
Inhibition of miR-155 Promotes TGF-β Mediated Suppression of HIV Release in the Cervical Epithelial Cells

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Virus Used for In Vitro Assays
2.2. Co-Culture of ME-180 with HIV-1 Infected T-Cells
2.3. TGF-β Neutralization in ME-180 Cells
2.4. Transfection of miR-155 Inhibitor in ME-180 Cells
2.5. Total RNA Isolation, cDNA Synthesis and Quantitative Real-Time PCR (q-PCR)
2.6. ELISA
2.7. miRNA-mRNA Target Prediction
2.8. Statistics
3. Results
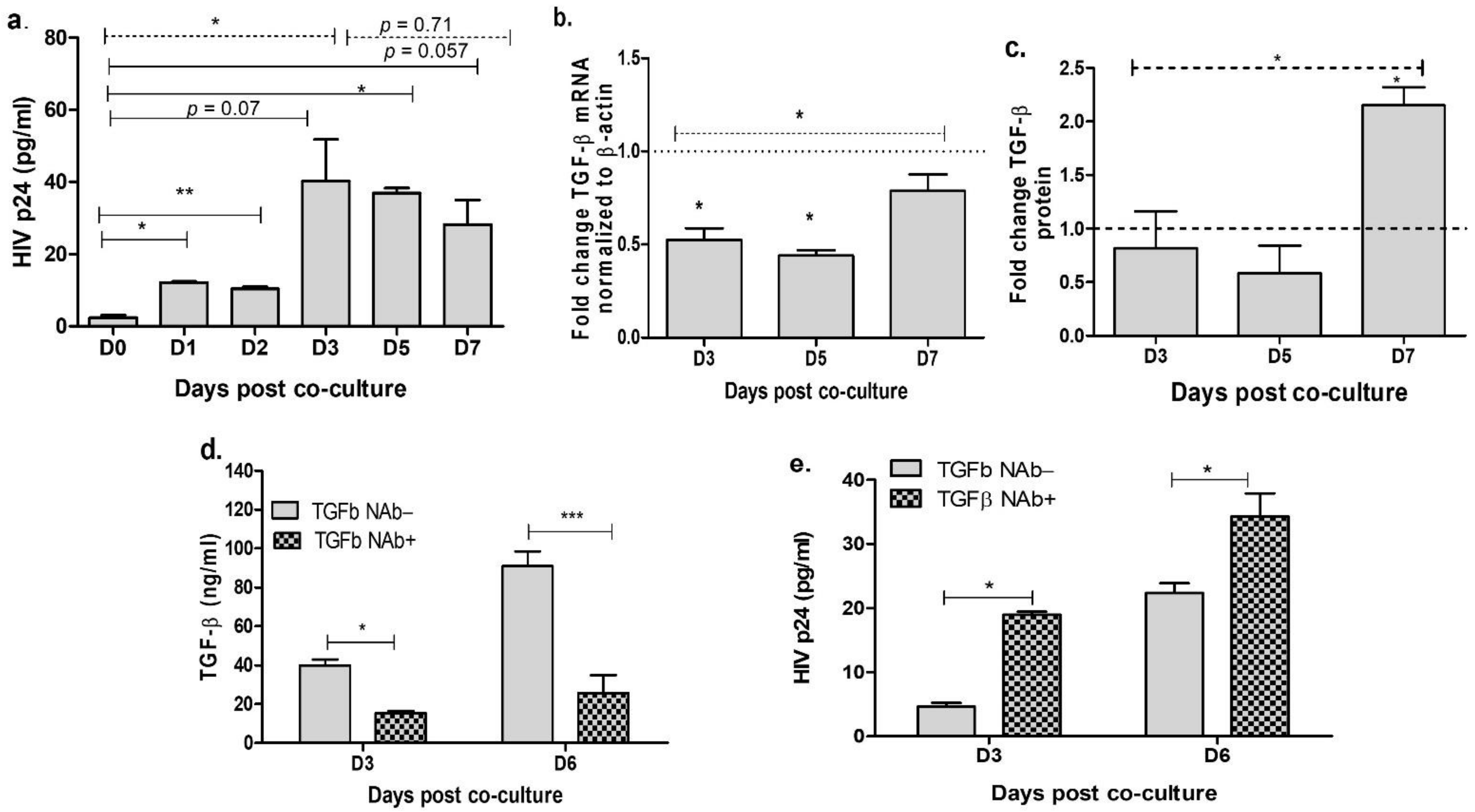
3.1. Neutralization of TGF-β in the Cervical Cells Increases HIV Release
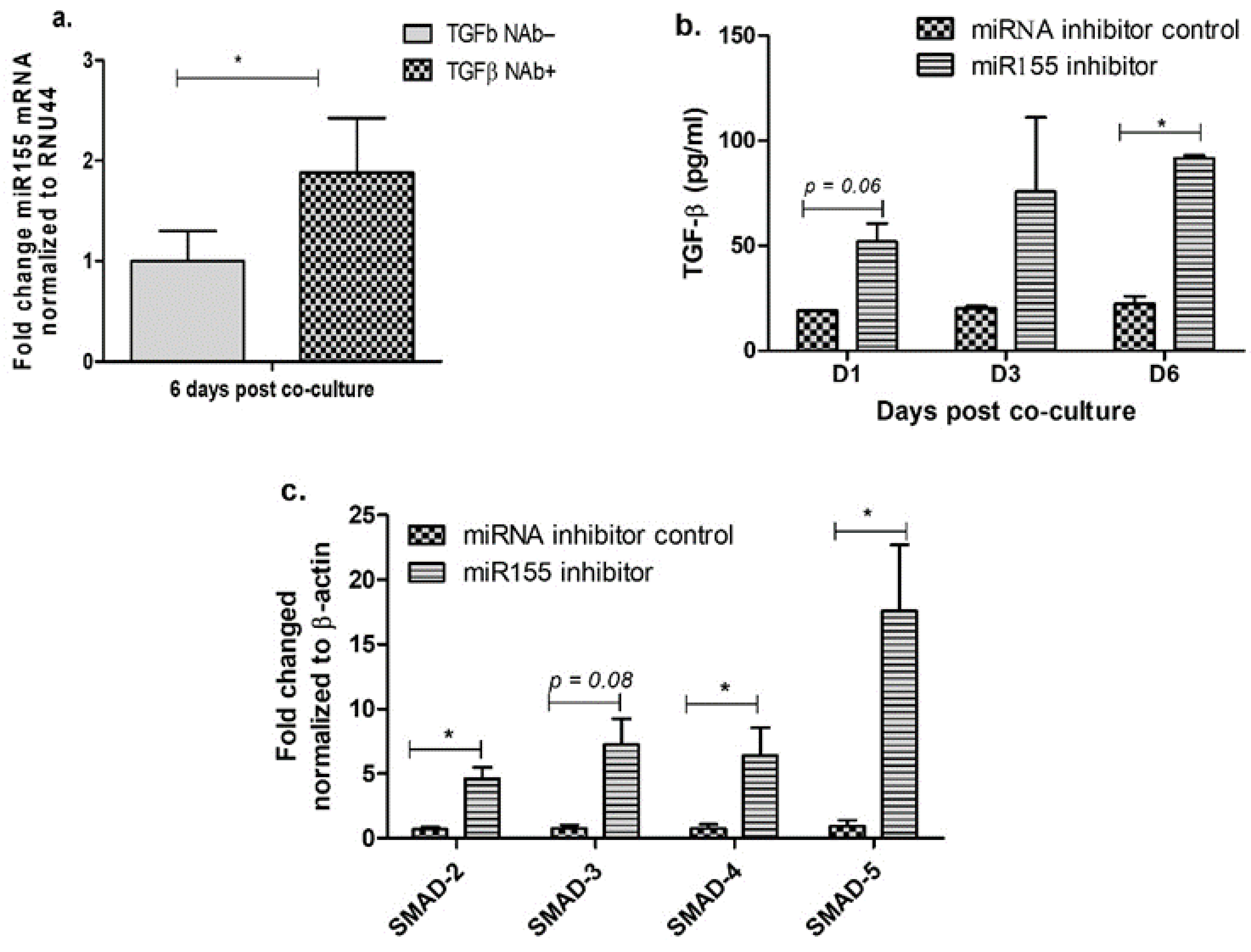
3.2. Inhibition of miR-155 Increases TGF-β Signaling in the Cervical Epithelial Cells
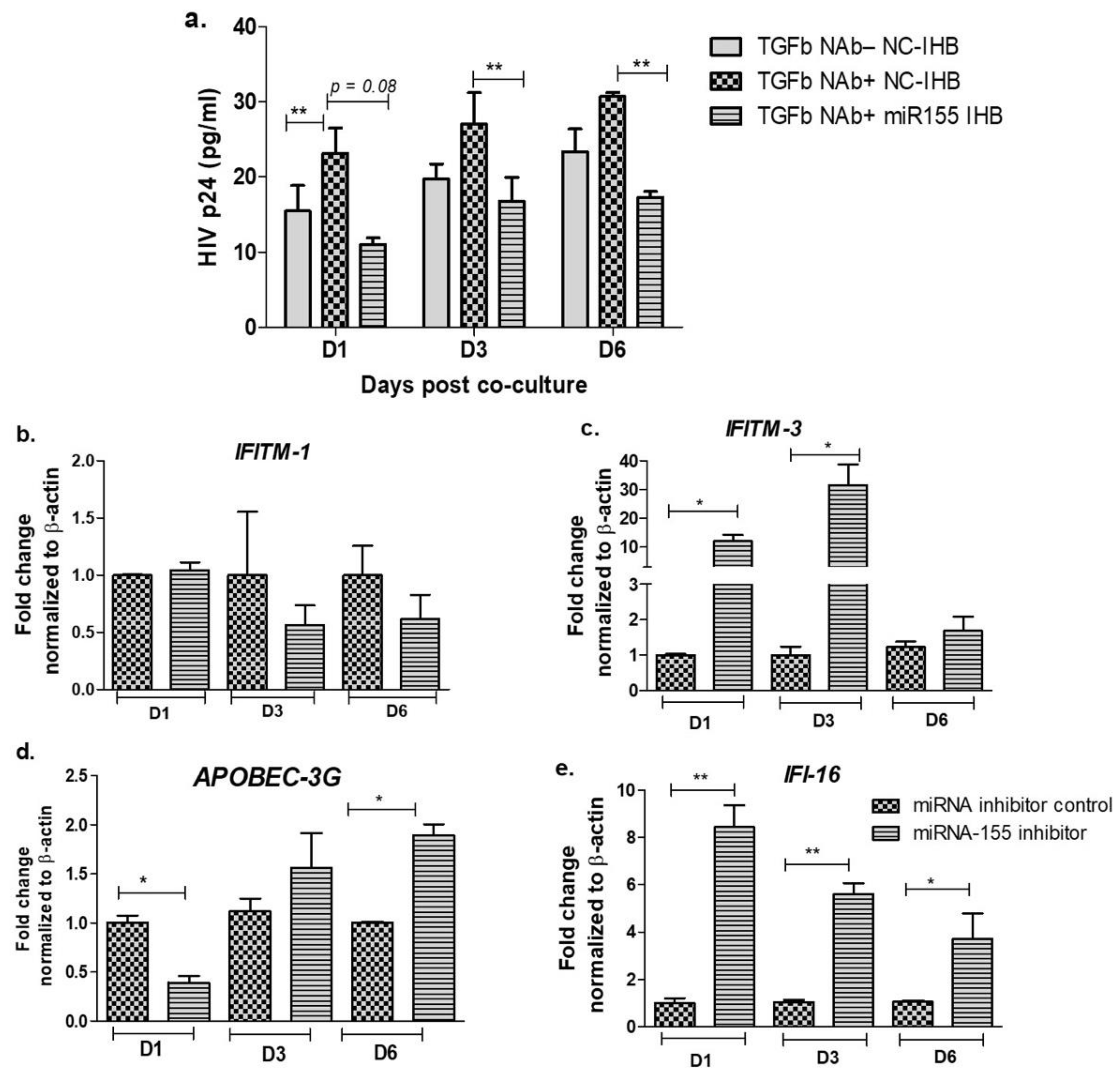
3.3. Inhibition of miR-155 Increases the Expression of Host Restriction Factors and Suppresses HIV Release in the Cervical Epithelial Cells
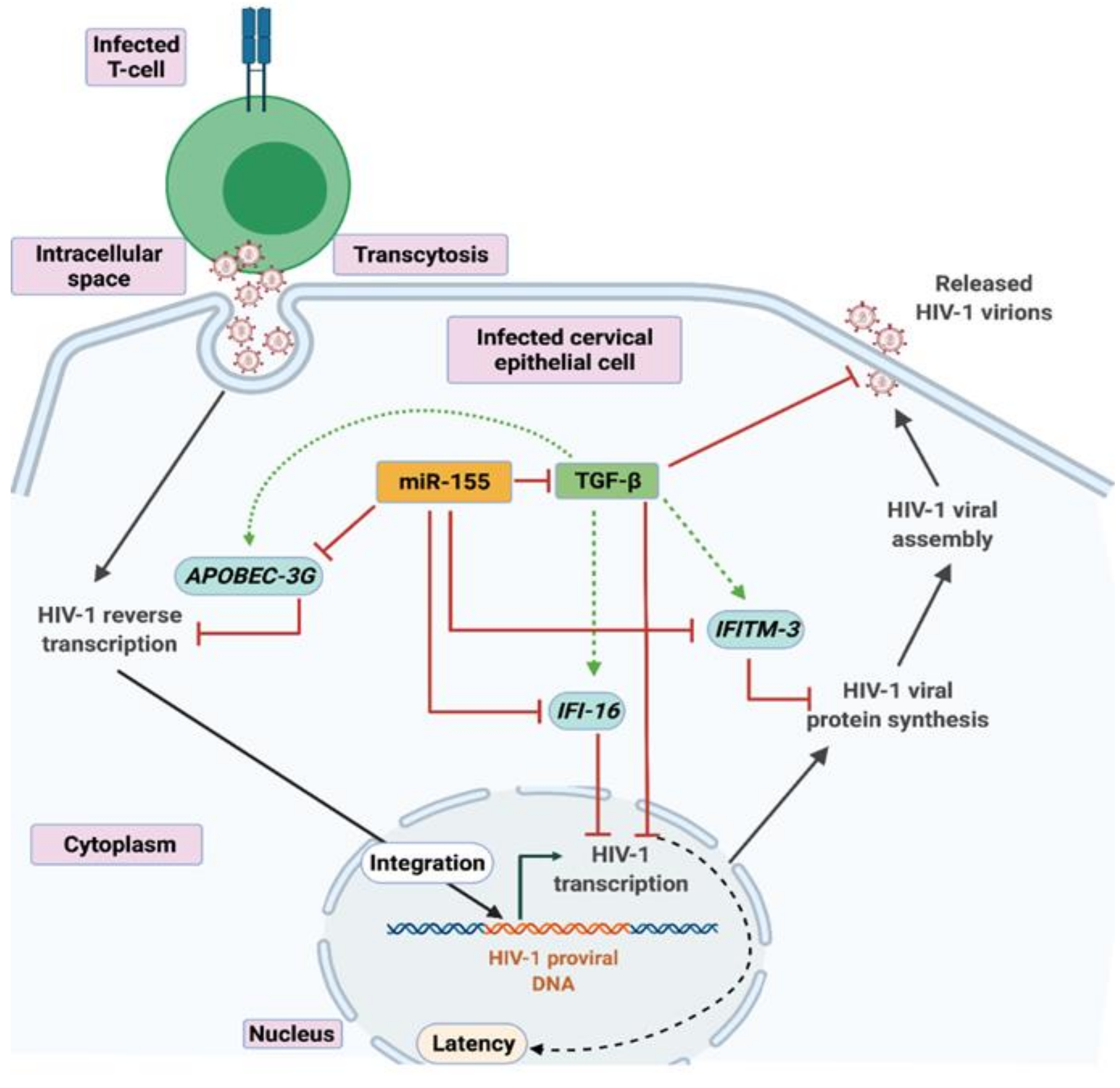
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poli, G.; Kinter, A.L.; Justement, J.S.; Bressler, P.; Kehrl, J.H.; Fauci, A.S. Transforming Growth Factor Beta Suppresses Human Immunodeficiency Virus Expression and Replication in Infected Cells of the Monocyte/Macrophage Lineage. J. Exp. Med. 1991, 173, 589–597. [Google Scholar] [CrossRef]
- Prud’homme, G.J. Pathobiology of Transforming Growth Factor b in Cancer, Fibrosis and Immunologic Disease, and Therapeutic Considerations. Lab. Invest. 2007, 87, 15. [Google Scholar] [CrossRef]
- Theron, A.J.; Anderson, R.; Rossouw, T.M.; Steel, H.C. The Role of Transforming Growth Factor Beta-1 in the Progression of HIV/AIDS and Development of Non-AIDS-Defining Fibrotic Disorders. Front. Immunol. 2017, 8, 1461. [Google Scholar] [CrossRef]
- Kekow, J.; Wachsman, W.; McCutchan, J.A.; Cronin, M.; Carson, D.A.; Lotz, M. Transforming Growth Factor Beta and Noncytopathic Mechanisms of Immunodeficiency in Human Immunodeficiency Virus Infection. Proc. Natl. Acad. Sci. USA 1990, 87, 8321–8325. [Google Scholar] [CrossRef]
- Maina, E.K.; Abana, C.Z.; Bukusi, E.A.; Sedegah, M.; Lartey, M.; Ampofo, W.K. Plasma Concentrations of Transforming Growth Factor Beta 1 in Non-Progressive HIV-1 Infection Correlates with Markers of Disease Progression. Cytokine 2016, 81, 109–116. [Google Scholar] [CrossRef]
- Hu, R.; Oyaizu, N.; Than, S.; Kalyanaraman, V.S.; Wang, X.P.; Pahwa, S. HIV-1 Gp160 Induces Transforming Growth Factor-Beta Production in Human PBMC. Clin. Immunol. Immunopathol. 1996, 80, 283–289. [Google Scholar] [CrossRef]
- Patel, P.; Khan, N.; Rani, M.; Gupta, D.; Jameel, S. The Expression of HIV-1 Vpu in Monocytes Causes Increased Secretion of TGF-β That Activates Profibrogenic Genes in Hepatic Stellate Cells. PLoS ONE 2014, 9, e88934. [Google Scholar] [CrossRef]
- Poggi, A.; Zocchi, M.R. HIV-1 Tat Triggers TGF-β Production and NK Cell Apoptosis That Is Prevented by Pertussis Toxin B. Clin. Dev. Immunol. 2006, 13, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Moriuchi, M.; Moriuchi, H. Cell-Type-Dependent Effect of Transforming Growth Factor β, a Major Cytokine in Breast Milk, on Human Immunodeficiency Virus Type 1 Infection of Mammary Epithelial MCF-7 Cells or Macrophages. J. Virol. 2004, 78, 13046–13052. [Google Scholar] [CrossRef] [PubMed]
- Chinnapaiyan, S.; Dutta, R.K.; Nair, M.; Chand, H.S.; Rahman, I.; Unwalla, H.J. TGF-Β1 Increases Viral Burden and Promotes HIV-1 Latency in Primary Differentiated Human Bronchial Epithelial Cells. Sci. Rep. 2019, 9, 12552. [Google Scholar] [CrossRef] [PubMed]
- Lazdins, J.K.; Klimkait, T.; Woods-Cook, K.; Walker, M.; Alteri, E.; Cox, D.; Cerletti, N.; Shipman, R.; Bilbe, G.; McMaster, G. In Vitro Effect of Transforming Growth Factor-Beta on Progression of HIV-1 Infection in Primary Mononuclear Phagocytes. J. Immunol. 1991, 147, 1201–1207. [Google Scholar] [PubMed]
- Cheung, K.-W.; Wu, T.; Ho, S.F.; Wong, Y.C.; Liu, L.; Wang, H.; Chen, Z. A4β7+ CD4+ Effector/Effector Memory T Cells Differentiate into Productively and Latently Infected Central Memory T Cells by Transforming Growth Factor Β1 during HIV-1 Infection. J. Virol. 2018, 92, e01510-17. [Google Scholar] [CrossRef]
- Iqbal, S.M.; Kaul, R. Review Article: Mucosal Innate Immunity as a Determinant of HIV Susceptibility: HIV and Mucosal innate immunity. Am. J. Reprod. Immunol. 2007, 59, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Liang, Y.; Hur, H.; Villegas, G.; Calenda, G.; Reis, A.; Millen, L.; Barnable, P.; Mamkina, L.; Kumar, N.; et al. Comparative Transcriptome Analysis of the Human Endocervix and Ectocervix during the Proliferative and Secretory Phases of the Menstrual Cycle. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Reis Machado, J.; da Silva, M.V.; Cavellani, C.L.; Antônia dos Reis, M.; dos Monteiro, M.L.G.R.; de Teixeira, V.P.A.; Rosa Miranda Corrêa, R. Mucosal Immunity in the Female Genital Tract, HIV/AIDS. BioMed Res. Int. 2014, 2014, 350195. [Google Scholar] [CrossRef]
- Shattock, R.J.; Moore, J.P. Inhibiting Sexual Transmission of HIV-1 Infection. Nat. Rev. Microbiol. 2003, 1, 25–34. [Google Scholar] [CrossRef]
- Moreno de Lara, L.; Parthasarathy, R.S.; Rodriguez-Garcia, M. Mucosal Immunity and HIV Acquisition in Women. Curr. Opin. Physiol. 2021, 19, 32–38. [Google Scholar] [CrossRef]
- Fulcher, J.A.; Romas, L.; Hoffman, J.C.; Elliott, J.; Saunders, T.; Burgener, A.D.; Anton, P.A.; Yang, O.O. Highly Human Immunodeficiency Virus-Exposed Seronegative Men Have Lower Mucosal Innate Immune Reactivity. AIDS Res. Hum. Retrovir. 2017, 33, 788–795. [Google Scholar] [CrossRef]
- Heron, S.E.; Elahi, S. HIV Infection and Compromised Mucosal Immunity: Oral Manifestations and Systemic Inflammation. Front. Immunol. 2017, 8, 241. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Noble, N.A.; Miller, D.E.; Gold, L.I.; Hishida, A.; Nagase, M.; Cohen, A.H.; Border, W.A. Increased Levels of Transforming Growth Factor-β in HIV-Associated Nephropathy. Kidney Int. 1999, 55, 579–592. [Google Scholar] [CrossRef]
- Francis, S.C.; Hou, Y.; Baisley, K.; van de Wijgert, J.; Watson-Jones, D.; Ao, T.T.; Herrera, C.; Maganja, K.; Andreasen, A.; Kapiga, S.; et al. Immune Activation in the Female Genital Tract: Expression Profiles of Soluble Proteins in Women at High Risk for HIV Infection. PLoS ONE 2016, 11, e0143109. [Google Scholar] [CrossRef]
- Gumbi, P.P.; Nkwanyana, N.N.; Bere, A.; Burgers, W.A.; Gray, C.M.; Williamson, A.-L.; Hoffman, M.; Coetzee, D.; Denny, L.; Passmore, J.-A.S. Impact of Mucosal Inflammation on Cervical Human Immunodeficiency Virus (HIV-1)-Specific CD8 T-Cell Responses in the Female Genital Tract during Chronic HIV Infection. J. Virol. 2008, 82, 8529–8536. [Google Scholar] [CrossRef] [PubMed]
- Saxena, V.; Ghate, M.; Bichare, S.; Khan, I.; Majumdar, R.; Angadi, M.; Kulkarni, S.; Paranjape, R.; Thakar, M.R. Increased Degranulation of Immune Cells Is Associated with Higher Cervical Viral Load in HIV-Infected Women. AIDS 2018, 32, 1939–1949. [Google Scholar] [CrossRef]
- Thakar, M.; Patil, R.; Shukre, S.; Bichare, S.; Kadam, P.; Khopkar, P.; Ghate, M.; Paranjape, R. Short Communication: Genital Tumor Growth Factor-Β1 Levels in HIV-Infected Indian Women Are Associated with Reduced Levels of Innate Antimicrobial Products and Increased HIV Shedding. AIDS Res. Hum. Retrovir. 2014, 30, 648–653. [Google Scholar] [CrossRef]
- Thibodeau, V.; Fourcade, L.; Labbé, A.-C.; Alary, M.; Guédou, F.; Poudrier, J.; Roger, M. Highly-Exposed HIV-1 Seronegative Female Commercial Sex Workers Sustain in Their Genital Mucosa Increased Frequencies of Tolerogenic Myeloid and Regulatory T-Cells. Sci. Rep. 2017, 7, 43857. [Google Scholar] [CrossRef]
- George, J.; Lewis, M.G.; Renne, R.; Mattapallil, J.J. Suppression of Transforming Growth Factor β Receptor 2 and Smad5 Is Associated with High Levels of MicroRNA MiR-155 in the Oral Mucosa during Chronic Simian Immunodeficiency Virus Infection. J. Virol. 2015, 89, 2972–2978. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Broering, R.; Trippler, M.; Wu, J.; Zhang, E.; Zhang, X.; Gerken, G.; Lu, M.; Schlaak, J.F. MicroRNA-155 Controls Toll-like Receptor 3- and Hepatitis C Virus-Induced Immune Responses in the Liver. J. Viral Hepat. 2014, 21, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Yang, H.; He, L.; Zhao, J.; Coppola, D.; Dalton, W.S.; Cheng, J.Q. MicroRNA-155 Is Regulated by the Transforming Growth Factor β/Smad Pathway and Contributes to Epithelial Cell Plasticity by Targeting RhoA. Mol. Cell. Biol. 2008, 28, 6773–6784. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chi, X.; Li, R.; Ouyang, J.; Chen, Y. HIV-1-Infected Cell-Derived Exosomes Promote the Growth and Progression of Cervical Cancer. Int. J. Biol. Sci. 2019, 15, 2438–2447. [Google Scholar] [CrossRef] [PubMed]
- Louafi, F.; Martinez-Nunez, R.T.; Sanchez-Elsner, T. MicroRNA-155 Targets SMAD2 and Modulates the Response of Macrophages to Transforming Growth Factor-Β*. J. Biol. Chem. 2010, 285, 41328–41336. [Google Scholar] [CrossRef]
- Rai, D.; Kim, S.-W.; McKeller, M.R.; Aguiar, R.C. Targeting of SMAD5 Links MicroRNA-155 to the TGFβ Pathway and Lymphomagenesis. Blood 2009, 114, 315. [Google Scholar] [CrossRef]
- Kutsch, O.; Levy, D.N.; Bates, P.J.; Decker, J.; Kosloff, B.R.; Shaw, G.M.; Priebe, W.; Benveniste, E.N. Bis-Anthracycline Antibiotics Inhibit Human Immunodeficiency Virus Type 1 Transcription. Antimicrob. Agents Chemother. 2004, 48, 1652–1663. [Google Scholar] [CrossRef] [PubMed]
- Ochsenbauer-Jambor, C.; Jones, J.; Heil, M.; Zammit, K.P.; Kutsch, O. T-Cell Line for HIV Drug Screening Using EGFP as a Quantitative Marker of HIV-1 Replication. BioTechniques 2006, 40, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Adachi, A.; Gendelman, H.E.; Koenig, S.; Folks, T.; Willey, R.; Rabson, A.; Martin, M.A. Production of Acquired Immunodeficiency Syndrome-Associated Retrovirus in Human and Nonhuman Cells Transfected with an Infectious Molecular Clone. J. Virol. 1986, 59, 284. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. MiRBase: MicroRNA Sequences, Targets and Gene Nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef]
- Krüger, J.; Rehmsmeier, M. RNAhybrid: MicroRNA Target Prediction Easy, Fast and Flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef]
- Rehmsmeier, M.; Steffen, P.; Höchsmann, M.; Giegerich, R. Fast and Effective Prediction of MicroRNA/Target Duplexes. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.M.; Tan, X.; Pearce-Pratt, R.; Zacharopoulos, V.R. An Assay for HIV Infection of Cultured Human Cervix-Derived Cells. J. Virol. Methods 1995, 52, 1–13. [Google Scholar] [CrossRef]
- Hata, A.; Chen, Y.-G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-β—An Excellent Servant but a Bad Master. J. Transl. Med. 2012, 10, 183. [Google Scholar] [CrossRef]
- Nakao, A.; Imamura, T.; Souchelnytskyi, S.; Kawabata, M.; Ishisaki, A.; Oeda, E.; Tamaki, K.; Hanai, J.; Heldin, C.H.; Miyazono, K.; et al. TGF-Beta Receptor-Mediated Signalling through Smad2, Smad3 and Smad4. EMBO J. 1997, 16, 5353–5362. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-Y.J.; Fu, R.M.; Liang, C.; Sloan, R.D. IFITM Proteins Inhibit HIV-1 Protein Synthesis. Sci. Rep. 2018, 8, 14551. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.F.; Koyama, T.; Kinomoto, M.; Tokunaga, K. Retroelements versus APOBEC3 Family Members: No Great Escape from the Magnificent Seven. Front. Microbiol. 2012, 3, 275. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Dudley, J.P. APOBECs and Virus Restriction. Virology 2015, 479–480, 131–145. [Google Scholar] [CrossRef]
- Schröfelbauer, B.; Chen, D.; Landau, N.R. A Single Amino Acid of APOBEC3G Controls Its Species-Specific Interaction with Virion Infectivity Factor (Vif). Proc. Natl. Acad. Sci. USA 2004, 101, 3927–3932. [Google Scholar] [CrossRef] [PubMed]
- Hotter, D.; Bosso, M.; Jønsson, K.L.; Krapp, C.; Stürzel, C.M.; Das, A.; Littwitz-Salomon, E.; Berkhout, B.; Russ, A.; Wittmann, S.; et al. IFI16 Targets the Transcription Factor Sp1 to Suppress HIV-1 Transcription and Latency Reactivation. Cell Host Microbe 2019, 25, 858–872.e13. [Google Scholar] [CrossRef]
- Nchioua, R.; Bosso, M.; Kmiec, D.; Kirchhoff, F. Cellular Factors Targeting HIV-1 Transcription and Viral RNA Transcripts. Viruses 2020, 12, 495. [Google Scholar] [CrossRef]
- Wong, J.K.; Yukl, S.A. Tissue Reservoirs of HIV. Curr. Opin. HIV AIDS 2016, 11, 362–370. [Google Scholar] [CrossRef]
- Miller, C.J.; Shattock, R.J. Target Cells in Vaginal HIV Transmission. Microbes Infect. 2003, 5, 59–67. [Google Scholar] [CrossRef]
- Bobardt, M.D.; Chatterji, U.; Selvarajah, S.; Van der Schueren, B.; David, G.; Kahn, B.; Gallay, P.A. Cell-Free Human Immunodeficiency Virus Type 1 Transcytosis through Primary Genital Epithelial Cells. J. Virol. 2007, 81, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Micsenyi, A.M.; Zony, C.; Alvarez, R.A.; Durham, N.D.; Chen, B.K.; Klotman, M.E. Postintegration HIV-1 Infection of Cervical Epithelial Cells Mediates Contact-Dependent Productive Infection of T Cells. J. Infect. Dis. 2013, 208, 1756–1767. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tan, X.; Pearce-Pratt, R.; Phillips, D.M. Productive Infection of a Cervical Epithelial Cell Line with Human Immunodeficiency Virus: Implications for Sexual Transmission. J. Virol. 1993, 67, 6447–6452. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Chen, Z.; Phillips, D.M. Human Genital Epithelial Cells Capture Cell-Free Human Immunodeficiency Virus Type 1 and Transmit the Virus to CD4+ Cells: Implications for Mechanisms of Sexual Transmission. J. Infect. Dis. 2003, 188, 1473–1482. [Google Scholar] [CrossRef]
- Lai, W.; Huang, L.; Zhu, L.; Ferrari, G.; Chan, C.; Li, W.; Lee, K.-H.; Chen, C.-H. Gnidimacrin, a Potent Anti-HIV Diterpene, Can Eliminate Latent HIV-1 Ex Vivo by Activation of Protein Kinase C β. J. Med. Chem. 2015, 58, 8638–8646. [Google Scholar] [CrossRef]
- Palermo, E.; Acchioni, C.; Di Carlo, D.; Zevini, A.; Muscolini, M.; Ferrari, M.; Castiello, L.; Virtuoso, S.; Borsetti, A.; Antonelli, G.; et al. Activation of Latent HIV-1 T Cell Reservoirs with a Combination of Innate Immune and Epigenetic Regulators. J. Virol. 2019, 93, e01194-19. [Google Scholar] [CrossRef]
- Wei, D.G.; Chiang, V.; Fyne, E.; Balakrishnan, M.; Barnes, T.; Graupe, M.; Hesselgesser, J.; Irrinki, A.; Murry, J.P.; Stepan, G.; et al. Histone Deacetylase Inhibitor Romidepsin Induces HIV Expression in CD4 T Cells from Patients on Suppressive Antiretroviral Therapy at Concentrations Achieved by Clinical Dosing. PLoS Pathog. 2014, 10, e1004071. [Google Scholar] [CrossRef]
- Ait-Ammar, A.; Kula, A.; Darcis, G.; Verdikt, R.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Rohr, O.; Van Lint, C. Current Status of Latency Reversing Agents Facing the Heterogeneity of HIV-1 Cellular and Tissue Reservoirs. Front. Microbiol. 2020, 10, 3060. [Google Scholar] [CrossRef]
- Cantero-Pérez, J.; Grau-Expósito, J.; Serra-Peinado, C.; Rosero, D.A.; Luque-Ballesteros, L.; Astorga-Gamaza, A.; Castellví, J.; Sanhueza, T.; Tapia, G.; Lloveras, B.; et al. Resident Memory T Cells Are a Cellular Reservoir for HIV in the Cervical Mucosa. Nat. Commun. 2019, 10, 4739. [Google Scholar] [CrossRef]
- Iversen, A.K.N.; Attermann, J.; Gerstoft, J.; Fugger, L.; Mullins, J.I.; Skinhøj, P. Longitudinal and Cross-Sectional Studies of HIV-1 RNA and DNA Loads in Blood and the Female Genital Tract. Eur. J. Obstet. Gynecol. Reprod. Biol. 2004, 117, 227–235. [Google Scholar] [CrossRef]
- Billeter, A.T.; Hellmann, J.; Roberts, H.; Druen, D.; Gardner, S.A.; Sarojini, H.; Galandiuk, S.; Chien, S.; Bhatnagar, A.; Spite, M.; et al. MicroRNA-155 Potentiates the Inflammatory Response in Hypothermia by Suppressing IL-10 Production. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 5322–5336. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Aguiar, R.C.T. MicroRNA-155 Controls RB Phosphorylation in Normal and Malignant B Lymphocytes via the Noncanonical TGF-Β1/SMAD5 Signaling Module. Blood 2014, 123, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Vigorito, E.; Clare, S.; Warren, M.V.; Couttet, P.; Soond, D.R.; van Dongen, S.; Grocock, R.J.; Das, P.P.; Miska, E.A.; et al. Requirement of Bic/MicroRNA-155 for Normal Immune Function. Science 2007, 316, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; McBride, J.; Fewell, C.; Lacey, M.; Wang, X.; Lin, Z.; Cameron, J.; Flemington, E.K. MicroRNA-155 Is an Epstein-Barr Virus-Induced Gene That Modulates Epstein-Barr Virus-Regulated Gene Expression Pathways. J. Virol. 2008, 82, 5295–5306. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular Mechanisms of Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Taura, M.; Song, E.; Ho, Y.-C.; Iwasaki, A. Apobec3A Maintains HIV-1 Latency through Recruitment of Epigenetic Silencing Machinery to the Long Terminal Repeat. Proc. Natl. Acad. Sci. USA 2019, 116, 2282–2289. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.L.; Wilson, H.; Iyer, S.S.; Coss, K.; Doores, K.; Smith, S.; Kellam, P.; Finzi, A.; Borrow, P.; Hahn, B.H.; et al. Resistance of Transmitted Founder HIV-1 to IFITM-Mediated Restriction. Cell Host Microbe 2016, 20, 429–442. [Google Scholar] [CrossRef]
- Yu, J.; Li, M.; Wilkins, J.; Ding, S.; Swartz, T.H.; Esposito, A.M.; Zheng, Y.-M.; Freed, E.O.; Liang, C.; Chen, B.K.; et al. IFITM Proteins Restrict HIV-1 Infection by Antagonizing the Envelope Glycoprotein. Cell Rep. 2015, 13, 145–156. [Google Scholar] [CrossRef]
- Farberov, L.; Herzig, E.; Modai, S.; Isakov, O.; Hizi, A.; Shomron, N. MicroRNA-Mediated Regulation of P21 and TASK1 Cellular Restriction Factors Enhances HIV-1 Infection. J. Cell Sci. 2015, 128, 1607–1616. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Position | Minimum Free Energy (mfe) (kcal/mol) | Binding Region | Most Optimal miRNA-mRNA Duplex Hybrid Structures |
|---|---|---|---|---|
| APOBEC-3G | 6–30 | −19.1 |  |  |
| IFI-16 | 122–144 | −19.2 |  |  |
| IFITM-3 | 25–47 | −17.9 |  |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gokavi, J.; Sadawarte, S.; Shelke, A.; Kulkarni-Kale, U.; Thakar, M.; Saxena, V. Inhibition of miR-155 Promotes TGF-β Mediated Suppression of HIV Release in the Cervical Epithelial Cells. Viruses 2021, 13, 2266. https://doi.org/10.3390/v13112266
Gokavi J, Sadawarte S, Shelke A, Kulkarni-Kale U, Thakar M, Saxena V. Inhibition of miR-155 Promotes TGF-β Mediated Suppression of HIV Release in the Cervical Epithelial Cells. Viruses. 2021; 13(11):2266. https://doi.org/10.3390/v13112266
Chicago/Turabian StyleGokavi, Jyotsna, Sharwari Sadawarte, Anant Shelke, Urmila Kulkarni-Kale, Madhuri Thakar, and Vandana Saxena. 2021. "Inhibition of miR-155 Promotes TGF-β Mediated Suppression of HIV Release in the Cervical Epithelial Cells" Viruses 13, no. 11: 2266. https://doi.org/10.3390/v13112266
APA StyleGokavi, J., Sadawarte, S., Shelke, A., Kulkarni-Kale, U., Thakar, M., & Saxena, V. (2021). Inhibition of miR-155 Promotes TGF-β Mediated Suppression of HIV Release in the Cervical Epithelial Cells. Viruses, 13(11), 2266. https://doi.org/10.3390/v13112266