
In Vivo Modelling of Hepatitis B Virus Subgenotype A1 Replication Using Adeno-Associated Viral Vectors

 , , and
, , and 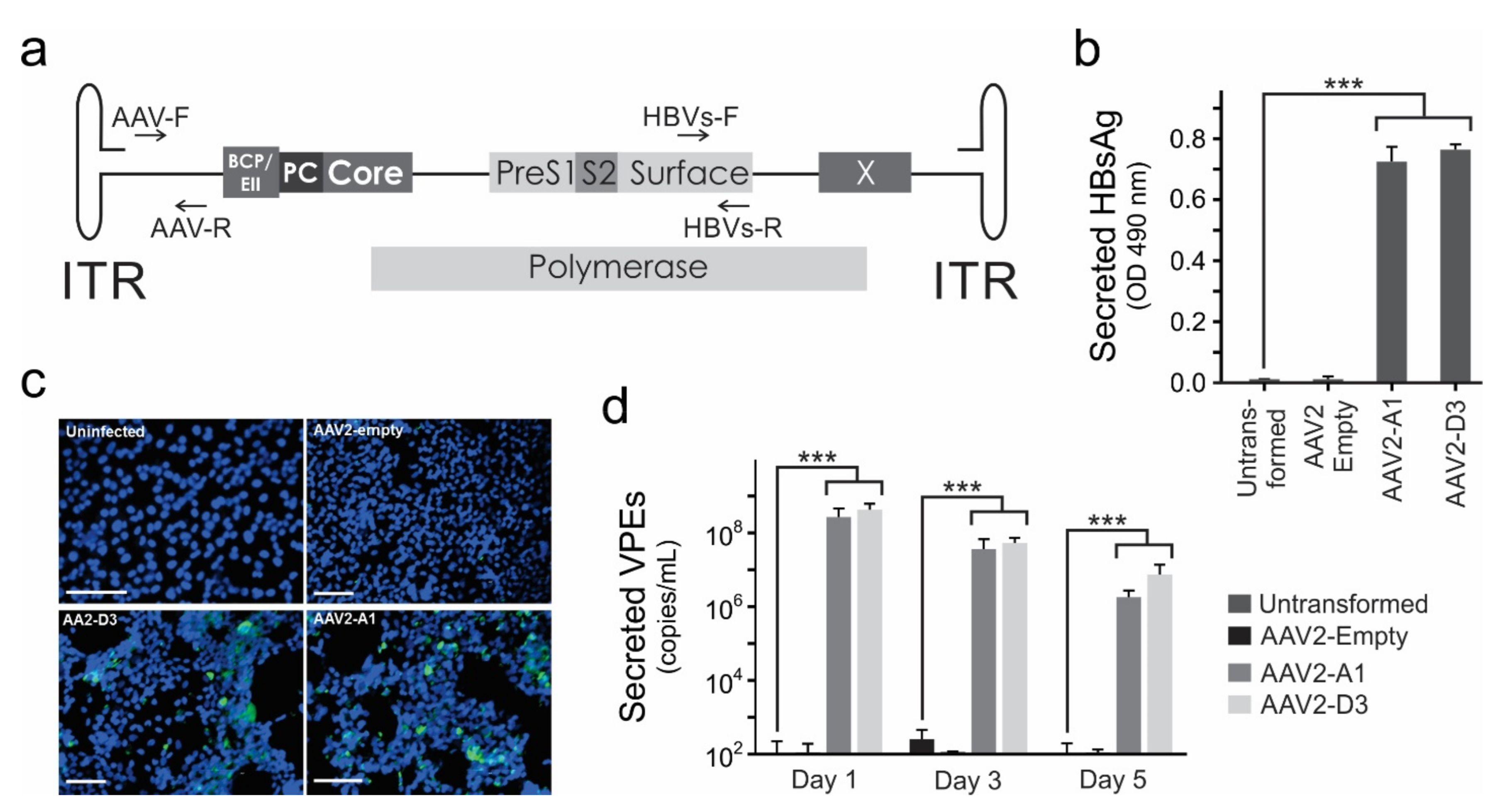{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Construction and Characterisation of AAV Plasmids Containing Greater-Than-Genome-Length HBV Sequences
2.2. Production of AAV Vectors
2.3. Assessment of HBV Gene Expression and Production of HBV Particles following Transduction of Cultured Cells with Recombinant AAV-HBV Vectors
2.4. Treatment of Mice with AAVs and Assessment of HBV Replication In Vivo
2.5. Evaluation of AAV-Induced Innate Immune Stimulation in Mice
2.6. Measurement of HBV Surface Antigen, HBV Core Antigen and HBV e Antigen Expression in Mice
2.7. Detection of HBV Particle Equivalents, DNA Intermediates and RNA in Mice
2.8. Assessment of Hepatotoxicity, Inflammatory Cell Infiltration and Fibrosis in Mice
2.9. Assessment of Artificial Primary Micro-RNAs Efficacy against
HBV in AAV-HBV Model
3. Results
3.1. AAV2-HBV Vectors Mediate HBV Gene Expression and Replication In Vitro
3.2. Administration of Recombinant AAV8-HBV Vectors to Mice Does Not Result in an Immunostimulatory Response
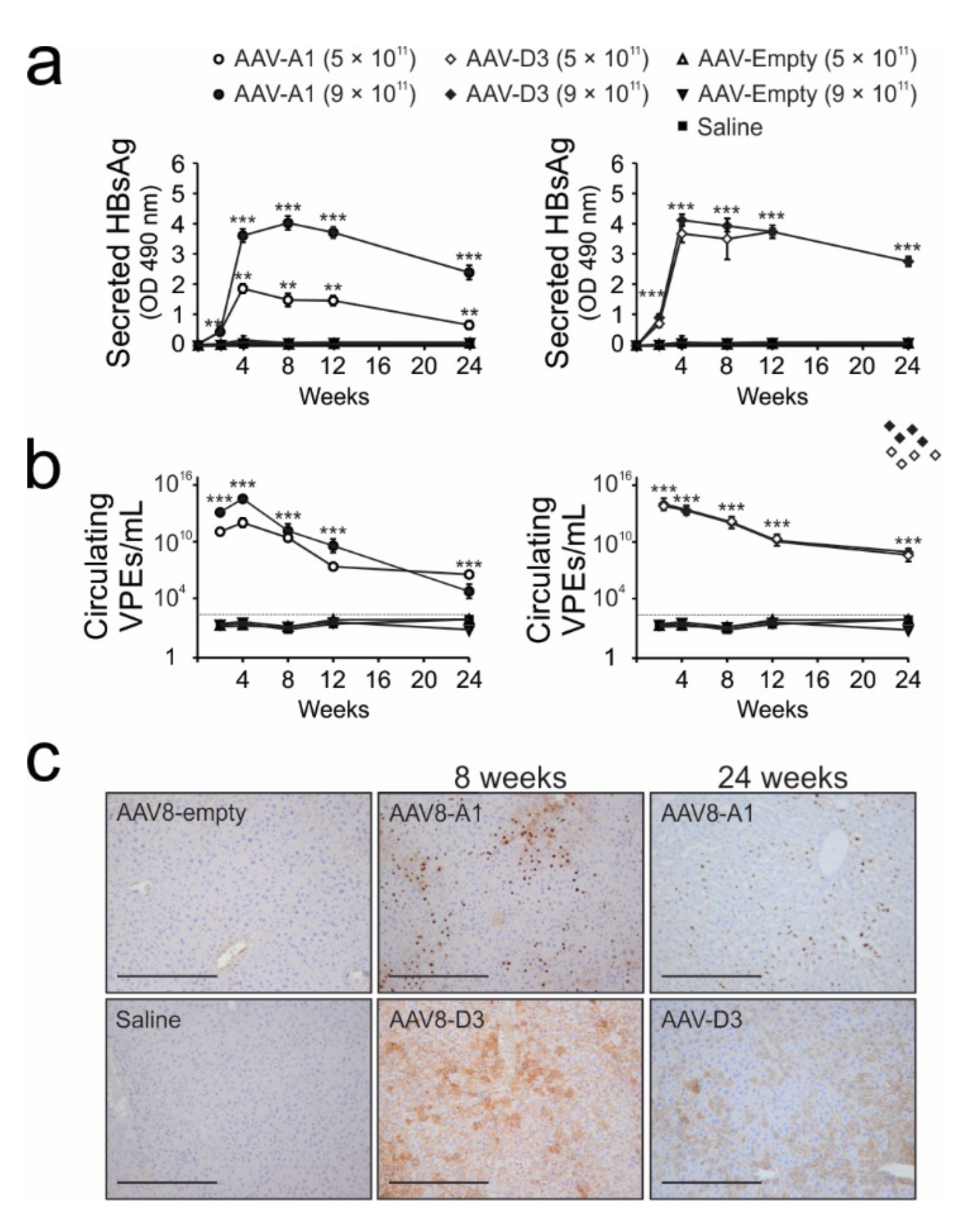
3.3. Transduction of Mice with AAV8-HBV Vectors Results in HBV Gene Expression and Replication
3.4. Injection of Mice with AAV8 Vectors Bearing HBV Sequences Does Not Cause Hepatotoxicity or Fibrosis
3.5. Successful Use of AAV8-HBV Model to Asess the Efficacy of Anti-HBV Pri-miR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Asim, M.; Malik, A.; Sarma, M.P.; Polipalli, S.K.; Begum, N.; Ahmad, I.; Khan, L.A.; Husain, S.; Akhtar, N.; Husain, S. Hepatitis B virus BCP, Precore/core, X gene mutations/genotypes and the risk of hepatocellular carcinoma in India. J. Med. Virol. 2010, 82, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Valaydon, Z.S.; Locarnini, S.A. The virological aspects of hepatitis B. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 257–264. [Google Scholar] [CrossRef]
- Revill, P.; Testoni, B.; Locarnini, S.; Zoulim, F. Global strategies are required to cure and eliminate HBV infection. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 239–248. [Google Scholar] [CrossRef]
- WHO. Interim Guidance for Country Validation of Viral Hepatitis Elimination; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Kramvis, A. Genotypes and genetic variability of hepatitis B virus. Intervirology 2014, 57, 141–150. [Google Scholar] [CrossRef]
- Spearman, C.; Sonderup, M.W. Preventing hepatitis B and hepatocellular carcinoma in South Africa: The case for a birth-dose vaccine. SAMJ S. Afr. Med. J. 2014, 104, 610–612. [Google Scholar] [CrossRef] [PubMed]
- Kimbi, G.C.; Kramvis, A.; Kew, M.C. Distinctive sequence characteristics of subgenotype A1 isolates of hepatitis B virus from South Africa. J. Gen. Virol. 2004, 85 Pt 5, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Kew, M.C.; Kramvis, A.; Yu, M.C.; Arakawa, K.; Hodkinson, J. Increased hepatocarcinogenic potential of hepatitis B virus genotype A in Bantu-speaking sub-saharan Africans. J. Med. Virol. 2005, 75, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Kramvis, A.; Paraskevis, D. Subgenotype A1 of HBV—Tracing human migrations in and out of Africa. Antivir. Ther. 2013, 18 Pt B, 513–521. [Google Scholar] [CrossRef]
- Kramvis, A.; Kew, M.; Francois, G. Hepatitis B virus genotypes. Vaccine 2005, 23, 2409–2423. [Google Scholar] [CrossRef]
- Lampertico, P.; Liaw, Y.F. New perspectives in the therapy of chronic hepatitis B. Gut 2012, 61 (Suppl. 1), i18–i24. [Google Scholar] [CrossRef]
- Scaglione, S.J.; Lok, A.S. Effectiveness of hepatitis B treatment in clinical practice. Gastroenterology 2012, 142, 1360–1368.e1. [Google Scholar] [CrossRef] [PubMed]
- Colagrossi, L.; Hermans, L.E.; Salpini, R.; Di Carlo, D.; Pas, S.D.; Alvarez, M.; Ben-Ari, Z.; Boland, G.; Bruzzone, B.; Coppola, N. Immune-escape mutations and stop-codons in HBsAg develop in a large proportion of patients with chronic HBV infection exposed to anti-HBV drugs in Europe. BMC Infect. Dis. 2018, 18, 251. [Google Scholar] [CrossRef]
- Maepa, M.B.; Roelofse, I.; Ely, A.; Arbuthnot, P. Progress and prospects of anti-HBV gene therapy development. Int. J. Mol. Sci. 2015, 16, 17589–17610. [Google Scholar] [CrossRef]
- Bloom, K.; Maepa, M.B.; Ely, A.; Arbuthnot, P. Gene Therapy for Chronic HBV—Can We Eliminate cccDNA? Genes 2018, 9, 207. [Google Scholar] [CrossRef] [PubMed]
- Revill, P.A.; Chisari, F.V.; Block, J.M.; Dandri, M.; Gehring, A.J.; Guo, H.; Hu, J.; Kramvis, A.; Lampertico, P.; Janssen, H.L.A.; et al. A global scientific strategy to cure hepatitis B. Lancet Gastroenterol. Hepatol. 2019, 4, 545–558. [Google Scholar] [CrossRef]
- Burwitz, B.J.; Wettengel, J.M.; Muck-Hausl, M.A.; Ringelhan, M.; Ko, C.; Festag, M.M.; Hammond, K.B.; Northrup, M.; Bimber, B.N.; Jacob, T.; et al. Hepatocytic expression of human sodium-taurocholate cotransporting polypeptide enables hepatitis B virus infection of macaques. Nat. Commun. 2017, 8, 2146. [Google Scholar] [CrossRef]
- Inuzuka, T.; Takahashi, K.; Chiba, T.; Marusawa, H. Mouse models of hepatitis B virus infection comprising host-virus immunologic interactions. Pathogens 2014, 3, 377–389. [Google Scholar] [CrossRef] [PubMed]
- von Freyend, M.J.; Untergasser, A.; Arzberger, S.; Oberwinkler, H.; Drebber, U.; Schirmacher, P.; Protzer, U. Sequential control of hepatitis B virus in a mouse model of acute, self-resolving hepatitis B. J. Viral Hepat. 2011, 18, 216–226. [Google Scholar] [CrossRef]
- Sprinzl, M.F.; Oberwinkler, H.; Schaller, H.; Protzer, U. Transfer of hepatitis B virus genome by adenovirus vectors into cultured cells and mice: Crossing the species barrier. J. Virol. 2001, 75, 5108–5118. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.R.; Gabel, Y.A.; Graf, S.; Arzberger, S.; Kurts, C.; Heikenwalder, M.; Knolle, P.A.; Protzer, U. Transfer of HBV genomes using low doses of adenovirus vectors leads to persistent infection in immune competent mice. Gastroenterology 2012, 142, 1447–1450. [Google Scholar] [CrossRef]
- Huang, Y.H.; Fang, C.C.; Tsuneyama, K.; Chou, H.Y.; Pan, W.Y.; Shih, Y.M.; Wu, P.Y.; Chen, Y.; Leung, P.S.; Gershwin, M.E.; et al. A murine model of hepatitis B-associated hepatocellular carcinoma generated by adeno-associated virus-mediated gene delivery. Int. J. Oncol. 2011, 39, 1511–1519. [Google Scholar]
- Yang, D.; Liu, L.; Zhu, D.; Peng, H.; Su, L.; Fu, Y.X.; Zhang, L. A mouse model for HBV immunotolerance and immunotherapy. Cell. Mol. Immunol. 2014, 11, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Yu, H.; Li, C.; Hirsch, M.L.; Zhang, L.; Samulski, R.J.; Li, W.; Liu, Z. Adeno-associated virus vector mediated delivery of the HBV genome induces chronic hepatitis B virus infection and liver fibrosis in mice. PLoS ONE 2015, 10, e0130052. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Cheng, T.; Cai, Y.J.; Yuan, Q.; Liu, C.; Zhang, T.; Xia, D.Z.; Li, R.Y.; Yang, L.W.; Wang, Y.B. RNA Interference inhibits hepatitis B virus of different genotypes in vitro and in vivo. BMC Microbiol. 2010, 10, 214. [Google Scholar] [CrossRef] [PubMed]
- Bhoola, N.H.; Reumann, K.; Kew, M.C.; Will, H.; Kramvis, A. Construction of replication competent plasmids of hepatitis B virus subgenotypes A1, A2 and D3 with authentic endogenous promoters. J. Virol. Methods 2014, 203, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J. Virol. 1992, 66, 4107–4116. [Google Scholar] [CrossRef]
- Maepa, M.B.; Ely, A.; Grayson, W.; Arbuthnot, P. Sustained Inhibition of HBV Replication In Vivo after Systemic Injection of AAVs Encoding Artificial Antiviral Primary MicroRNAs. Mol. Ther. Nucleic Acids 2017, 7, 190–199. [Google Scholar] [CrossRef]
- Bourhill, T.; Arbuthnot, P.; Ely, A. Successful disabling of the 5′ UTR of HCV using adeno-associated viral vectors to deliver modular multimeric primary microRNA mimics. J. Virol. Methods 2016, 235, 26–33. [Google Scholar] [CrossRef]
- Grimm, D.; Streetz, K.L.; Jopling, C.L.; Storm, T.A.; Pandey, K.; Davis, C.R.; Marion, P.; Salazar, F.; Kay, M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006, 441, 537–541. [Google Scholar] [CrossRef]
- Crowther, C.; Mowa, B.; Arbuthnot, P. Hepatic delivery of artificial micro RNAs using helper-dependent adenoviral vectors. In SiRNA Delivery Methods; Springer Nature: Heidelberg, Germany, 2016; pp. 249–260. [Google Scholar]
- Bloom, K.; Kaldine, H.; Cathomen, T.; Mussolino, C.; Ely, A.; Arbuthnot, P. Inhibition of replication of hepatitis B virus using transcriptional repressors that target the viral DNA. BMC Infect. Dis. 2019, 19, 802. [Google Scholar] [CrossRef]
- Moore, D.D.; Dowhan, D. Preparation and Analysis of DNA. In Current Protocols in Molecular Biology; Wiley Online Library: Hoboken, NJ, USA, 2002; pp. 2.1.1–2.1.3. [Google Scholar]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Wang, L.; Lee, J.S.; Schurmann, N.; Gu, S.; Borner, K.; Storm, T.A.; Kay, M.A. Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver. J. Clin. Investig. 2010, 120, 3106–3119. [Google Scholar] [CrossRef] [PubMed]
- Kramvis, A.; Kew, M.C. Relationship of genotypes of hepatitis B virus to mutations, disease progression and response to antiviral therapy. J. Viral Hepat. 2005, 12, 456–464. [Google Scholar] [CrossRef]
- Tanaka, Y.; Hasegawa, I.; Kato, T.; Orito, E.; Hirashima, N.; Acharya, S.K.; Gish, R.G.; Kramvis, A.; Kew, M.C.; Yoshihara, N.; et al. A case-control study for differences among hepatitis B virus infections of genotypes A (subtypes Aa and Ae) and D. Hepatology 2004, 40, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Bannister, E.; Sozzi, V.; Mason, H.; Locarnini, S.; Hardikar, W.; Revill, P.A. Analysis of the in vitro replication phenotype of African hepatitis B virus (HBV) genotypes and subgenotypes present in Australia identifies marked differences in DNA and protein expression. Virology 2020, 540, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Bhoola, N.H.; Kramvis, A. Hepatitis B e Antigen Expression by Hepatitis B Virus Subgenotype A1 Relative to Subgenotypes A2 and D3 in Cultured Hepatocellular Carcinoma (Huh7) Cells. Intervirology 2016, 59, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.; Peppa, D.; Khanna, P.; Nebbia, G.; Jones, M.; Brendish, N.; Lascar, R.M.; Brown, D.; Gilson, R.J.; Tedder, R.J.; et al. Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology 2009, 137, 1289–1300. [Google Scholar] [CrossRef]
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6669–6674. [Google Scholar] [CrossRef]
- Ronzitti, G.; Gross, D.A.; Mingozzi, F. Human Immune Responses to Adeno-Associated Virus (AAV) Vectors. Front. Immunol. 2020, 11, 670. [Google Scholar] [CrossRef] [PubMed]
- Martino, A.T.; Suzuki, M.; Markusic, D.M.; Zolotukhin, I.; Ryals, R.C.; Moghimi, B.; Ertl, H.C.; Muruve, D.A.; Lee, B.; Herzog, R.W. The genome of self-complementary adeno-associated viral vectors increases Toll-like receptor 9–dependent innate immune responses in the liver. Blood. J. Am. Soc. Hematol. 2011, 117, 6459–6468. [Google Scholar] [CrossRef] [PubMed]
- Hordeaux, J.; Wang, Q.; Katz, N.; Buza, E.L.; Bell, P.; Wilson, J.M. The neurotropic properties of AAV-PHP. B are limited to C57BL/6J mice. Mol. Ther. 2018, 26, 664–668. [Google Scholar] [CrossRef]
- Rabe, B.; Vlachou, A.; Pante, N.; Helenius, A.; Kann, M. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc. Natl. Acad. Sci. USA 2003, 100, 9849–9854. [Google Scholar] [CrossRef]
- Deroubaix, A.; Osseman, Q.; Cassany, A.; Begu, D.; Ragues, J.; Kassab, S.; Laine, S.; Kann, M. Expression of viral polymerase and phosphorylation of core protein determine core and capsid localization of the human hepatitis B virus. J. Gen. Virol. 2015, 96 Pt 1, 183–195. [Google Scholar] [CrossRef]
- Kawai, K.; Horiike, N.; Michitaka, K.; Onji, M. The effects of hepatitis B virus core promoter mutations on hepatitis B core antigen distribution in hepatocytes as detected by laser-assisted microdissection. J. Hepatol. 2003, 38, 635–641. [Google Scholar] [CrossRef]
- Jazayeri, M.S.; Dornan, E.S.; Boner, W.; Fattovich, G.; Hadziyannis, S.; Carman, W.F. Intracellular distribution of hepatitis B virus core protein expressed in vitro depends on the sequence of the isolate and the serologic pattern. J. Infect. Dis. 2004, 189, 1634–1645. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kramvis, A.; Arakawa, K.; Yu, M.C.; Nogueira, R.; Stram, D.O.; Kew, M. Relationship of serological subtype, basic core promoter and precore mutations to genotypes/subgenotypes of hepatitis B virus. J. Med. Virol. 2008, 80, 27–46. [Google Scholar] [CrossRef] [PubMed]
- Li, H.C.; Huang, E.Y.; Su, P.Y.; Wu, S.Y.; Yang, C.C.; Lin, Y.S.; Chang, W.C.; Shih, C. Nuclear export and import of human hepatitis B virus capsid protein and particles. PLoS Pathog. 2010, 6, e1001162. [Google Scholar] [CrossRef]
- Yeh, C.T.; Wong, S.W.; Fung, Y.K.; Ou, J.H. Cell cycle regulation of nuclear localization of hepatitis B virus core protein. Proc. Natl. Acad. Sci. USA 1993, 90, 6459–6463. [Google Scholar] [CrossRef]
- Chu, C.M.; Yeh, C.T.; Sheen, I.S.; Liaw, Y.F. Subcellular localization of hepatitis B core antigen in relation to hepatocyte regeneration in chronic hepatitis B. Gastroenterology 1995, 109, 1926–1932. [Google Scholar] [CrossRef]
- Chu, C.M.; Liaw, Y.F. Intrahepatic distribution of hepatitis B surface and core antigens in chronic hepatitis B virus infection. Hepatocyte with cytoplasmic/membranous hepatitis B core antigen as a possible target for immune hepatocytolysis. Gastroenterology 1987, 92, 220–225. [Google Scholar] [CrossRef]
- Hsu, H.C.; Su, I.J.; Lai, M.Y.; Chen, D.S.; Chang, M.H.; Chuang, S.M.; Sung, J.L. Biologic and prognostic significance of hepatocyte hepatitis B core antigen expressions in the natural course of chronic hepatitis B virus infection. J. Hepatol. 1987, 5, 45–50. [Google Scholar] [CrossRef]
- Lucifora, J.; Salvetti, A.; Marniquet, X.; Mailly, L.; Testoni, B.; Fusil, F.; Inchauspe, A.; Michelet, M.; Michel, M.L.; Levrero, M.; et al. Detection of the hepatitis B virus (HBV) covalently-closed-circular DNA (cccDNA) in mice transduced with a recombinant AAV-HBV vector. Antiviral Res. 2017, 145, 14–19. [Google Scholar] [CrossRef]
- Ko, C.; Su, J.; Festag, J.; Bester, R.; Kosinska, A.D.; Protzer, U. Intramolecular recombination enables the formation of hepatitis B virus (HBV) cccDNA in mice after HBV genome transfer using recombinant AAV vectors. Antiviral Res. 2021, 194, 105140. [Google Scholar] [CrossRef]
- Dion, S.; Bourgine, M.; Godon, O.; Levillayer, F.; Michel, M.L. Adeno-associated virus-mediated gene transfer leads to persistent hepatitis B virus replication in mice expressing HLA-A2 and HLA-DR1 molecules. J. Virol. 2013, 87, 5554–5563. [Google Scholar] [CrossRef] [PubMed]
- Saeidi, A.; Zandi, K.; Cheok, Y.Y.; Saeidi, H.; Wong, W.F.; Lee, C.Y.Q.; Cheong, H.C.; Yong, Y.K.; Larsson, M.; Shankar, E.M. T-cell exhaustion in chronic infections: Reversing the state of exhaustion and reinvigorating optimal protective immune responses. Front. Immunol. 2018, 9, 2569. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Chandler, R.J.; LaFave, M.C.; Varshney, G.K.; Trivedi, N.S.; Carrillo-Carrasco, N.; Senac, J.S.; Wu, W.; Hoffmann, V.; Elkahloun, A.G.; Burgess, S.M.; et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J. Clin. Investig. 2015, 125, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Logan, G.J.; Dane, A.P.; Hallwirth, C.V.; Smyth, C.M.; Wilkie, E.E.; Amaya, A.K.; Zhu, E.; Khandekar, N.; Ginn, S.L.; Liao, S.H.Y.; et al. Identification of liver-specific enhancer-promoter activity in the 3’ untranslated region of the wild-type AAV2 genome. Nat. Genet. 2017, 49, 1267–1273. [Google Scholar] [CrossRef]
- Nault, J.C.; Datta, S.; Imbeaud, S.; Franconi, A.; Mallet, M.; Couchy, G.; Letouze, E.; Pilati, C.; Verret, B.; Blanc, J.F.; et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 2015, 47, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Vo, T.T.; Poovorawan, K.; Charoen, P.; Soonthornworasiri, N.; Nontprasert, A.; Kittitrakul, C.; Phumratanaprapin, W.; Tangkijvanich, P. Association between Hepatitis B Surface Antigen Levels and the Risk of Hepatocellular Carcinoma in Patients with Chronic Hepatitis B Infection: Systematic Review and Meta-Analysis. Asian Pac. J. Cancer Prev. 2019, 20, 2239–2246. [Google Scholar]
- Ely, A.; Naidoo, T.; Arbuthnot, P. Efficient silencing of gene expression with modular trimeric Pol II expression cassettes comprising microRNA shuttles. Nucleic Acids Res. 2009, 37, e91. [Google Scholar] [CrossRef] [PubMed]
- Ely, A.; Naidoo, T.; Mufamadi, S.; Crowther, C.; Arbuthnot, P. Expressed anti-HBV primary microRNA shuttles inhibit viral replication efficiently in vitro and in vivo. Mol. Ther. 2008, 16, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Rösler, C.; Kidd-Ljunggren, K.; Nassal, M. Quantitative assessment of the antiviral potencies of 21 shRNA vectors targeting conserved, including structured, hepatitis B virus sites. J. Hepatol. 2010, 52, 817–826. [Google Scholar] [CrossRef]
- Decorsiere, A.; Mueller, H.; van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Limani, S.W.; Mnyandu, N.; Ely, A.; Wadee, R.; Kramvis, A.; Arbuthnot, P.; Maepa, M.B. In Vivo Modelling of Hepatitis B Virus Subgenotype A1 Replication Using Adeno-Associated Viral Vectors. Viruses 2021, 13, 2247. https://doi.org/10.3390/v13112247
Limani SW, Mnyandu N, Ely A, Wadee R, Kramvis A, Arbuthnot P, Maepa MB. In Vivo Modelling of Hepatitis B Virus Subgenotype A1 Replication Using Adeno-Associated Viral Vectors. Viruses. 2021; 13(11):2247. https://doi.org/10.3390/v13112247
Chicago/Turabian StyleLimani, Shonisani Wendy, Njabulo Mnyandu, Abdullah Ely, Reubina Wadee, Anna Kramvis, Patrick Arbuthnot, and Mohube Betty Maepa. 2021. "In Vivo Modelling of Hepatitis B Virus Subgenotype A1 Replication Using Adeno-Associated Viral Vectors" Viruses 13, no. 11: 2247. https://doi.org/10.3390/v13112247
APA StyleLimani, S. W., Mnyandu, N., Ely, A., Wadee, R., Kramvis, A., Arbuthnot, P., & Maepa, M. B. (2021). In Vivo Modelling of Hepatitis B Virus Subgenotype A1 Replication Using Adeno-Associated Viral Vectors. Viruses, 13(11), 2247. https://doi.org/10.3390/v13112247