pathogenesis of rabies virus. Vet. Med. Sci. 2016, 7, 1089–1098. [Google Scholar] [CrossRef]
- Dietzschold, B.; Li, J.; Faber, M.; Schnell, M. Concepts in the pathogenesis of rabies. Future Virol. 2008, 3, 481–490. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Clark, H.F. Cell to cell transmission of virus in the central nervous system. II. Experimental rabies in mouse. Lab. Investig. 1975, 33, 391–399. [Google Scholar]
- Potratz, M.; Zaeck, L.; Christen, M.; Kamp, V.; Klein, A.; Freuling, C.M.; Müller, T.; Finke, S. Astrocyte Infection during Rabies Encephalitis Depends on the Virus Strain and Infection Route as Demonstrated by Novel Quantitative 3D Analysis of Cell Tropism. Cells 2020, 9, 412. [Google Scholar] [CrossRef]
- Potratz, M.; Zaeck, L.M.; Weigel, C.; Klein, A.; Freuling, C.M.; Müller, T.; Finke, S. Neuroglia infection by rabies virus after anterograde virus spread in peripheral neurons. Acta Neuropathol. Commun. 2020, 8, 199. [Google Scholar] [CrossRef]
- Sung, J.H.; Hayano, M.; Mastri, A.R.; Okagaki, T. A case of human rabies and ultrastructure of the Negri body. J. Neuropathol. Exp. Neurol. 1976, 35, 541–559. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.C.; Phelan, C.C.; Rossiter, J.P. Infection of Bergmann glia in the cerebellum of a skunk experimentally infected with street rabies virus. Can. J. Vet. Res. 2000, 64, 226–228. [Google Scholar] [PubMed]
- Prosniak, M.; Zborek, A.; Scott, G.S.; Roy, A.; Phares, T.W.; Koprowski, H.; Hooper, D.C. Differential expression of growth factors at the cellular level in virus-infected brain. Proc. Natl. Acad. Sci. USA 2003, 100, 6765–6770. [Google Scholar] [CrossRef] [PubMed]
- Ray, N.B.; Power, C.; Lynch, W.P.P.; Ewalt, L.C.C.; Lodmell, D.L.L. Rabies viruses infect primary cultures of murine, feline, and human microglia and astrocytes. Arch. Virol. 1997, 142, 1011–1019. [Google Scholar] [CrossRef]
- Sugamata, M.; Miyazawa, M.; Mori, S.; Spangrude, G.J.; Ewalt, L.C.; Lodmell, D.L. Paralysis of street rabies virus-infected mice is dependent on T lymphocytes. J. Virol. 1992, 66, 1252–1260. [Google Scholar] [CrossRef]
- Lafon, M. Immune evasion, a critical strategy for rabies virus. Dev. Biol. 2008, 131, 413–419. [Google Scholar]
- Thoulouze, M.I.; Lafage, M.; Schachner, M.; Hartmann, U.; Cremer, H.; Lafon, M. The neural cell adhesion molecule is a receptor for rabies virus. J. Virol. 1998, 72, 7181–7190. [Google Scholar] [CrossRef]
- Tuffereau, C.; Bénéjean, J.; Blondel, D.; Kieffer, B.; Flamand, A. Low-affinity nerve-growth factor receptor (P75NTR) can serve as a receptor for rabies virus. EMBO J. 1998, 17, 7250–7259. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z.; Liu, R.; Shuai, L.; Wang, X.; Luo, J.; Wang, C.; Chen, W.; Wang, X.; Ge, J.; et al. Metabotropic glutamate receptor subtype 2 is a cellular receptor for rabies virus. PLoS Pathog. 2018, 2, e1007189. [Google Scholar] [CrossRef]
- Lentz, T.L.; Burrage, T.G.; Smith, A.L.; Crick, J.; Tignor, G.H. Is the acetylcholine receptor a rabies virus receptor? Science 1982, 215, 182–184. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sloan, S.A.A.; Clarke, L.E.E.; Caneda, C.; Plaza, C.A.A.; Blumenthal, P.D.D.; Vogel, H.; Steinberg, G.K.K.; Edwards, M.S.B.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Hawrylycz, M.J.; Lein, E.S.; Guillozet-Bongaarts, A.L.; Shen, E.H.; Ng, L.; Miller, J.A.; van de Lagemaat, L.N.; Smith, K.A.; Ebbert, A.; Riley, Z.L.; et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 2012, 489, 391–399. [Google Scholar] [CrossRef]
- Campadelli-Fiume, G.; Cocchi, F.; Menotti, L.; Lopez, M. The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells. Rev. Med. Virol 2000, 10, 305–319. [Google Scholar] [CrossRef]
- Takahashi, K.; Nakanishi, H.; Miyahara, M.; Mandai, K.; Satoh, K.; Satoh, A.; Nishioka, H.; Aoki, J.; Nomoto, A.; Mizoguchi, A.; et al. Nectin/PRR: An immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with Afadin, a PDZ domain-containing protein. J. Cell Biol. 1999, 145, 539–549. [Google Scholar] [CrossRef]
- Kopp, S.J.; Banisadr, G.; Glajch, K.; Maurer, U.E.; Grunewald, K.; Miller, R.J.; Osten, P.; Spear, P.G. Infection of neurons and encephalitis after intracranial inoculation of herpes simplex virus requires the entry receptor nectin-1. Proc. Natl. Acad. Sci. USA 2009, 106, 17916–17920. [Google Scholar] [CrossRef]
- Steinberg, M.W.; Cheung, T.C.; Ware, C.F. The signaling networks of the herpesvirus entry mediator (TNFRSF14) in immune regulation. Immunol. Rev. 2011, 244, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Clement, C.; Scanlan, P.M.; Kowlessur, D.; Yue, B.Y.; Shukla, D. A role for herpesvirus entry mediator as the receptor for herpes simplex virus 1 entry into primary human trabecular meshwork cells. J. Virol. 2005, 79, 13173–13179. [Google Scholar] [CrossRef]
- Guzman, G.; Oh, S.; Shukla, D.; Engelhard, H.H.; Valyi-Nagy, T. Expression of entry receptor nectin-1 of herpes simplex virus 1 and/or herpes simplex virus 2 in normal and neoplastic human nervous system tissues. Acta Virol. 2006, 50, 59–66. [Google Scholar]
- Haarr, L.; Shukla, D.; Rodahl, E.; Dal Canto, M.C.; Spear, P.G. Transcription from the gene encoding the herpesvirus entry receptor nectin-1 (HveC) in nervous tissue of adult mouse. Virology 2001, 287, 301–309. [Google Scholar] [CrossRef]
- Tian, B.; Zhou, M.; Yang, Y.; Yu, L.; Luo, Z.; Tian, D.; Wang, K.; Cui, M.; Chen, H.; Fu, Z.F.; et al. Lab-attenuated rabies virus causes abortive infection and induces cytokine expression in astrocytes by activating mitochondrial antiviral-signaling protein signaling pathway. Front. Immunol. 2018, 8, 2011. [Google Scholar] [CrossRef]
- Balachandran, A.; Charlton, K. Experimental rabies infection of non-nervous tissues in skunks (Mephitis mephitis) and foxes (Vulpes vulpes). Vet. Pathol. 1994, 31, 93–102. [Google Scholar] [CrossRef]
- Charlton, K.M.; Nadin-Davis, S.; Casey, G.A.; Wandeler, A.I. The long incubation period in rabies: Delayed progression of infection in muscle at the site of exposure. Acta Neuropathol. 1997, 94, 73–77. [Google Scholar] [CrossRef]
- Charlton, K.M.; Casey, G.A. Experimental rabies in skunks: Immunofluorescence light and electron microscopic studies. Lab. Investig. 1979, 41, 36–44. [Google Scholar]
- Tsiang, H.; Koulakoff, A.; Bizzini, B.; Berwald-Netter, Y. Neurotropism of rabies virus. An in vitro study. J. Neuropathol. Exp. Neurol. 1983, 42, 439–452. [Google Scholar] [CrossRef]
- Brzózka, K.; Finke, S.; Conzelmann, K.-K. Inhibition of Interferon Signaling by Rabies Virus Phosphoprotein P: Activation-Dependent Binding of STAT1 and STAT2. J. Virol. 2006, 80, 2675–2683. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Moseley, G.W.; Blondel, D.; Shimizu, K.; Rowe, C.L.; Ito, Y.; Masatani, T.; Nakagawa, K.; Jans, D.A.; Sugiyama, M. Role of interferon antagonist activity of rabies virus phosphoprotein in viral pathogenicity. J. Virol. 2010, 84, 6699–6710. [Google Scholar] [CrossRef] [PubMed]
- Vidy, A.; Chelbi-Alix, M.; Blondel, D. Rabies Virus P Protein Interacts with STAT1 and Inhibits Interferon Signal Transduction Pathways. J. Virol. 2005, 79, 14411–14420. [Google Scholar] [CrossRef] [PubMed]
- Vidy, A.; El Bougrini, J.; Chelbi-Alix, M.K.; Blondel, D. The Nucleocytoplasmic Rabies Virus P Protein Counteracts Interferon Signaling by Inhibiting both Nuclear Accumulation and DNA Binding of STAT1. J. Virol. 2007, 81, 4255–4263. [Google Scholar] [CrossRef]
- Rieder, M.; Brzozka, K.; Pfaller, C.K.; Cox, J.H.; Stitz, L.; Conzelmann, K. Genetic Dissection of Interferon-Antagonistic Functions of Rabies Virus Phosphoprotein: Inhibition of Interferon Regulatory Factor 3 Activation Is Important for Pathogenicity. J. Virol. 2011, 85, 842–852. [Google Scholar] [CrossRef]
- Moseley, G.W.; Lahaye, X.; Roth, D.M.; Oksayan, S.; Filmer, R.P.; Rowe, C.L.; Blondel, D.; Jans, D.A. Dual modes of rabies P-protein association with microtubules: A novel strategy to suppress the antiviral response. J. Cell Sci. 2009, 122, 3652–3662. [Google Scholar] [CrossRef]
- Bertoune, M.; Nickl, B.; Krieger, T.; Wohlers, L.; Bonaterra, G.A.; Dietzschold, B.; Weihe, E.; Bette, M. The phenotype of the RABV glycoprotein determines cellular and global virus load in the brain and is decisive for the pace of the disease. Virology 2017, 511, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Sonthonnax, F.; Besson, B.B.; Bonnaud, E.; Jouvion, G.; Merino, D.; Larrous, F.; Bourhy, H. Lyssavirus matrix protein cooperates with phosphoprotein to modulate the Jak-Stat pathway. Sci. Rep. 2019, 9, 12171. [Google Scholar] [CrossRef]
- Zhao, P.; Yang, Y.; Feng, H.; Zhao, L.; Qin, J.; Zhang, T.; Wang, H.; Yang, S.; Xia, X. Global gene expression changes in BV2 microglial cell line during rabies virus infection. Infect. Genet. Evol. 2013, 20, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Q.; Xie, Y.; Gao, X.; Wang, Q.; Zhu, W.Y. Inflammatory response and MAPK and NF-κB pathway activation induced by natural street rabies virus infection in the brain tissues of dogs and humans. Virol. J. 2020, 17, 157. [Google Scholar] [CrossRef]
- Liu, S.Q.; Gao, X.; Xie, Y.; Wang, Q.; Zhu, W.Y. Rabies viruses of different virulence regulates inflammatory responses both in vivo and in vitro via MAPK and NF-κB pathway. Mol. Immunol. 2020, 125, 70–82. [Google Scholar] [CrossRef]
- Feige, L.; Kozaki, T.; Dias de Melo, G.; Guillemot, V.; Larrous, F.; Ginhoux, F.; Bourhy, H. Cell-type specific innate immune responses shape rabies virus tropism. Manuscr. Submitt. Publ. 2021. [Google Scholar] [CrossRef]
- Valério-Gomes, B.; Guimarães, D.M.; Szczupak, D.; Lent, R. The Absolute Number of Oligodendrocytes in the Adult Mouse Brain. Front. Neuroanat. 2018, 12, 90. [Google Scholar] [CrossRef]
- Osanai, Y.; Shimizu, T.; Mori, T.; Yoshimura, Y.; Hatanaka, N.; Nambu, A.; Kimori, Y.; Koyama, S.; Kobayashi, K.; Ikenaka, K. Rabies virus-mediated oligodendrocyte labeling reveals a single oligodendrocyte myelinates axons from distinct brain regions. Glia 2017, 65, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Archelos, J.J.; Hartung, H.P. Mechanisms of immune regulation in the peripheral nervous system. Brain Pathol. 1999, 9, 343–360. [Google Scholar] [CrossRef]
- Ydens, E.; Lornet, G.; Smits, V.; Goethals, S.; Timmerman, V.; Janssens, S. The neuroinflammatory role of Schwann cells in disease. Neurobiol. Dis. 2013, 55, 95–103. [Google Scholar] [CrossRef]
- Jenson, A.B.; Rabin, E.R.; Bentinck, D.C.; Melnick, J.L. Rabies virus neuronitis. J. Virol. 1969, 3, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Charlton, K.M.; Casey, G.A.; Wandeler, A.I.; Nadin-Davis, S. Early events in rabies virus infection of the central nervous system in skunks (Mephitis mephitis). Acta Neuropathol. 1995, 91, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Zaeck, L.; Potratz, M.; Freuling, C.M.; Müller, T.; Finke, S. High-Resolution 3D Imaging of Rabies Virus Infection in Solvent-Cleared Brain Tissue. JoVE 2019, e59402. [Google Scholar] [CrossRef]
- Whitley, R.; Baines, J. Clinical management of herpes simplex virus infections: Past, present, and future. F1000Res 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Obara, Y.; Furuta, Y.; Takasu, T.; Suzuki, S.; Suzuki, H.; Matsukawa, S.; Fujioka, Y.; Takahashi, H.; Kurata, T.; Nagashima, K. Distribution of herpes simplex virus types 1 and 2 genomes in human spinal ganglia studied by PCR and in situ hybridization. J. Med. Virol. 1997, 52, 136–142. [Google Scholar] [CrossRef]
- Richter, E.R.; Dias, J.K.; Gilbert, J.E.; Atherton, S.S. Distribution of herpes simplex virus type 1 and varicella zoster virus in ganglia of the human head and neck. J. Infect. Dis. 2009, 200, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.E.; Johnson, R.T. An explanation for the localization of herpes simplex encephalitis? Ann. Neurol. 1979, 5, 2–5. [Google Scholar] [CrossRef]
- Johnson, R.T.; Mims, C.A. Pathogenesis of viral infections of the nervous system. N. Engl. J. Med. 1968, 278, 23–30. [Google Scholar] [CrossRef]
- Esiri, M.M. Herpes-Simplex Encephalitis—an Immunohistological Study of the Distribution of Viral-Antigen within the Brain. J. Neurol. Sci. 1982, 54, 209–226. [Google Scholar] [CrossRef]
- Kennedy, P.G.; Clements, G.B.; Brown, S.M. Differential susceptibility of human neural cell types in culture to infection with herpes simplex virus. Brain 1983, 106 Pt 1, 101–119. [Google Scholar] [CrossRef]
- Bansode, Y.D.; Chattopadhyay, D.; Saha, B. Innate immune response in astrocytes infected with herpes simplex virus 1. Arch. Virol. 2019, 164, 1433–1439. [Google Scholar] [CrossRef]
- Bansode, Y.D.; Chattopadhyay, D.; Saha, B. Transcriptomic Analysis of Interferon Response in Toll-Like Receptor 2 Ligand-Treated and Herpes Simplex Virus 1-Infected Neurons and Astrocytes. Viral Immunol. 2020. [Google Scholar] [CrossRef]
- Liu, Z.; Guan, Y.; Sun, X.; Shi, L.; Liang, R.; Lv, X.; Xin, W. HSV-1 activates NF-kappaB in mouse astrocytes and increases TNF-alpha and IL-6 expression via Toll-like receptor 3. Neurol. Res. 2013, 35, 755–762. [Google Scholar] [CrossRef]
- Gumenyuk, A.V.; Tykhomyrov, A.A.; Savosko, S.I.; Guzyk, M.M.; Rybalko, S.L.; Ryzha, A.O.; Chaikovsky, Y.B. State of Astrocytes in the Mice Brain under Conditions of Herpes Viral Infection and Modeled Stroke. Neurophysiology 2018, 50, 326–331. [Google Scholar] [CrossRef]
- Hensel, N.; Raker, V.; Forthmann, B.; Detering, N.T.; Kubinski, S.; Buch, A.; Katzilieris-Petras, G.; Spanier, J.; Gudi, V.; Wagenknecht, S.; et al. HSV-1 triggers paracrine fibroblast growth factor response from cortical brain cells via immediate-early protein ICP0. J. Neuroinflamm. 2019, 16, 248. [Google Scholar] [CrossRef] [PubMed]
- Fiacco, T.A.; Agulhon, C.; McCarthy, K.D. Sorting Out Astrocyte Physiology from Pharmacology. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 151–174. [Google Scholar] [CrossRef] [PubMed]
- Aravalli, R.N.; Hu, S.; Rowen, T.N.; Palmquist, J.M.; Lokensgard, J.R. TLR2-mediated proinflammatory cytokine and chemokine production by microglial cells in response to herpes simplex virus. J. Immunol. 2005, 175, 4189–4193. [Google Scholar] [CrossRef]
- Fekete, R.; Cserep, C.; Lenart, N.; Toth, K.; Orsolits, B.; Martinecz, B.; Mehes, E.; Szabo, B.; Nemeth, V.; Gonci, B.; et al. Microglia control the spread of neurotropic virus infection via P2Y12 signalling and recruit monocytes through P2Y12-independent mechanisms. Acta Neuropathol. 2018, 136, 461–482. [Google Scholar] [CrossRef]
- Bello-Morales, R.; Fedetz, M.; Alcina, A.; Tabares, E.; Lopez-Guerrero, J.A. High susceptibility of a human oligodendroglial cell line to herpes simplex type 1 infection. J. Neurovirol. 2005, 11, 190–198. [Google Scholar] [CrossRef]
- Kastrukoff, L.F.; Kim, S.U. Oligodendrocytes from human donors differ in resistance to herpes simplex virus 1 (HSV-1). Glia 2002, 38, 87–92. [Google Scholar] [CrossRef]
- Ugolini, G.; Kuypers, H.G.; Simmons, A. Retrograde transneuronal transfer of herpes simplex virus type 1 (HSV 1) from motoneurones. Brain Res. 1987, 422, 242–256. [Google Scholar] [CrossRef]
- Boukhvalova, M.S.; Mortensen, E.; Mbaye, A.; Lopez, D.; Kastrukoff, L.; Blanco, J.C.G. Herpes Simplex Virus 1 Induces Brain Inflammation and Multifocal Demyelination in the Cotton Rat Sigmodon hispidus. J. Virol. 2019, 94. [Google Scholar] [CrossRef] [PubMed]
- Townsend, J.J.; Collins, P.K. Peripheral nervous system demyelination with herpes simplex virus. J. Neuropathol. Exp. Neurol. 1986, 45, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Shimeld, C.; Efstathiou, S.; Hill, T. Tracking the spread of a lacZ-tagged herpes simplex virus type 1 between the eye and the nervous system of the mouse: Comparison of primary and recurrent infection. J. Virol. 2001, 75, 5252–5262. [Google Scholar] [CrossRef]
- Hill, T.J.; Field, H.J. The interaction of herpes simplex virus with cultures of peripheral nervous tissue: An electron microscopic study. J. Gen. Virol. 1973, 21, 123–133. [Google Scholar] [CrossRef]
- Wilkinson, R.; Leaver, C.; Simmons, A.; Pereira, R.A. Restricted replication of herpes simplex virus in satellite glial cell cultures clonally derived from adult mice. J. Neurovirol. 1999, 5, 384–391. [Google Scholar] [CrossRef]
- Zerboni, L.; Che, X.; Reichelt, M.; Qiao, Y.; Gu, H.; Arvin, A. Herpes simplex virus 1 tropism for human sensory ganglion neurons in the severe combined immunodeficiency mouse model of neuropathogenesis. J. Virol. 2013, 87, 2791–2802. [Google Scholar] [CrossRef]
- Suja, M.S.; Mahadevan, A.; Madhusudana, S.N.; Shankar, S.K. Role of Apoptosis in Rabies Viral Encephalitis: A Comparative Study in Mice, Canine, and Human Brain with a Review of Literature. Patholog. Res. Int. 2011, 2011, 1–13. [Google Scholar] [CrossRef]
- Lokensgard, J.R.; Hu, S.; Sheng, W.; van Oijen, M.; Cox, D.; Cheeran, M.C.-J.J.; Peterson, P.K.; Lokensgard, J.R.; Hu, S.; Wen, J.R.; et al. Robust expression of TNF-, IL-1, RANTES, and IP-10 by human microglial cells during nonproductive infection with herpes simplex virus. J. Neurovirol. 2001, 7, 208–219. [Google Scholar] [CrossRef]
- Rosenberg, G.A. Neurological diseases in relation to the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1139–1151. [Google Scholar] [CrossRef]
- Schnell, M.J.; McGettigan, J.P.; Wirblich, C.; Papaneri, A. The cell biology of rabies virus: Using stealth to reach the brain. Nat. Rev. Microbiol. 2009, 8, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Pape, K.; Tamouza, R.; Leboyer, M.; Zipp, F. Immunoneuropsychiatry—Novel perspectives on brain disorders. Nat. Rev. Neurol. 2019, 15, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Scott, T.; Nel, L. Subversion of the Immune Response by Rabies Virus. Viruses 2016, 8, 231. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Hooper, D.C. Lethal Silver-Haired Bat Rabies Virus Infection Can Be Prevented by Opening the Blood-Brain Barrier. J. Virol. 2007, 81, 7993–7998. [Google Scholar] [CrossRef] [PubMed]
- Phares, T.W.; Kean, R.B.; Mikheeva, T.; Hooper, D.C. Regional Differences in Blood-Brain Barrier Permeability Changes and Inflammation in the Apathogenic Clearance of Virus from the Central Nervous System. J. Immunol. 2006, 176, 7666–7675. [Google Scholar] [CrossRef]
- Roy, A.; Phares, T.W.; Koprowski, H.; Hooper, D.C. Failure to open the blood-brain barrier and deliver immune effectors to central nervous system tissues leads to the lethal outcome of silver-haired bat rabies virus infection. J. Virol. 2007, 81, 1110–1118. [Google Scholar] [CrossRef]
- Roy, A.; Hooper, D.C. Immune evasion by rabies viruses through the maintenance of blood-brain barrier integrity. J. Neurovirol. 2008, 14, 401–411. [Google Scholar] [CrossRef]
- Hooper, D.C.; Roy, A.; Barkhouse, D.A.; Li, J.; Kean, R.B. Rabies Virus Clearance from the Central Nervous System, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2011; Volume 79, ISBN 9780123870407. [Google Scholar]
- Alvarez, L.; Fajardo, R.; Lopez, E.; Pedroza, R.; Hemachudha, T.; Kamolvarin, N.; Cortes, G.; Baer, G.M. Partial recovery from rabies in a nine-year-old boy. Pediatr. Infect. Dis. J. 1994, 13, 1154–1155. [Google Scholar] [CrossRef]
- Hattwick, M.A.; Weis, T.T.; Stechschulte, C.J.; Baer, G.M.; Gregg, M.B. Recovery from rabies. A case report. Ann. Intern. Med. 1972, 76, 931–942. [Google Scholar] [CrossRef]
- Porras, C.; Barboza, J.J.; Fuenzalida, E.; Adaros, H.L.; Oviedo, A.M.; Furst, J. Recovery from rabies in man. Ann. Intern. Med. 1976, 85, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Madhusudana, S.N.; Nagaraj, D.; Uday, M.; Ratnavalli, E.; Kumar, M.V. Partial recovery from rabies in a six-year-old girl. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 2002, 6, 85–86. [Google Scholar] [CrossRef]
- Hu, W.T.; Willoughby, R.E.J.; Dhonau, H.; Mack, K.J. Long-term follow-up after treatment of rabies by induction of coma. N. Engl. J. Med. 2007, 357, 945–946. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Recovery of a patient from clinical rabies--Wisconsin, 2004. MMWR. Morb. Mortal. Wkly. Rep. 2004, 53, 1171–1173. [Google Scholar]
- Centers for Disease Control and Prevention (CDC). Recovery of a patient from clinical rabies--California, 2011. MMWR. Morb. Mortal. Wkly. Rep. 2012, 61, 61–65. [Google Scholar]
- Dietzschold, B.; Kao, M.; Zheng, Y.M.; Chen, Z.Y.; Maul, G.; Fu, Z.F.; Rupprecht, C.E.; Koprowski, H. Delineation of putative mechanisms involved in antibody-mediated clearance of rabies virus from the central nervous system. Proc. Natl. Acad. Sci. USA 1992, 89, 7252–7256. [Google Scholar] [CrossRef]
- Rowell, J.F.; Griffin, D.E. The inflammatory response to nonfatal Sindbis virus infection of the nervous system is more severe in SJL than in BALB/c mice and is associated with low levels of IL-4 mRNA and high levels of IL-10-producing CD4+ T cells. J. Immunol. 1999, 162, 1624–1632. [Google Scholar]
- Singh, A.K.; Yang, J.-Q.; Parekh, V.V.; Wei, J.; Wang, C.-R.; Joyce, S.; Singh, R.R.; Van Kaer, L. The natural killer T cell ligand alpha-galactosylceramide prevents or promotes pristane-induced lupus in mice. Eur. J. Immunol. 2005, 35, 1143–1154. [Google Scholar] [CrossRef]
- Lodmell, D.L.; Ewalt, L.C. Pathogenesis of street rabies virus infections in resistant and susceptible strains of mice. J. Virol. 1985, 55, 788–795. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, G.; Wen, Y.; Yang, S.; Xia, X.; Fu, Z.F. Intracerebral Administration of Recombinant Rabies Virus Expressing GM-CSF Prevents the Development of Rabies after Infection with Street Virus. PLoS ONE 2011, 6, e25414. [Google Scholar] [CrossRef]
- de Melo, G.D.; Sonthonnax, F.; Lepousez, G.; Jouvion, G.; Minola, A.; Zatta, F.; Larrous, F.; Kergoat, L.; Mazo, C.; Moigneu, C.; et al. A combination of two human monoclonal antibodies cures symptomatic rabies. EMBO Mol. Med. 2020, 12, e12628. [Google Scholar] [CrossRef] [PubMed]
- Hellert, J.; Buchrieser, J.; Larrous, F.; Minola, A.; de Melo, G.D.; Soriaga, L.; England, P.; Haouz, A.; Telenti, A.; Schwartz, O.; et al. Structure of the prefusion-locking broadly neutralizing antibody RVC20 bound to the rabies virus glycoprotein. Nat. Commun. 2020, 11, 596. [Google Scholar] [CrossRef]
- Liu, H.; Qiu, K.; He, Q.; Lei, Q.; Lu, W. Mechanisms of Blood-Brain Barrier Disruption in Herpes Simplex Encephalitis. J. Neuroimmune Pharmacol. 2019, 14, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, P.; Ramakrishna, C.; Brown, J.; Tyszka, J.M.; Hamamura, M.; Hinton, D.R.; Kovats, S.; Nalcioglu, O.; Weinberg, K.; Openshaw, H.; et al. The immune response to herpes simplex virus type 1 infection in susceptible mice is a major cause of central nervous system pathology resulting in fatal encephalitis. J. Virol. 2008, 82, 7078–7088. [Google Scholar] [CrossRef]
- Mancini, M.; Vidal, S.M. Insights into the pathogenesis of herpes simplex encephalitis from mouse models. Mamm. Genome 2018, 29, 425–445. [Google Scholar] [CrossRef]
- Chan, W.L.; Javanovic, T.; Lukic, M.L. Infiltration of immune T cells in the brain of mice with herpes simplex virus-induced encephalitis. J. Neuroimmunol. 1989, 23, 195–201. [Google Scholar] [CrossRef]
- Muller, W.A. Leukocyte-endothelial cell interactions in the inflammatory response. Lab. Investig. 2002, 82, 521–533. [Google Scholar] [CrossRef]
- Brundula, V.; Rewcastle, N.B.; Metz, L.M.; Bernard, C.C.; Yong, V.W. Targeting leukocyte MMPs and transmigration: Minocycline as a potential therapy for multiple sclerosis. Brain 2002, 125, 1297–1308. [Google Scholar] [CrossRef]
- Yong, V.W.; Power, C.; Edwards, D.R. Metalloproteinases in biology and pathology of the nervous system. Nat. Rev. Neurosci. 2001, 2, 502–511. [Google Scholar] [CrossRef]
- Subileau, E.A.; Rezaie, P.; Davies, H.A.; Colyer, F.M.; Greenwood, J.; Male, D.K.; Romero, I.A. Expression of Chemokines and Their Receptors by Human Brain Endothelium: Implications for Multiple Sclerosis. J. Neuropathol. Exp. Neurol. 2009, 68, 227–240. [Google Scholar] [CrossRef]
- Kim, Y.C.; Bang, D.; Lee, S.; Lee, K.H. The effect of herpesvirus infection on the expression of cell adhesion molecules on cultured human dermal microvascular endothelial cells. J. Dermatol. Sci. 2000, 24, 38–47. [Google Scholar] [CrossRef]
- Sobel, R.A.; Mitchell, M.E.; Fondren, G. Intercellular adhesion molecule-1 (ICAM-1) in cellular immune reactions in the human central nervous system. Am. J. Pathol. 1990, 136, 1309–1316. [Google Scholar] [PubMed]
- Brankin, B.; Hart, M.N.; Cosby, S.L.; Fabry, Z.; Allen, I.V. Adhesion molecule expression and lymphocyte adhesion to cerebral endothelium: Effects of measles virus and herpes simplex 1 virus. J. Neuroimmunol. 1995, 56, 1–8. [Google Scholar] [CrossRef]
- He, Q.; Liu, H.; Huang, C.; Wang, R.; Luo, M.; Lu, W. Herpes Simplex Virus 1-Induced Blood-Brain Barrier Damage Involves Apoptosis Associated With GM130-Mediated Golgi Stress. Front. Mol. Neurosci. 2020, 13, 2. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, Z.N.; Guo, Y.J.; Mei, Y.W. Favorable effects of MMP-9 knockdown in murine herpes simplex encephalitis using small interfering RNA. Neurol. Res. 2010, 32, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Dai, J.; Bai, F.; Kong, K.F.; Wong, S.J.; Montgomery, R.R.; Madri, J.A.; Fikrig, E. Matrix metalloproteinase 9 facilitates West Nile virus entry into the brain. J. Virol. 2008, 82, 8978–8985. [Google Scholar] [CrossRef]
- Hu, S.; Sheng, W.S.; Schachtele, S.J.; Lokensgard, J.R. Reactive oxygen species drive herpes simplex virus (HSV)-1-induced proinflammatory cytokine production by murine microglia. J. Neuroinflamm. 2011, 8, 123. [Google Scholar] [CrossRef]
- Roberts, T.K.; Eugenin, E.A.; Lopez, L.; Romero, I.A.; Weksler, B.B.; Couraud, P.O.; Berman, J.W. CCL2 disrupts the adherens junction: Implications for neuroinflammation. Lab. Investig. 2012, 92, 1213–1233. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Keep, R.F.; Kunkel, S.L.; Andjelkovic, A. V Potential role of MCP-1 in endothelial cell tight junction “opening”: Signaling via Rho and Rho kinase. J. Cell Sci. 2003, 116, 4615–4628. [Google Scholar] [CrossRef]
- Jung, J.S.; Bhat, R.V.; Preston, G.M.; Guggino, W.B.; Baraban, J.M.; Agre, P. Molecular characterization of an aquaporin cDNA from brain: Candidate osmoreceptor and regulator of water balance. Proc. Natl. Acad. Sci. USA 1994, 91, 13052–13056. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 2013, 14, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Safain, M.G.; Roguski, M.; Kryzanski, J.T.; Weller, S.J. A review of the combined medical and surgical management in patients with herpes simplex encephalitis. Clin. Neurol. Neurosurg. 2015, 128, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Armien, A.G.; Hu, S.; Little, M.R.; Robinson, N.; Lokensgard, J.R.; Low, W.C.; Cheeran, M.C. Chronic cortical and subcortical pathology with associated neurological deficits ensuing experimental herpes encephalitis. Brain Pathol. 2010, 20, 738–750. [Google Scholar] [CrossRef]
- Michinaga, S.; Koyama, Y. Dual Roles of Astrocyte-Derived Factors in Regulation of Blood-Brain Barrier Function after Brain Damage. Int. J. Mol. Sci. 2019, 20, 571. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.H.; Paludan, S.R. Viral evasion of DNA-stimulated innate immune responses. Cell. Mol. Immunol. 2017, 14, 4–13. [Google Scholar] [CrossRef]
- Guo, Y.J.; Luo, T.; Wu, F.; Mei, Y.W.; Peng, J.; Liu, H.; Li, H.R.; Zhang, S.L.; Dong, J.H.; Fang, Y.; et al. Involvement of TLR2 and TLR9 in the anti-inflammatory effects of chlorogenic acid in HSV-1-infected microglia. Life Sci. 2015, 127, 12–18. [Google Scholar] [CrossRef]
- Zhou, Y.; Ye, L.; Wan, Q.; Zhou, L.; Wang, X.; Li, J.; Hu, S.; Zhou, D.; Ho, W. Activation of Toll-like receptors inhibits herpes simplex virus-1 infection of human neuronal cells. J. Neurosci. Res. 2009, 87, 2916–2925. [Google Scholar] [CrossRef]
- Furr, S.R.; Marriott, I. Viral CNS infections: Role of glial pattern recognition receptors in neuroinflammation. Front. Microbiol. 2012, 3, 201. [Google Scholar] [CrossRef]
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef]
- Katz, I.S.S.; Guedes, F.; Fernandes, E.R.; dos Ramos Silva, S. Immunological aspects of rabies: A literature review. Arch. Virol. 2017, 162, 3251–3268. [Google Scholar] [CrossRef]
- Yordy, B.; Iijima, N.; Huttner, A.; Leib, D.; Iwasaki, A. A neuron-specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe 2012, 12, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhu, S.; Hu, L.; Ye, P.; Wang, Y.; Tian, Q.; Mei, M.; Chen, H.; Guo, X. Wild-type rabies virus induces autophagy in human and mouse neuroblastoma cell lines. Autophagy 2016, 12, 1704–1720. [Google Scholar] [CrossRef] [PubMed]
- Yakoub, A.M.; Shukla, D. Autophagy stimulation abrogates herpes simplex virus-1 infection. Sci. Rep. 2015, 5, 9730. [Google Scholar] [CrossRef]
- Nakamichi, K.; Saiki, M.; Sawada, M.; Takayama-Ito, M.; Yamamuro, Y.; Morimoto, K.; Kurane, I. Rabies Virus-Induced Activation of Mitogen-Activated Protein Kinase and NF- B Signaling Pathways Regulates Expression of CXC and CC Chemokine Ligands in Microglia. J. Virol. 2005, 79, 11801–11812. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Akaike, T.; Maeda, H. Role of nitric oxide in pathogenesis of herpes simplex virus encephalitis in rats. Virology 1999, 256, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Cagnin, A.; Myers, R.; Gunn, R.N.; Lawrence, A.D.; Stevens, T.; Kreutzberg, G.W.; Jones, T.; Banati, R.B. In Vivo visualization of activated glia by [11C] (R)-PK11195-PET following herpes encephalitis reveals projected neuronal damage beyond the primary focal lesion. Brain 2001, 124, 2014–2027. [Google Scholar] [CrossRef]
- Reinert, L.S.; Harder, L.; Holm, C.K.; Iversen, M.B.; Horan, K.A.; Dagnæs-Hansen, F.; Ulhøi, B.P.; Holm, T.H.; Mogensen, T.H.; Owens, T.; et al. TLR3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates establishment of CNS infection in mice. J. Clin. Investig. 2012, 122, 1368–1376. [Google Scholar] [CrossRef]
- Conrady, C.D.; Drevets, D.A.; Carr, D.J. Herpes simplex type I (HSV-1) infection of the nervous system: Is an immune response a good thing? J. Neuroimmunol. 2010, 220, 1–9. [Google Scholar] [CrossRef]
- Li, J.; Faber, M.; Dietzschold, B.; Hooper, D.C. The Role of Toll-Like Receptors in the Induction of Immune Responses during Rabies Virus Infection, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2011; Volume 79, ISBN 9780123870407. [Google Scholar]
- Faul, E.J.; Wanjalla, C.N.; Suthar, M.S.; Gale, M.; Wirblich, C.; Schnell, M.J. Rabies Virus Infection Induces Type I Interferon Production in an IPS-1 Dependent Manner While Dendritic Cell Activation Relies on IFNAR Signaling. PLoS Pathog. 2010, 6, e1001016. [Google Scholar] [CrossRef]
- Prehaud, C.; Mégret, F.; Lafage, M.; Lafon, M. Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J. Virol. 2005, 79, 12893–12904. [Google Scholar] [CrossRef] [PubMed]
- Ménager, P.; Roux, P.; Mégret, F.; Bourgeois, J.-P.; Le Sourd, A.-M.; Danckaert, A.; Lafage, M.; Préhaud, C.; Lafon, M. Toll-like receptor 3 (TLR3) plays a major role in the formation of rabies virus Negri Bodies. PLoS Pathog. 2009, 5, e1000315. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.C.; Rossiter, J.P.; Lafon, M. Expression of Toll-like receptor 3 in the human cerebellar cortex in rabies, herpes simplex encephalitis, and other neurological diseases. J. Neurovirol. 2006, 12, 229–234. [Google Scholar] [CrossRef]
- Luo, Z.; Li, Y.; Zhou, M.; Lv, L.; Wu, Q.; Chen, C.; Zhang, Y.; Sui, B.; Tu, C.; Cui, M.; et al. Toll-like receptor 7 enhances rabies virus-induced humoral immunity by facilitating the formation of germinal centers. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Liu, R.; Wang, J.; Yang, Y.; Khan, I.; Zhu, N. Rabies virus lipopeptide conjugated to a TLR7 agonist improves the magnitude and quality of the Th1-biased humoral immune response in mice. Virology 2016, 497, 102–110. [Google Scholar] [CrossRef]
- Luo, Z.; Lv, L.; Li, Y.; Sui, B.; Wu, Q.; Zhang, Y.; Pei, J.; Li, M.; Zhou, M.; Hooper, D.C.; et al. Dual Role of Toll-Like Receptor 7 in the Pathogenesis of Rabies Virus in a Mouse Model. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Mercer, J.; Greber, U.F. Virus interactions with endocytic pathways in macrophages and dendritic cells. Trends Microbiol. 2013, 21, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Lytle, A.G.; Shen, S.; McGettigan, J.P. Lymph Node but Not Intradermal Injection Site Macrophages Are Critical for Germinal Center Formation and Antibody Responses to Rabies Vaccination. J. Virol. 2015, 89, 2842–2848. [Google Scholar] [CrossRef][Green Version]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.-K.; Schlee, M.; et al. 5’-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef]
- Yang, Y.; Huang, Y.; Gnanadurai, C.W.; Cao, S.; Liu, X.; Cui, M.; Fu, Z.F. The Inability of Wild-Type Rabies Virus to Activate Dendritic Cells Is Dependent on the Glycoprotein and Correlates with Its Low Level of the De Novo -Synthesized Leader RNA. J. Virol. 2015, 89, 2157–2169. [Google Scholar] [CrossRef]
- Lafon, M. Modulation of the immune response in the nervous system by rabies virus. Curr. Top. Microbiol. Immunol. 2005, 289, 239–258. [Google Scholar] [CrossRef] [PubMed]
- Sui, B.; Chen, D.; Liu, W.; Tian, B.; Lv, L.; Pei, J.; Wu, Q.; Zhou, M.; Fu, Z.F.; Zhang, Y.; et al. Comparison of lncRNA and mRNA expression in mouse brains infected by a wild-type and a lab-attenuated Rabies lyssavirus. J. Gen. Virol. 2021, 102. [Google Scholar] [CrossRef]
- Ménager, P.; Roux, P.; Mégret, F.; Préhaud, C.; Bourgeois, J.-P.; Le Sourd, A.-M.; Lafage, M.; Lafon, M. TLR3 is a key component of rabies virus induced Negri bodies. BMC Proc. 2008, 2, 3. [Google Scholar] [CrossRef]
- Matsumoto, M.; Oshiumi, H.; Seya, T. Antiviral responses induced by the TLR3 pathway. Rev. Med. Virol. 2011, 21, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Lahaye, X.; Vidy, A.; Pomier, C.; Obiang, L.; Harper, F.; Gaudin, Y.; Blondel, D. Functional Characterization of Negri Bodies (NBs) in Rabies Virus-Infected Cells: Evidence that NBs Are Sites of Viral Transcription and Replication. J. Virol. 2009, 83, 7948–7958. [Google Scholar] [CrossRef]
- Nikolic, J.; Le Bars, R.; Lama, Z.; Scrima, N.; Lagaudrière-Gesbert, C.; Gaudin, Y.; Blondel, D. Negri bodies are viral factories with properties of liquid organelles. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Salaun, B.; Coste, I.; Rissoan, M.-C.; Lebecque, S.J.; Renno, T. TLR3 Can Directly Trigger Apoptosis in Human Cancer Cells. J. Immunol. 2006, 176, 4894–4901. [Google Scholar] [CrossRef]
- Plioplys, A.V.; Massimini, N. Alpha/Beta Interferon Is a Neuronal Growth Factor. Neuroimmunomodulation 1995, 2, 31–35. [Google Scholar] [CrossRef]
- Meucci, O.; Fatatis, A.; Simen, A.A.; Bushell, T.J.; Gray, P.W.; Miller, R.J. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc. Natl. Acad. Sci. USA 1998, 95, 14500–14505. [Google Scholar] [CrossRef]
- Pizzi, M.; Sarnico, I.; Boroni, F.; Benarese, M.; Dreano, M.; Garotta, G.; Valerio, A.; Spano, P. Prevention of neuron and oligodendrocyte degeneration by interleukin-6 (IL-6) and IL-6 receptor/IL-6 fusion protein in organotypic hippocampal slices. Mol. Cell. Neurosci. 2004, 25, 301–311. [Google Scholar] [CrossRef]
- Hirota, H.; Kiyama, H.; Kishimoto, T.; Taga, T. Accelerated nerve regeneration in mice by upregulated expression of interleukin (IL) 6 and IL-6 receptor after trauma. J. Exp. Med. 1996, 183, 2627–2634. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Tobita, M. Pathology. In Rabies; Jackson, A.C., Fooks, A.R., Eds.; Elsevier: San Diego, CA, USA, 2002; pp. 283–306. [Google Scholar]
- Kassis, R.; Larrous, F.; Estaquier, J.; Bourhy, H. Lyssavirus Matrix Protein Induces Apoptosis by a TRAIL-Dependent Mechanism Involving Caspase-8 Activation. J. Virol. 2004, 78, 6543–6555. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Hooper, D.C.; Spitsin, S.; Koprowski, H.; Dietzschold, B. Pathogenicity of different rabies virus variants inversely correlates with apoptosis and rabies virus glycoprotein expression in infected primary neuron cultures. J. Virol. 1999, 73, 510–518. [Google Scholar] [CrossRef]
- Kojima, I.; Izumi, F.; Ozawa, M.; Fujimoto, Y.; Okajima, M.; Ito, N.; Sugiyama, M.; Masatani, T. Analyses of cell death mechanisms related to amino acid substitution at position 95 in the rabies virus matrix protein. J. Gen. Virol. 2021, 102. [Google Scholar] [CrossRef]
- Gholami, A.; Kassis, R.; Real, E.; Delmas, O.; Guadagnini, S.; Larrous, F.; Obach, D.; Prevost, M.-C.; Jacob, Y.; Bourhy, H. Mitochondrial dysfunction in lyssavirus-induced apoptosis. J. Virol. 2008, 82, 4774–4784. [Google Scholar] [CrossRef] [PubMed]
- Dolman, C.L.; Charlton, K.M. Massive necrosis of the brain in rabies. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 1987, 14, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, L.P.; Gamon, T.H.M.; Cuevas, S.E.C.; Asano, K.M.; de Oliveira Fahl, W.; Iamamoto, K.; Scheffer, K.C.; Achkar, S.M.; Zanatto, D.A.; Mori, C.M.C.; et al. A rabies virus vampire bat variant shows increased neuroinvasiveness in mice when compared to a carnivore variant. Arch. Virol. 2017, 162, 3671–3679. [Google Scholar] [CrossRef]
- Li, X.-Q.; Sarmento, L.; Fu, Z.F. Degeneration of neuronal processes after infection with pathogenic, but not attenuated, rabies viruses. J. Virol. 2005, 79, 10063–10068. [Google Scholar] [CrossRef]
- Yan, X.; Prosniak, M.; Curti, M.T.; Weiss, M.L.; Faber, M.; Dietzschold, B.; Fu, Z.F. Silver-haired bat rabies virus variant does not induce apoptosis in the brain of experimentally infected mice. J. Neurovirol. 2001, 7, 518–527. [Google Scholar] [CrossRef]
- Scott, C.A.; Rossiter, J.P.; Andrew, R.D.; Jackson, A.C. Structural Abnormalities in Neurons Are Sufficient to Explain the Clinical Disease and Fatal Outcome of Experimental Rabies in Yellow Fluorescent Protein-Expressing Transgenic Mice. J. Virol. 2008, 82, 513–521. [Google Scholar] [CrossRef]
- Lindqvist, R.; Mundt, F.; Gilthorpe, J.D.; Wölfel, S.; Gekara, N.O.; Kröger, A.; Överby, A.K. Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J. Neuroinflamm. 2016, 13, 277. [Google Scholar] [CrossRef]
- Bsibsi, M.; Persoon-Deen, C.; Verwer, R.W.H.; Meeuwsen, S.; Ravid, R.; Van Noort, J.M. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia 2006, 53, 688–695. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Chauhan, V.S.; Furr, S.R.; Sterka, D.G.; Nelson, D.A.; Moerdyk-Schauwecker, M.; Marriott, I.; Grdzelishvili, V.Z. Vesicular stomatitis virus infects resident cells of the central nervous system and induces replication-dependent inflammatory responses. Virology 2010, 400, 187–196. [Google Scholar] [CrossRef]
- Zhao, P.; Zhao, L.; Zhang, K.; Feng, H.; Wang, H.; Wang, T.; Xu, T.; Feng, N.; Wang, C.; Gao, Y.; et al. Infection with street strain rabies virus induces modulation of the microRNA profile of the mouse brain. Virol. J. 2012, 9, 159. [Google Scholar] [CrossRef]
- Diniz, L.P.; Matias, I.; Siqueira, M.; Stipursky, J.; Gomes, F.C.A. Astrocytes and the TGF-β1 Pathway in the Healthy and Diseased Brain: A Double-Edged Sword. Mol. Neurobiol. 2019, 56, 4653–4679. [Google Scholar] [CrossRef] [PubMed]
- Ceyzériat, K.; Abjean, L.; Carrillo-de Sauvage, M.A.; Ben Haim, L.; Escartin, C. The complex STATes of astrocyte reactivity: How are they controlled by the JAK-STAT3 pathway? Neuroscience 2016, 330, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Nakamichi, K.; Inoue, S.; Takasaki, T.; Morimoto, K.; Kurane, I. Rabies Virus Stimulates Nitric Oxide Production and CXC Chemokine Ligand 10 Expression in Macrophages through Activation of Extracellular Signal-Regulated Kinases 1 and 2. J. Virol. 2004, 78, 9376–9388. [Google Scholar] [CrossRef]
- Michlmayr, D.; McKimmie, C.S.; Pingen, M.; Haxton, B.; Mansfield, K.; Johnson, N.; Fooks, A.R.; Graham, G.J. Defining the Chemokine Basis for Leukocyte Recruitment during Viral Encephalitis. J. Virol. 2014, 88, 9553–9567. [Google Scholar] [CrossRef] [PubMed]
- Besson, B.; Kim, S.; Kim, T.; Ko, Y.; Lee, S.; Larrous, F.; Song, J.; Shum, D.; Grailhe, R.; Bourhy, H. Kinome-Wide RNA Interference Screening Identifies Mitogen-Activated Protein Kinases and Phosphatidylinositol Metabolism as Key Factors for Rabies Virus Infection. mSphere 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Kalita, J.; Srivastava, R.; Mishra, M.K.; Basu, A.; Misra, U.K. Cytokines and chemokines in viral encephalitis: A clinicoradiological correlation. Neurosci. Lett. 2010, 473, 48–51. [Google Scholar] [CrossRef]
- Chen, C.-J.; Chen, J.-H.; Chen, S.-Y.; Liao, S.-L.; Raung, S.-L. Upregulation of RANTES Gene Expression in Neuroglia by Japanese Encephalitis Virus Infection. J. Virol. 2004, 78, 12107–12119. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, S.; Duseja, R.; Das, S.; Appaiahgiri, M.B.; Vrati, S.; Basu, A. Induction of IP-10 (CXCL10) in astrocytes following Japanese encephalitis. Neurosci. Lett. 2007, 414, 45–50. [Google Scholar] [CrossRef]
- McKimmie, C.; Michlmayr, D. Role of CXCL10 in central nervous system inflammation. Int. J. Interf. Cytokine Mediat. Res. 2014, 6, 1. [Google Scholar] [CrossRef]
- Ma, Y.; He, B. Recognition of herpes simplex viruses: Toll-like receptors and beyond. J. Mol. Biol. 2014, 426, 1133–1147. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef]
- Perry, A.K.; Chen, G.; Zheng, D.; Tang, H.; Cheng, G. The host type I interferon response to viral and bacterial infections. Cell Res. 2005, 15, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Wu, C.; Zhang, Y.-J. Interplay between Janus Kinase/Signal Transducer and Activator of Transcription Signaling Activated by Type I Interferons and Viral Antagonism. Front. Immunol. 2017, 8, 1758. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.-H.; Macmillan, J.B.; Chen, Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef]
- Piret, J.; Boivin, G. Innate immune response during herpes simplex virus encephalitis and development of immunomodulatory strategies. Rev. Med. Virol. 2015, 25, 300–319. [Google Scholar] [CrossRef]
- Wang, Y.; Jia, J.; Wang, Y.; Li, F.; Song, X.; Qin, S.; Wang, Z.; Kitazato, K.; Wang, Y. Roles of HSV-1 infection-induced microglial immune responses in CNS diseases: Friends or foes? Crit. Rev. Microbiol. 2019, 45, 581–594. [Google Scholar] [CrossRef]
- Kurt-Jones, E.A.; Chan, M.; Zhou, S.; Wang, J.; Reed, G.; Bronson, R.; Arnold, M.M.; Knipe, D.M.; Finberg, R.W. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. USA 2004, 101, 1315–1320. [Google Scholar] [CrossRef]
- Wang, J.P.; Bowen, G.N.; Zhou, S.; Cerny, A.; Zacharia, A.; Knipe, D.M.; Finberg, R.W.; Kurt-Jones, E.A. Role of specific innate immune responses in herpes simplex virus infection of the central nervous system. J. Virol. 2012, 86, 2273–2281. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, L.N.; Reinert, L.S.; Malmgaard, L.; Bartholdy, C.; Thomsen, A.R.; Paludan, S.R. TLR2 and TLR9 synergistically control herpes simplex virus infection in the brain. J. Immunol. 2008, 181, 8604–8612. [Google Scholar] [CrossRef] [PubMed]
- Menasria, R.; Boivin, N.; Lebel, M.; Piret, J.; Gosselin, J.; Boivin, G. Both TRIF and IPS-1 Adaptor Proteins Contribute to the Cerebral Innate Immune Response against Herpes Simplex Virus 1 Infection. J. Virol. 2013, 87, 7301–7308. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.H.; Kwon, K.M.; Kim, Y.-E.; Kim, K.K.; Ahn, J.-H. DNA sensing-independent inhibition of herpes simplex virus 1 replication by DAI/ZBP1. J. Virol. 2013, 87, 3076–3086. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Goulet, M.-L.; Sze, A.; Hadj, S.B.; Belgnaoui, S.M.; Lababidi, R.R.; Zheng, C.; Fritz, J.H.; Olagnier, D.; Lin, R. RIG-I-Mediated STING Upregulation Restricts Herpes Simplex Virus 1 Infection. J. Virol. 2016, 90, 9406–9419. [Google Scholar] [CrossRef] [PubMed]
- Lafaille, F.G.; Pessach, I.M.; Zhang, S.Y.; Ciancanelli, M.J.; Herman, M.; Abhyankar, A.; Ying, S.W.; Keros, S.; Goldstein, P.A.; Mostoslavsky, G.; et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 2012, 491, 769–773. [Google Scholar] [CrossRef]
- Rosato, P.C.; Leib, D.A. Neuronal Interferon Signaling Is Required for Protection against Herpes Simplex Virus Replication and Pathogenesis. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef]
- Cho, H.; Proll, S.C.; Szretter, K.J.; Katze, M.G.; Gale, M.; Diamond, M.S. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat. Med. 2013, 19, 458–464. [Google Scholar] [CrossRef]
- Farmer, J.R.; Altschaefl, K.M.; O’Shea, K.S.; Miller, D.J. Activation of the Type I Interferon Pathway Is Enhanced in Response to Human Neuronal Differentiation. PLoS ONE 2013, 8, e58813. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.L.W.; Vernon, P.S.; Griffin, D.E. Differentiation of Neurons Restricts Arbovirus Replication and Increases Expression of the Alpha Isoform of IRF-7. J. Virol. 2015, 89, 48–60. [Google Scholar] [CrossRef]
- Lokensgard, J.R.; Cheeran, M.C.; Hu, S.; Gekker, G.; Peterson, P.K. Glial cell responses to herpesvirus infections: Role in defense and immunopathogenesis. J. Infect. Dis. 2002, 186 (Suppl. S2), S171–S179. [Google Scholar] [CrossRef]
- Cédile, O.; Wlodarczyk, A.; Owens, T. CCL2 recruits T cells into the brain in a CCR2-independent manner. Apmis 2017, 125, 945–956. [Google Scholar] [CrossRef]
- Robichon, K.; Patel, V.; Connor, B.; La Flamme, A.C. Clozapine reduces infiltration into the CNS by targeting migration in experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2020, 17, 53. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Hamilton, T.A.; Tani, M.; Stoler, M.H.; Shick, H.E.; Major, J.A.; Estes, M.L.; Thomas, D.M.; Tuohy, V.K. Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1993, 7, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Broad, A.; Kirby, J.A.; Jones, D.E.; Applied, I.; Transplantation Research, G. Toll-like receptor interactions: Tolerance of MyD88-dependent cytokines but enhancement of MyD88-independent interferon-beta production. Immunology 2007, 120, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Chucair-Elliott, A.J.; Conrady, C.; Zheng, M.; Kroll, C.M.; Lane, T.E.; Carr, D.J. Microglia-induced IL-6 protects against neuronal loss following HSV-1 infection of neural progenitor cells. Glia 2014, 62, 1418–1434. [Google Scholar] [CrossRef]
- Marques, C.P.; Hu, S.; Sheng, W.; Lokensgard, J.R. Microglial cells initiate vigorous yet non-protective immune responses during HSV-1 brain infection. Virus Res. 2006, 121, 1–10. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feige, L.; Zaeck, L.M.; Sehl-Ewert, J.; Finke, S.; Bourhy, H. Innate Immune Signaling and Role of Glial Cells in Herpes Simplex Virus- and Rabies Virus-Induced Encephalitis. Viruses 2021, 13, 2364. https://doi.org/10.3390/v13122364
Feige L, Zaeck LM, Sehl-Ewert J, Finke S, Bourhy H. Innate Immune Signaling and Role of Glial Cells in Herpes Simplex Virus- and Rabies Virus-Induced Encephalitis. Viruses. 2021; 13(12):2364. https://doi.org/10.3390/v13122364
Chicago/Turabian StyleFeige, Lena, Luca M. Zaeck, Julia Sehl-Ewert, Stefan Finke, and Hervé Bourhy. 2021. "Innate Immune Signaling and Role of Glial Cells in Herpes Simplex Virus- and Rabies Virus-Induced Encephalitis" Viruses 13, no. 12: 2364. https://doi.org/10.3390/v13122364
APA StyleFeige, L., Zaeck, L. M., Sehl-Ewert, J., Finke, S., & Bourhy, H. (2021). Innate Immune Signaling and Role of Glial Cells in Herpes Simplex Virus- and Rabies Virus-Induced Encephalitis. Viruses, 13(12), 2364. https://doi.org/10.3390/v13122364