
African Swine Fever Virus pE199L Induces Mitochondrial-Dependent Apoptosis

 ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Facility and Animal Experiment
2.2. Cell Lines and Antibodies
2.3. Virus Titration
2.4. Construction of Recombinant Plasmids
2.5. Cell Viability Assay
2.6. Apoptosis Assay
2.7. Caspase Activity Assay
2.8. TUNEL Assay
2.9. Measurement of the Mitochondrial Membrane Potential
2.10. RNA Extraction and Quantitative Reverse-Transcription Polymerase Chain Reaction (qRT-PCR)
2.11. Immunofluorescence Assay (IFA) and Real-Time Confocal Analysis
2.12. Co-IP and Western Blotting Assay
2.13. Mitochondria Purification, Cytochrome C Release, BAK Cross-Linking, and BAX Activation
2.14. Small Interfering Rnas Assay
2.15. Electron Microscopy Imaging
2.16. Statistical Analysis
3. Results
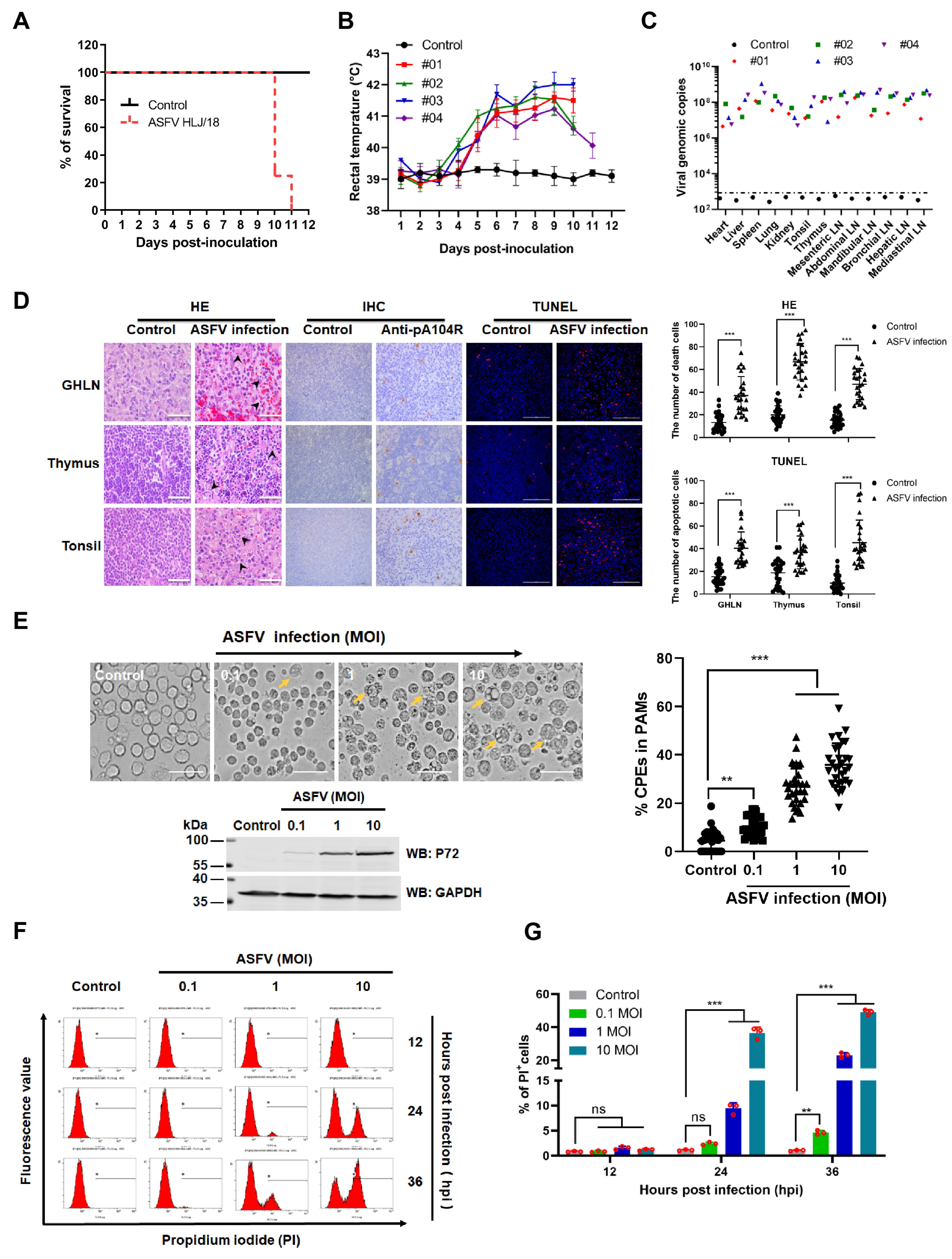
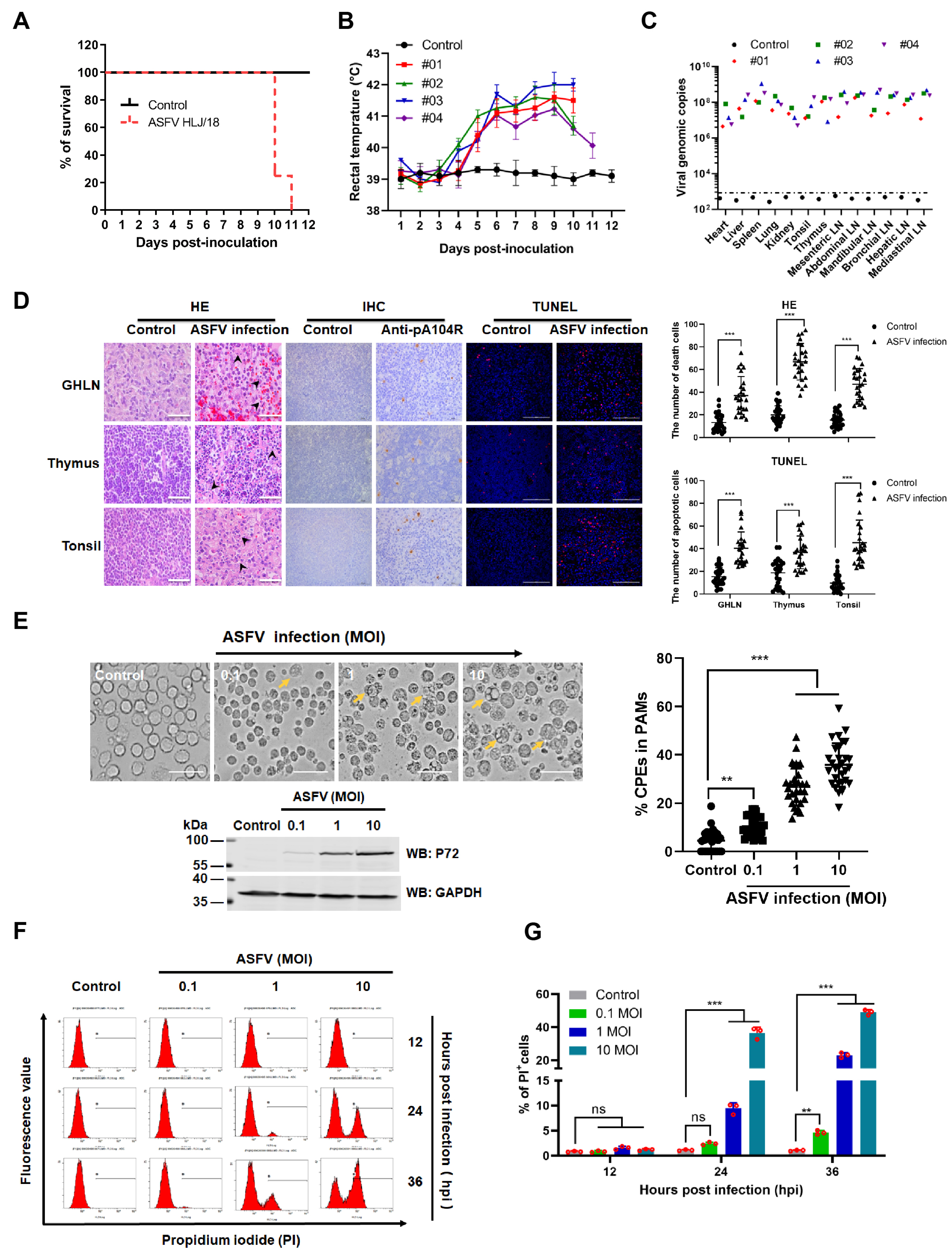
3.1. ASFV HLJ/18 Infection Induces Tissue Injury and Cell Death
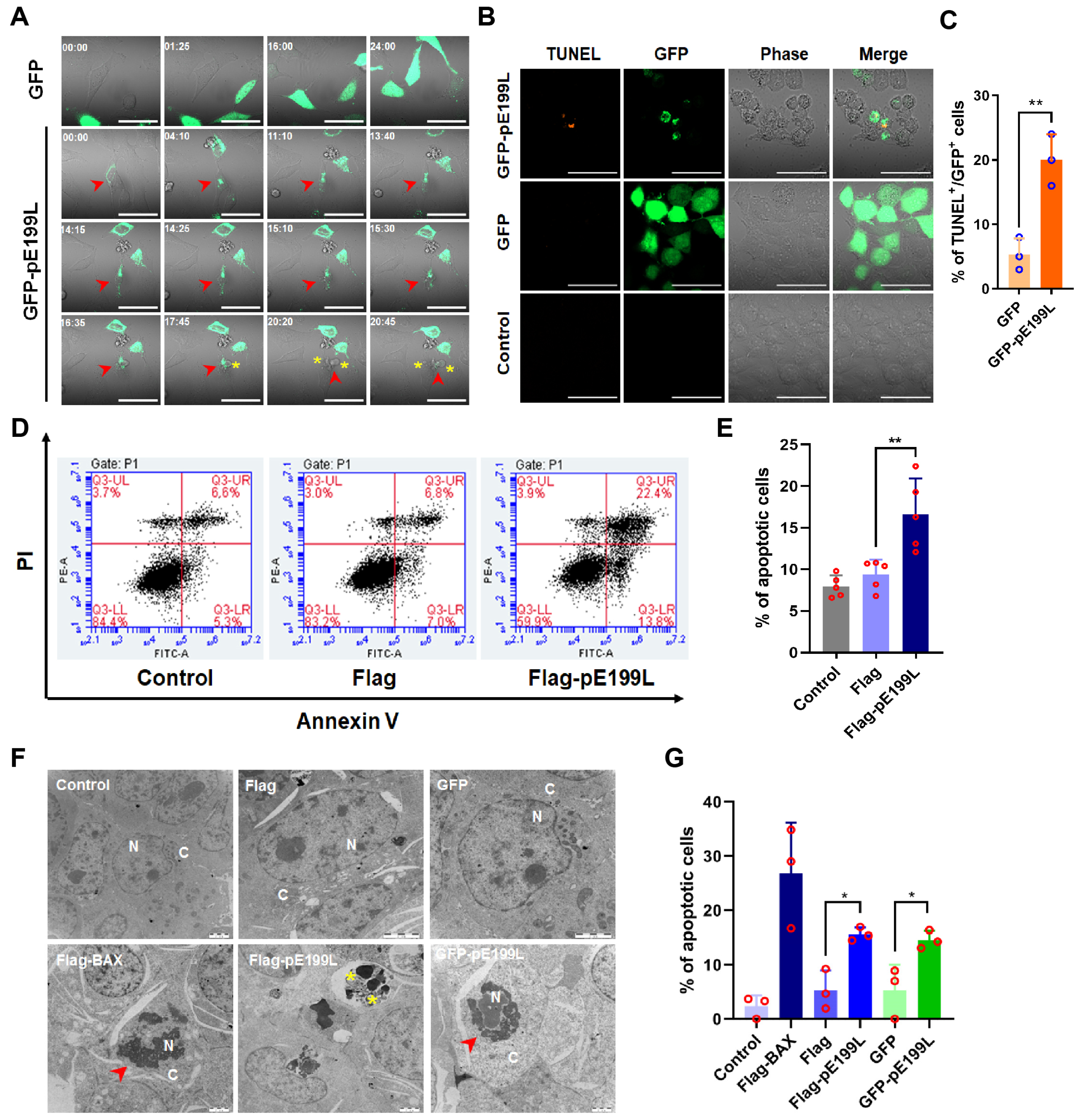
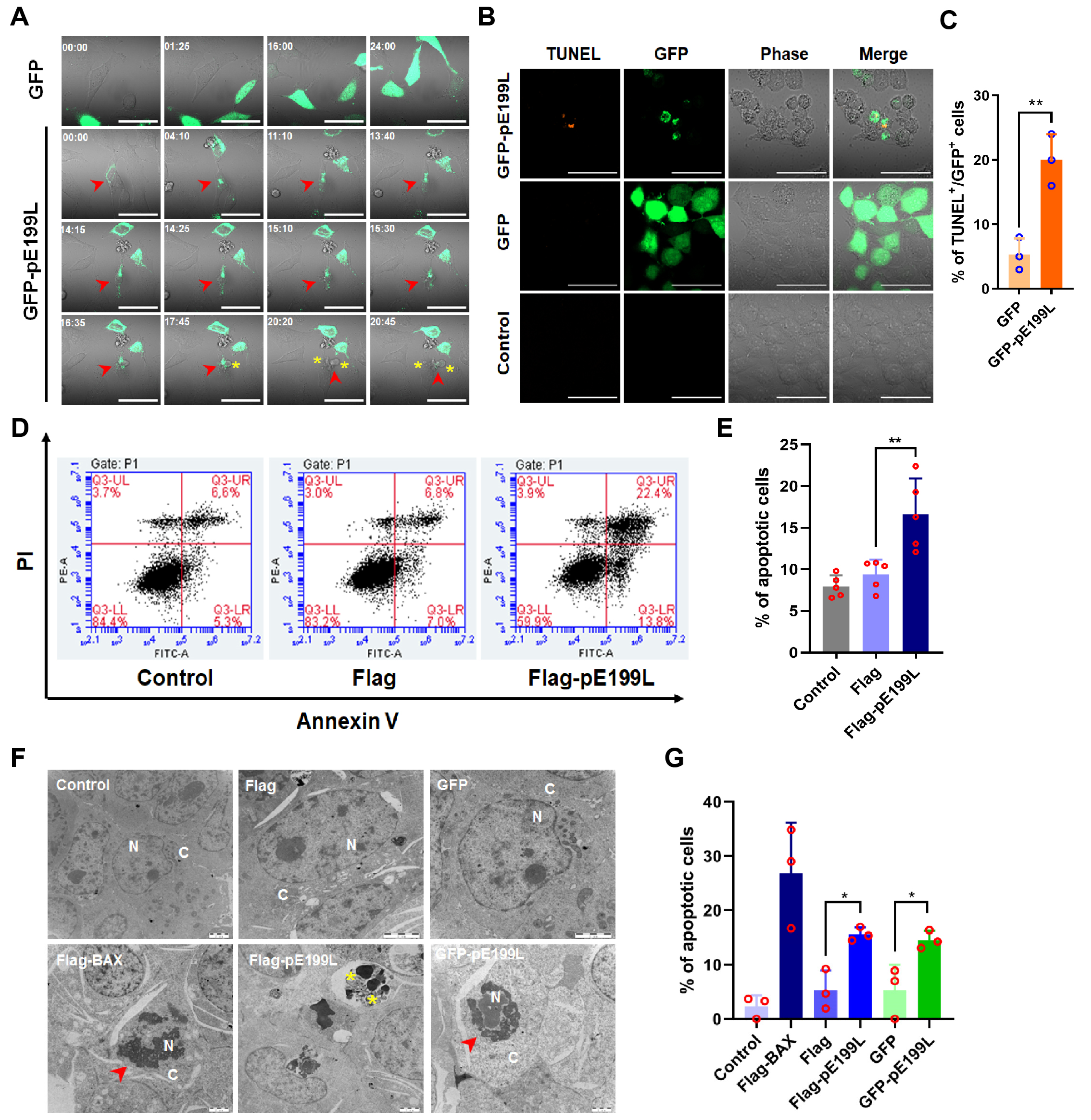
3.2. ASFV pE199L Induces Cell Death
3.3. ASFV pE199L Induces Apoptosis
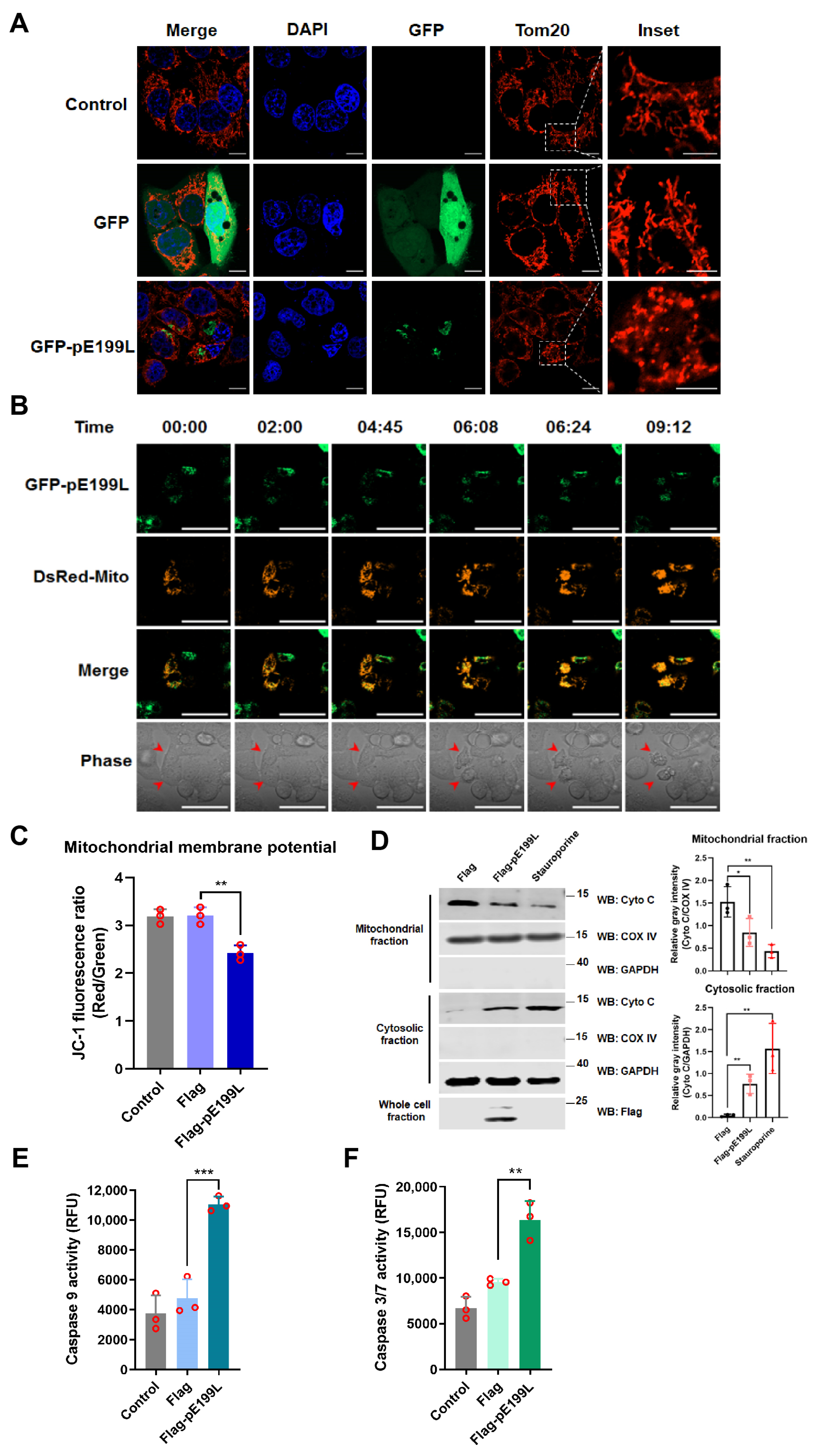
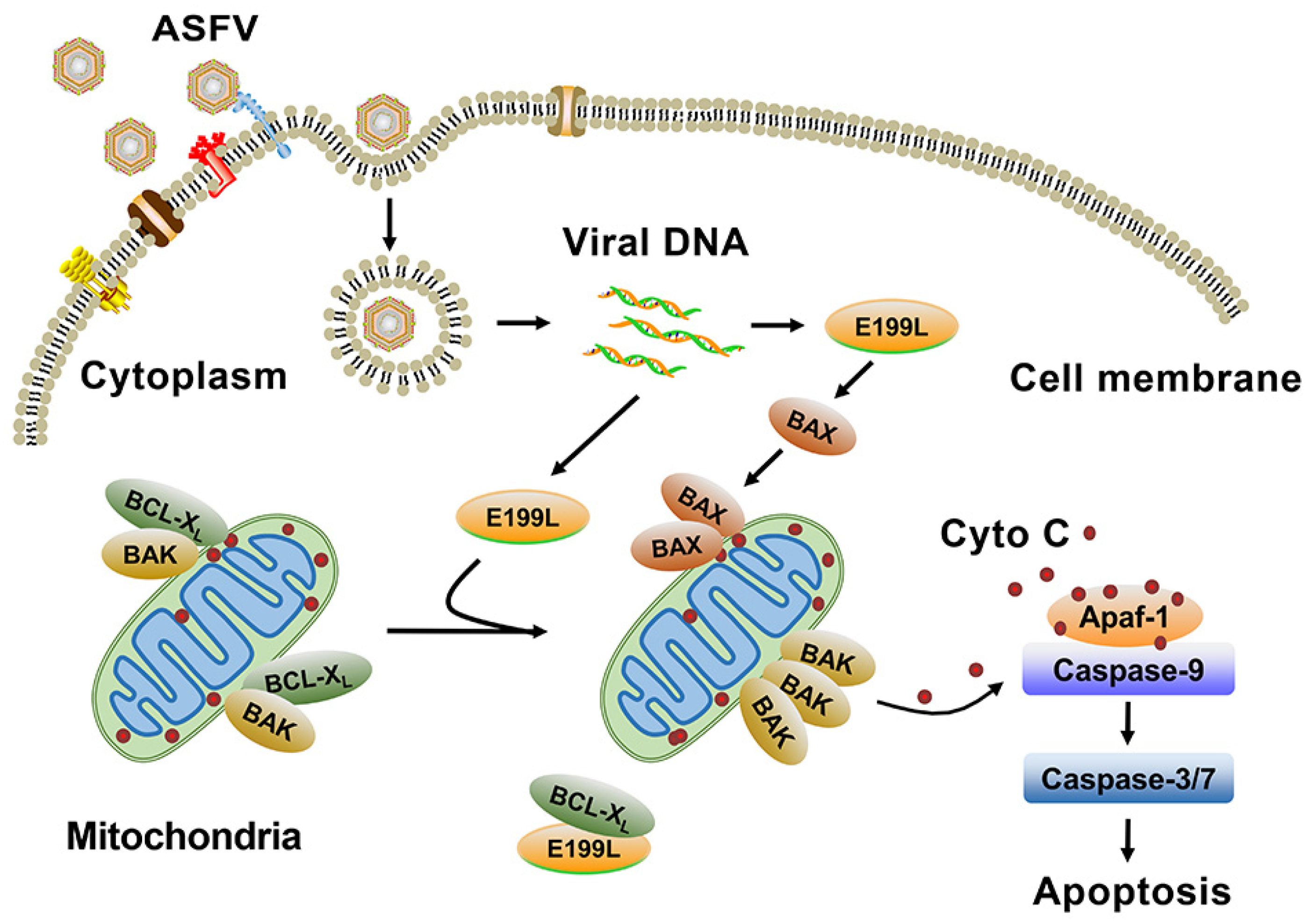
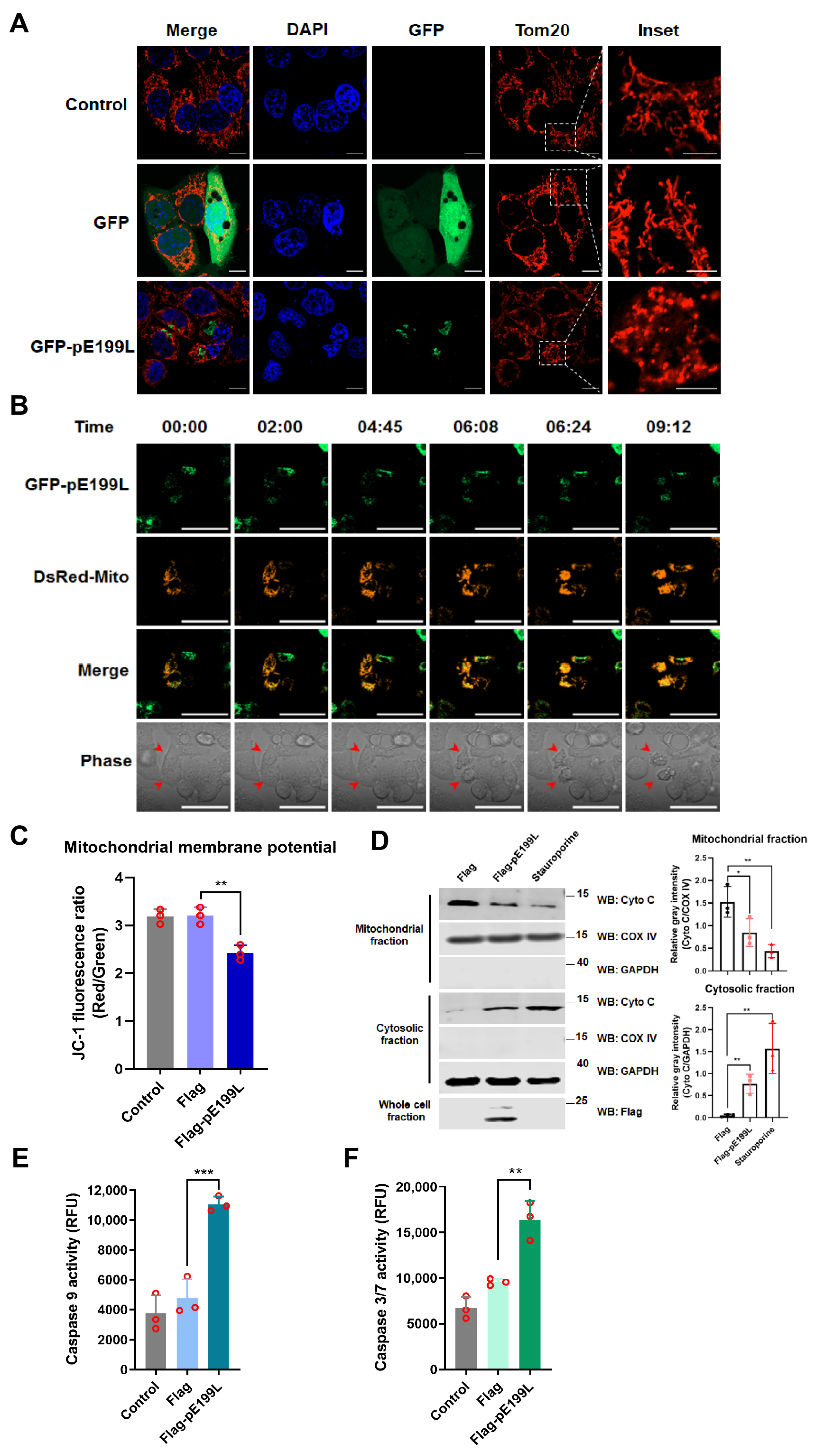
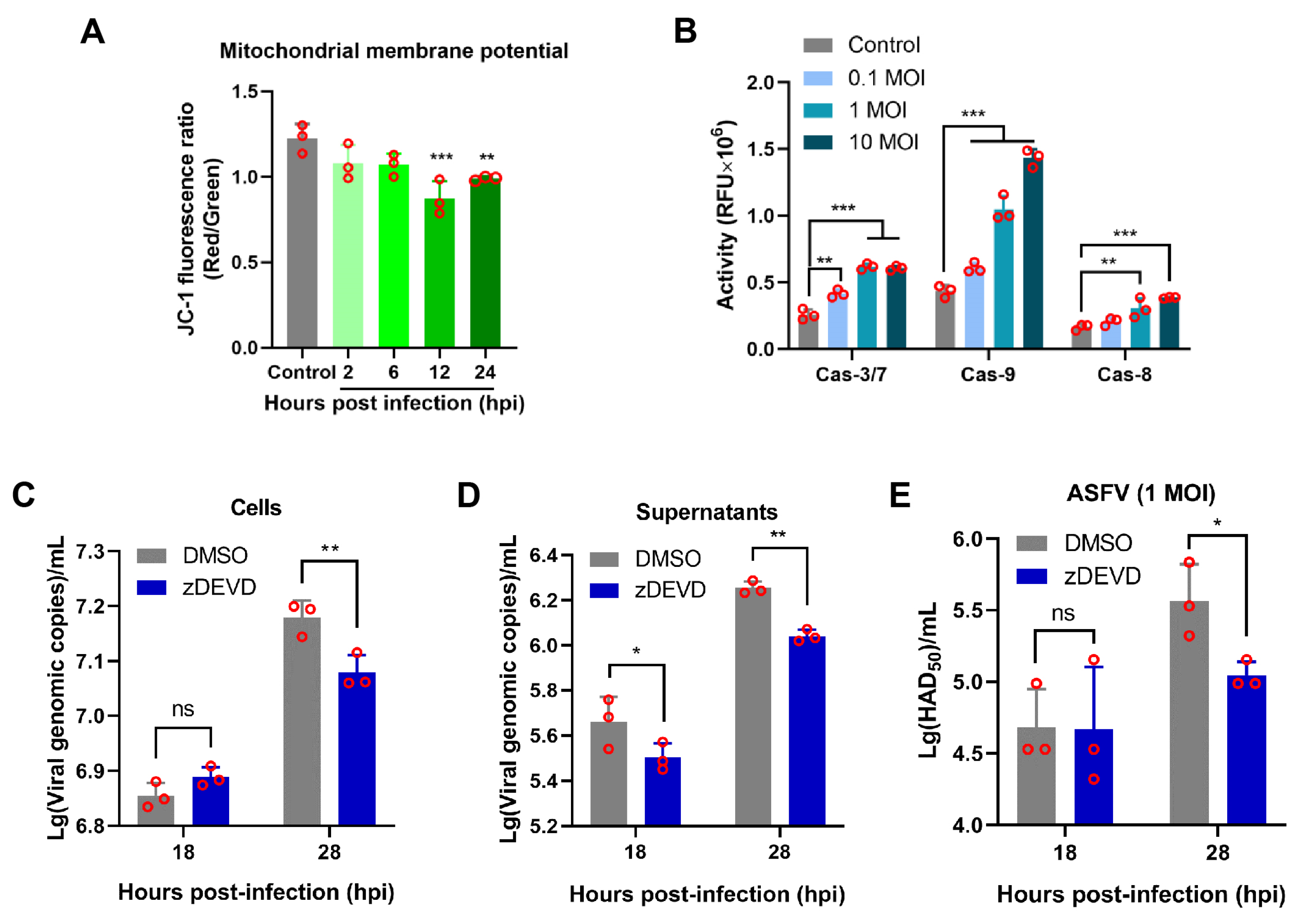
3.4. ASFV pE199L Triggers the Mitochondrial Apoptotic Pathway
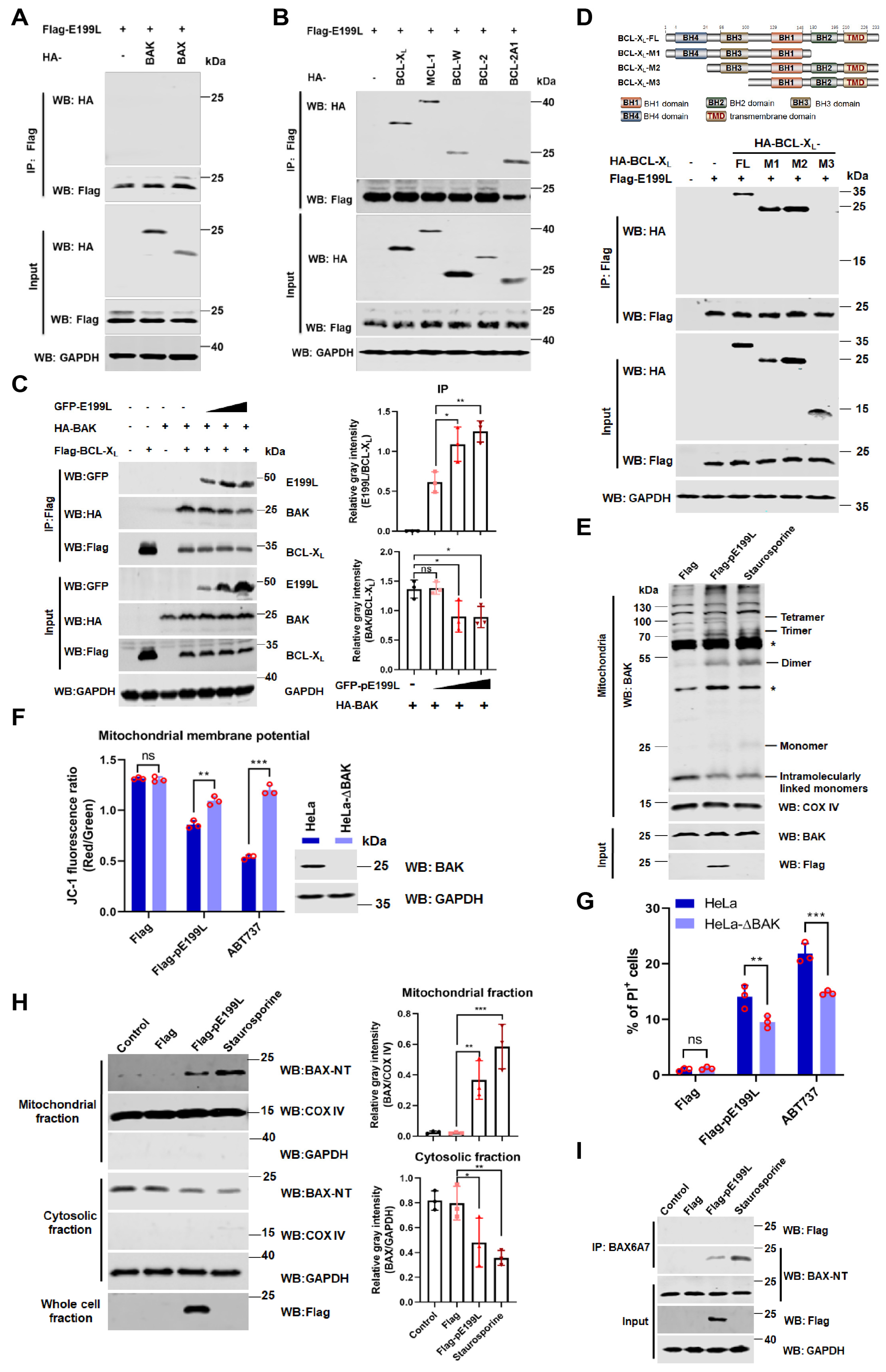
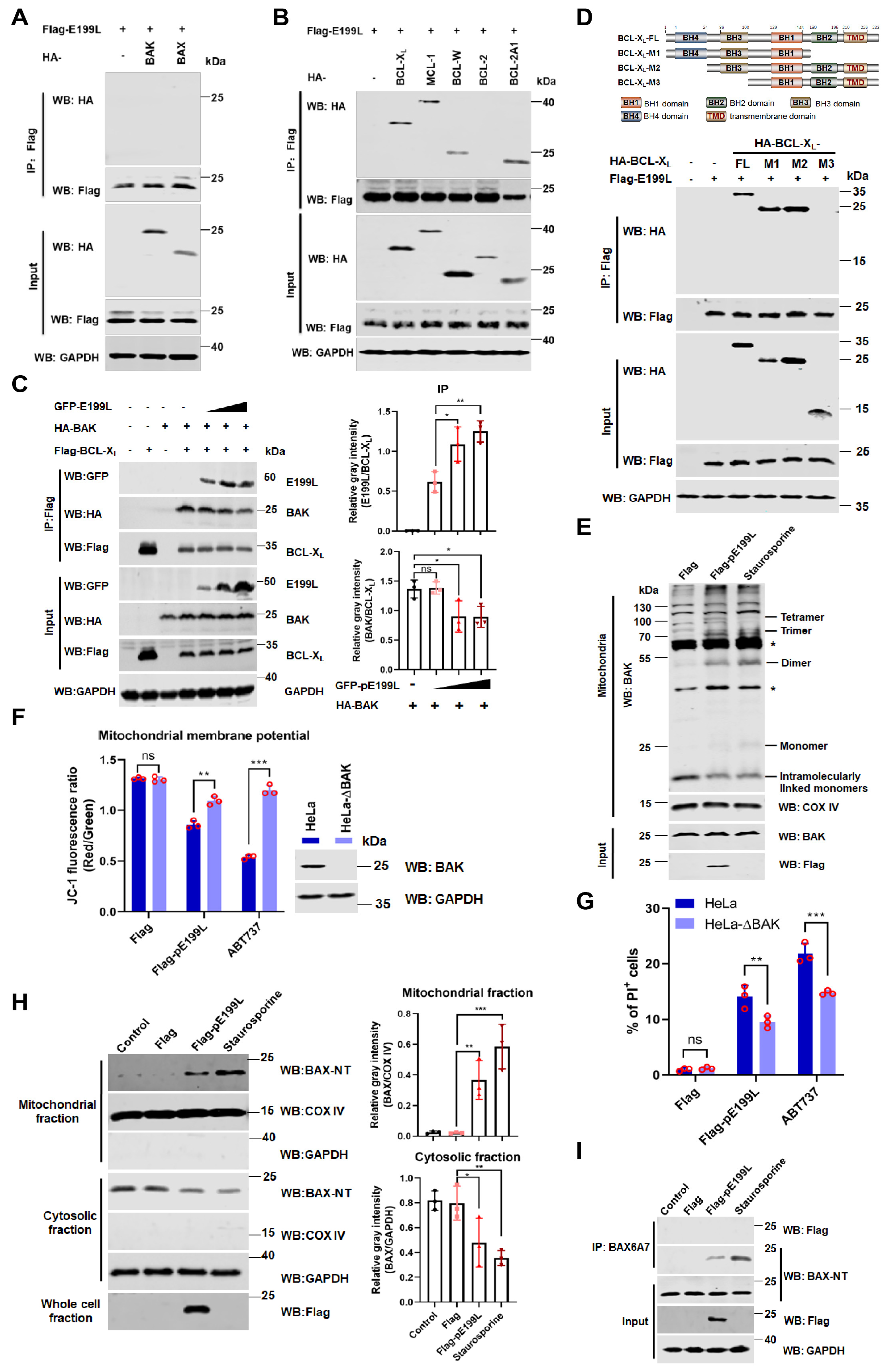
3.5. ASFV pE199L Promotes BAK Activation by Competing with BAK for BCL-XL
3.6. Apoptosis Induced by ASFV Infection Contributes to Viral Growth
4. Discussion
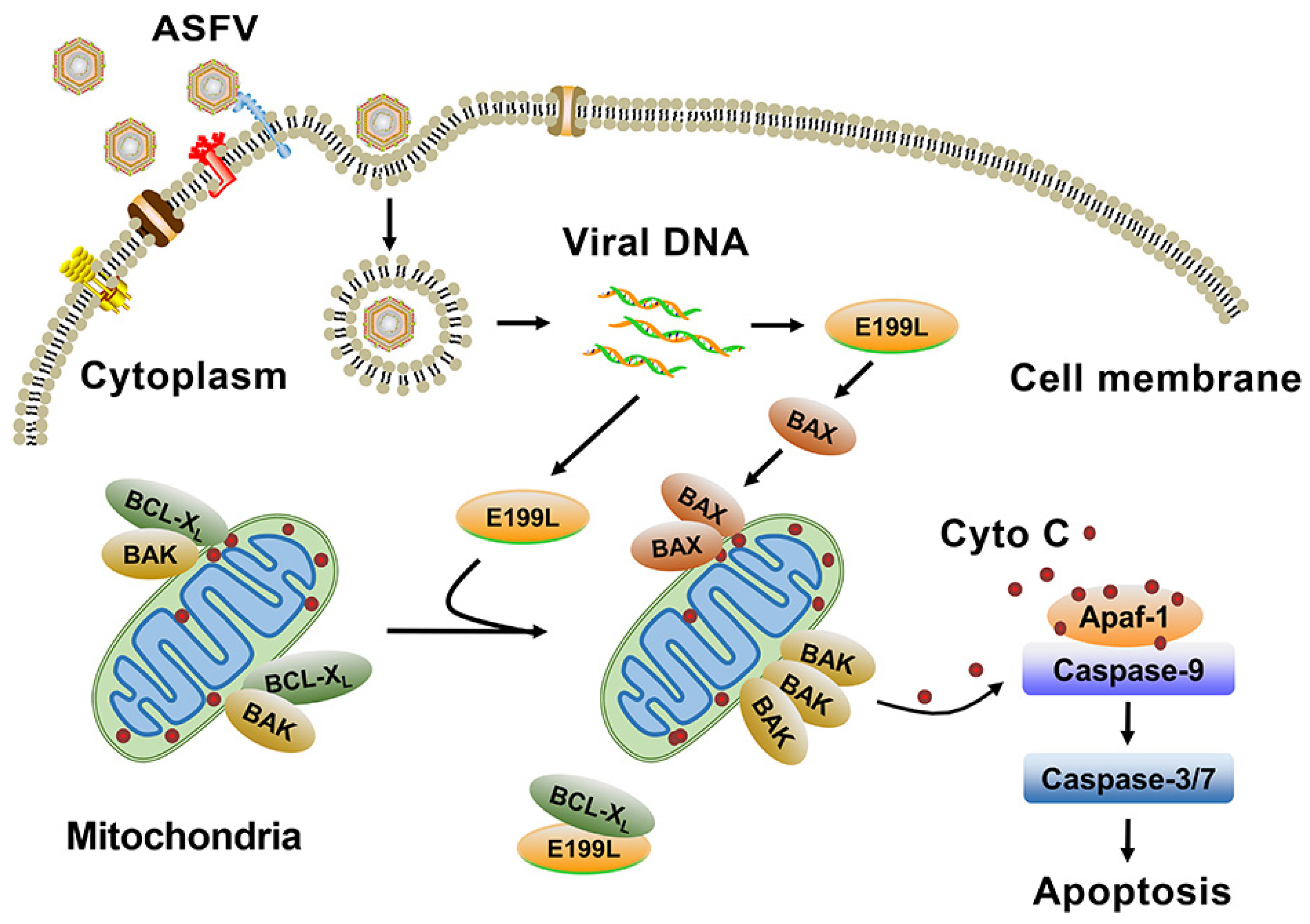
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mighell, E.; Ward, M.P. African Swine Fever spread across Asia, 2018–2019. Transbound. Emerg. Dis. 2021, 68, 2722–2732. [Google Scholar] [CrossRef]
- Dixon, L.K.; Chapman, D.A.; Netherton, C.L.; Upton, C. African swine fever virus replication and genomics. Virus Res. 2013, 173, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Ramiro-Ibáñez, F.; Ortega, A.; Brun, A.; Escribano, J.M.; Alonso, C. Apoptosis: A mechanism of cell killing and lymphoid organ impairment during acute African swine fever virus infection. J. Gen. Virol. 1996, 77, 2209–2219. [Google Scholar] [CrossRef]
- Portugal, R.; Leitão, A.; Martins, C. Apoptosis in porcine macrophages infected in vitro with African swine fever virus (ASFV) strains with different virulence. Arch. Virol. 2009, 154, 1441–1450. [Google Scholar] [CrossRef] [PubMed]
- Franzoni, G.; Razzuoli, E.; Dei Giudici, S.; Carta, T.; Galleri, G.; Zinellu, S.; Ledda, M.; Angioi, P.; Modesto, P.; Graham, S.P.; et al. Acyclovir Comparison of Macrophage Responses to African Swine Fever Viruses Reveals that the NH/P68 Strain is Associated with Enhanced Sensitivity to Type I IFN and Cytokine Responses from Classically Activated Macrophages. Pathogens 2020, 9, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvansakul, M.; Caria, S.; Hinds, M.G. The Bcl-2 Family in Host-Virus Interactions. Viruses 2017, 9, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Reviews. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Birkinshaw, R.W.; Czabotar, P.E. The BCL-2 family of proteins and mitochondrial outer membrane permeabilisation. Semin. Cell Dev. Biol. 2017, 72, 152–162. [Google Scholar] [CrossRef]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [Green Version]
- Billen, L.P.; Kokoski, C.L.; Lovell, J.F.; Leber, B.; Andrews, D.W. Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol. 2008, 6, e147. [Google Scholar] [CrossRef]
- Chen, H.C.; Kanai, M.; Inoue-Yamauchi, A.; Tu, H.C.; Huang, Y.; Ren, D.; Kim, H.; Takeda, S.; Reyna, D.E.; Chan, P.M.; et al. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat. Cell Biol. 2015, 17, 1270–1281. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; O’Neill, K.L.; Li, J.; Zhou, W.; Han, N.; Pang, X.; Wu, W.; Struble, L.; Borgstahl, G.; Liu, Z.; et al. BH3-only proteins target BCL-xL/MCL-1, not BAX/BAK, to initiate apoptosis. Cell Res. 2019, 29, 942–952. [Google Scholar] [CrossRef]
- Zhou, L.; Chang, D.C. Dynamics and structure of the Bax-Bak complex responsible for releasing mitochondrial proteins during apoptosis. J. Cell Sci 2008, 121, 2186–2196. [Google Scholar] [CrossRef] [Green Version]
- Cuconati, A.; White, E. Viral homologs of BCL-2: Role of apoptosis in the regulation of virus infection. Genes Dev. 2002, 16, 2465–2478. [Google Scholar] [CrossRef] [Green Version]
- Jacotot, E.; Ravagnan, L.; Loeffler, M.; Ferri, K.F.; Vieira, H.L.; Zamzami, N.; Costantini, P.; Druillennec, S.; Hoebeke, J.; Briand, J.P.; et al. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J. Exp. Med. 2000, 191, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Nomura-Takigawa, Y.; Nagano-Fujii, M.; Deng, L.; Kitazawa, S.; Ishido, S.; Sada, K.; Hotta, H. Non-structural protein 4A of Hepatitis C virus accumulates on mitochondria and renders the cells prone to undergoing mitochondria-mediated apoptosis. J. Gen. Virol. 2006, 87, 1935–1945. [Google Scholar] [CrossRef]
- Tan, Y.X.; Tan, T.H.; Lee, M.J.; Tham, P.Y.; Gunalan, V.; Druce, J.; Birch, C.; Catton, M.; Fu, N.Y.; Yu, V.C.; et al. Induction of apoptosis by the severe acute respiratory syndrome coronavirus 7a protein is dependent on its interaction with the Bcl-XL protein. J. Virol. 2007, 81, 6346–6355. [Google Scholar] [CrossRef] [Green Version]
- Marshall, B.; Puthalakath, H.; Caria, S.; Chugh, S.; Doerflinger, M.; Colman, P.M.; Kvansakul, M. Variola virus F1L is a Bcl-2-like protein that unlike its vaccinia virus counterpart inhibits apoptosis independent of Bim. Cell Death Dis. 2015, 6, e1680. [Google Scholar] [CrossRef]
- Galindo, I.; Hernaez, B.; Diaz-Gil, G.; Escribano, J.M.; Alonso, C. A179L, a viral Bcl-2 homologue, targets the core Bcl-2 apoptotic machinery and its upstream BH3 activators with selective binding restrictions for Bid and Noxa. Virology 2008, 375, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Nogal, M.L.; González de Buitrago, G.; Rodríguez, C.; Cubelos, B.; Carrascosa, A.L.; Salas, M.L.; Revilla, Y. African swine fever virus IAP homologue inhibits caspase activation and promotes cell survival in mammalian cells. J. Virol. 2001, 75, 2535–2543. [Google Scholar] [CrossRef] [Green Version]
- Hurtado, C.; Granja, A.G.; Bustos, M.J.; Nogal, M.L.; González de Buitrago, G.; de Yébenes, V.G.; Salas, M.L.; Revilla, Y.; Carrascosa, A.L. The C-type lectin homologue gene (EP153R) of African swine fever virus inhibits apoptosis both in virus infection and in heterologous expression. Virology 2004, 326, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Moon, A.; Childs, K.; Goodbourn, S.; Dixon, L.K. The African swine fever virus DP71L protein recruits the protein phosphatase 1 catalytic subunit to dephosphorylate eIF2alpha and inhibits CHOP induction but is dispensable for these activities during virus infection. J. Virol. 2010, 84, 10681–10689. [Google Scholar] [CrossRef] [Green Version]
- Hernáez, B.; Díaz-Gil, G.; García-Gallo, M.; Ignacio Quetglas, J.; Rodríguez-Crespo, I.; Dixon, L.; Escribano, J.M.; Alonso, C. The African swine fever virus dynein-binding protein p54 induces infected cell apoptosis. FEBS Lett. 2004, 569, 224–228. [Google Scholar] [CrossRef] [Green Version]
- Alonso, C.; Miskin, J.; Hernáez, B.; Fernandez-Zapatero, P.; Soto, L.; Cantó, C.; Rodríguez-Crespo, I.; Dixon, L.; Escribano, J.M. African swine fever virus protein p54 interacts with the microtubular motor complex through direct binding to light-chain dynein. J. Virol. 2001, 75, 9819–9827. [Google Scholar] [CrossRef] [Green Version]
- Brun, A.; Rivas, C.; Esteban, M.; Escribano, J.M.; Alonso, C. African swine fever virus gene A179L, a viral homologue of bcl-2, protects cells from programmed cell death. Virology 1996, 225, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Jenson, J.; Dixon, L.K.; Parkhouse, M.E. Characterization of the African swine fever virion protein j18L. J. Gen. Virol. 1996, 77, 941–946. [Google Scholar] [CrossRef]
- Matamoros, T.; Alejo, A. African Swine Fever Virus Protein pE199L Mediates Virus Entry by Enabling Membrane Fusion and Core Penetration. mBio 2020, 11, e00789-20. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, X.; Nie, Y.; Li, H.; Chen, W.; Lin, W.; Chen, F.; Xie, Q. African Swine Fever Virus Protein E199L Promotes Cell Autophagy through the Interaction of PYCR2. Virol. Sin. 2021, 36, 196–206. [Google Scholar] [CrossRef]
- Weingartl, H.M.; Sabara, M.; Pasick, J.; van Moorlehem, E.; Babiuk, L. Continuous porcine cell lines developed from alveolar macrophages: Partial characterization and virus susceptibility. J. Virol. Methods 2002, 104, 203–216. [Google Scholar] [CrossRef]
- Malmquist, W.A.; Hay, D. Hemadsorption and cytopathic effect produced by African Swine Fever virus in swine bone marrow and buffy coat cultures. Am. J. Vet. Res. 1960, 21, 104–108. [Google Scholar]
- Nemeth, B.; Fasseeh, A.; Molnar, A.; Bitter, I.; Horvath, M.; Koczian, K.; Gotze, A.; Nagy, B. A systematic review of health economic models and utility estimation methods in schizophrenia. Expert Rev. Pharm. Outcomes Res. 2018, 18, 267–275. [Google Scholar] [CrossRef]
- Sarosiek, K.A.; Chi, X.; Bachman, J.A.; Sims, J.J.; Montero, J.; Patel, L.; Flanagan, A.; Andrews, D.W.; Sorger, P.; Letai, A. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol. Cell 2013, 51, 751–765. [Google Scholar] [CrossRef] [Green Version]
- Sivandzade, F.; Bhalerao, A.; Cucullo, L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio Protoc. 2019, 9, e3128. [Google Scholar] [CrossRef]
- Li, J.; Hu, L.; Liu, Y. DDX19A Senses Viral RNA and Mediates NLRP3-Dependent Inflammasome Activation. J. Immunol. 2015, 195, 5732–5749. [Google Scholar] [CrossRef] [Green Version]
- Snigirevskaya, E.S.; Komissarchik, Y.Y. Ultrastructural traits of apoptosis. Cell Biol. Int. 2019, 43, 728–738. [Google Scholar] [CrossRef]
- Imre, G. Cell death signalling in virus infection. Cell Signal. 2020, 76, 109772. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Dewson, G.; Kratina, T.; Czabotar, P.; Day, C.L.; Adams, J.M.; Kluck, R.M. Bak activation for apoptosis involves oligomerization of dimers via their alpha6 helices. Mol. Cell 2009, 36, 696–703. [Google Scholar] [CrossRef] [Green Version]
- Cong, H.; Du, N.; Yang, Y.; Song, L.; Zhang, W.; Tien, P. Enterovirus 71 2B Induces Cell Apoptosis by Directly Inducing the Conformational Activation of the Proapoptotic Protein Bax. J. Virol. 2016, 90, 9862–9877. [Google Scholar] [CrossRef] [Green Version]
- Galindo, I.; Hernáez, B.; Muñoz-Moreno, R.; Cuesta-Geijo, M.A.; Dalmau-Mena, I.; Alonso, C. The ATF6 branch of unfolded protein response and apoptosis are activated to promote African swine fever virus infection. Cell Death Dis. 2012, 3, e341. [Google Scholar] [CrossRef] [Green Version]
- Hernaez, B.; Escribano, J.M.; Alonso, C. Visualization of the African swine fever virus infection in living cells by incorporation into the virus particle of green fluorescent protein-p54 membrane protein chimera. Virology 2006, 350, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Villamandos, J.C.; Hervás, J.; Moreno, C.; Carrasco, L.; Bautista, M.J.; Caballero, J.M.; Wilkinson, P.J.; Sierra, M.A. Subcellular changes in the tonsils of pigs infected with acute African swine fever virus. Vet. Res. 1997, 28, 179–189. [Google Scholar]
- Schaecher, S.R.; Touchette, E.; Schriewer, J.; Buller, R.M.; Pekosz, A. Severe acute respiratory syndrome coronavirus gene 7 products contribute to virus-induced apoptosis. J. Virol. 2007, 81, 11054–11068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Liu, X.; Hu, B.; Fan, Z.; Song, Y.; Wei, H.; Qiu, R.; Xu, W.; Zhu, W.; Wang, F. Rabbit Hemorrhagic Disease Virus Non-structural Protein 6 Induces Apoptosis in Rabbit Kidney Cells. Front. Microbiol. 2018, 9, 3308. [Google Scholar] [CrossRef] [PubMed]
- Chaulagain, S.; Delhon, G.A.; Khatiwada, S.; Rock, D.L. African Swine Fever Virus CD2v Protein Induces beta-Interferon Expression and Apoptosis in Swine Peripheral Blood Mononuclear Cells. Viruses 2021, 13, 1480. [Google Scholar] [CrossRef]
- Dixon, L.K.; Islam, M.; Nash, R.; Reis, A.L. African swine fever virus evasion of host defences. Virus Res. 2019, 266, 25–33. [Google Scholar] [CrossRef]
- Ohta, A.; Nishiyama, Y. Mitochondria and viruses. Mitochondrion 2011, 11, 1–12. [Google Scholar] [CrossRef]
- Fan, T.J.; Han, L.H.; Cong, R.S.; Liang, J. Caspase family proteases and apoptosis. Acta Biochim. Biophys. Sin. 2005, 37, 719–727. [Google Scholar] [CrossRef] [Green Version]
- Sainski, A.M.; Natesampillai, S.; Cummins, N.W.; Bren, G.D.; Taylor, J.; Saenz, D.T.; Poeschla, E.M.; Badley, A.D. The HIV-1-specific protein Casp8p41 induces death of infected cells through Bax/Bak. J. Virol. 2011, 85, 7965–7975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohmer, M.; Weber, A.; Sutter, G.; Ehrhardt, K.; Zimmermann, A.; Hacker, G. Anti-apoptotic Bcl-XL but not Mcl-1 contributes to protection against virus-induced apoptosis. Cell Death Dis. 2016, 7, e2340. [Google Scholar] [CrossRef] [PubMed]
- Wyzewski, Z.; Switlik, W.; Mielcarska, M.B.; Gregorczyk-Zboroch, K.P. The Role of Bcl-xL Protein in Viral Infections. Int. J. Mol. Sci. 2021, 22, 1956. [Google Scholar] [CrossRef] [PubMed]
- Geng, X.; Huang, C.; Qin, Y.; McCombs, J.E.; Yuan, Q.; Harry, B.L.; Palmer, A.E.; Xia, N.S.; Xue, D. Hepatitis B virus X protein targets Bcl-2 proteins to increase intracellular calcium, required for virus replication and cell death induction. Proc. Natl. Acad. Sci. USA 2012, 109, 18471–18476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banjara, S.; Caria, S.; Dixon, L.K.; Hinds, M.G.; Kvansakul, M. Structural Insight into African Swine Fever Virus A179L-Mediated Inhibition of Apoptosis. J. Virol. 2017, 91, e02228-16. [Google Scholar] [CrossRef] [Green Version]
- Anasir, M.I.; Caria, S.; Skinner, M.A.; Kvansakul, M. Structural basis of apoptosis inhibition by the fowlpox virus protein FPV039. J. Biol. Chem. 2017, 292, 9010–9021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.Y.; Chen, H.Y.; Cao, J.L.; Xiong, H.L.; Mo, X.B.; Li, T.L.; Kang, X.Z.; Zhao, J.H.; Yin, B.; Zhao, X.; et al. Structural and functional analyses of hepatitis B virus X protein BH3-like domain and Bcl-xL interaction. Nat. Commun. 2019, 10, 3192. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Primer | Sequence |
|---|---|---|
| P1 | pGFP-E199L | TACAAGTCCGGACTCAGATCTATGTCTTGCATGCCAGTTTCCA |
| P2 | TTATCTAGATCCGGTGGATCCAAAATTGTTTAGGTTTGAAAAAATAAGAG | |
| P3 | pCAGGS-HA-E199L | GTTCCAGATTACGCTGAATTCTCTTGCATGCCAGTTTCCACG |
| P4 | ATTAAGATCTGCTAGCTCGAGTTAAAAATTGTTTAGGTTTGAAAAAATAAG | |
| P5 | pCAGGS-HA-BCL-XL-M1 | GTTCCAGATTACGCTGAATTCTCTCAGAGCAACCGGGAGC |
| P6 | ATTAAGATCTGCTAGCTCGAGTCACTCTAGGTGGTCATTCAGGTAA | |
| P7 | pCAGGS-HA-BCL-XL-M2 | GTTCCAGATTACGCTGAATTCAGTCAGTTTAGTGATGTGGAAG |
| P8 | ATTAAGATCTGCTAGCTCGAGTCATTTCCGACTGAAGAGTGAGC | |
| P9 | pCAGGS-HA-BCL-XL-M3 | GTTCCAGATTACGCTGAATTCTACCGGCGGGCATTCAGTGAC |
| P10 | ATTAAGATCTGCTAGCTCGAGTCATTTCCGACTGAAGAGTGAGC | |
| P11 | pEGFP-C1-E183L | GTACAAGTCCGGACTCAGATCTGATTCTGAATTTTTTCAACCG |
| P12 | GTTATCTAGATCCGGTGGATCCTTACAAGGAGTTTTCTAGGTC | |
| P13 | siRNA-E199L-1 | GGAAGACAUCAAACGGUAATT |
| P14 | UUACCGUUUGAUGUCUUCCTT | |
| P15 | siRNA-E199L-2 | GGUAUAGGUCGGAAAUAUUTT |
| P16 | AAUAUUUCCGACCUAUACCTT | |
| P17 | siRNA-NC | UUCUCCGAACGUGUCACGUTT |
| P18 | ACGUGACACGUUCGGAGAATT | |
| P19 | qPCR-E199L | GGGCAATATTTCCGACCTATAC |
| P20 | GGGCAACTTATCGTCATTGT | |
| P21 | HPRT | GCCGAGGATTTGGAAAAGG |
| P22 | GCACACAGAGGGCTACGATG | |
| P23 | ASFV-p72 | CTGCTCATGGTATCAATCTTATCGA |
| P22 | GATACCACAAGATCAGCCGT | |
| P23 | FAM-CCACGGGAGGAATACCAACCCAGTG-TAMRA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, T.; Zhao, G.; Zhang, T.; Zhang, Z.; Chen, X.; Song, J.; Wang, X.; Li, J.; Huang, L.; Wen, L.; et al. African Swine Fever Virus pE199L Induces Mitochondrial-Dependent Apoptosis. Viruses 2021, 13, 2240. https://doi.org/10.3390/v13112240
Li T, Zhao G, Zhang T, Zhang Z, Chen X, Song J, Wang X, Li J, Huang L, Wen L, et al. African Swine Fever Virus pE199L Induces Mitochondrial-Dependent Apoptosis. Viruses. 2021; 13(11):2240. https://doi.org/10.3390/v13112240
Chicago/Turabian StyleLi, Tingting, Gaihong Zhao, Taoqing Zhang, Zhaoxia Zhang, Xin Chen, Jie Song, Xiao Wang, Jiangnan Li, Li Huang, Lili Wen, and et al. 2021. "African Swine Fever Virus pE199L Induces Mitochondrial-Dependent Apoptosis" Viruses 13, no. 11: 2240. https://doi.org/10.3390/v13112240
APA StyleLi, T., Zhao, G., Zhang, T., Zhang, Z., Chen, X., Song, J., Wang, X., Li, J., Huang, L., Wen, L., Li, C., Zhao, D., He, X., Bu, Z., Zheng, J., & Weng, C. (2021). African Swine Fever Virus pE199L Induces Mitochondrial-Dependent Apoptosis. Viruses, 13(11), 2240. https://doi.org/10.3390/v13112240