
Human Papillomaviruses-Associated Cancers: An Update of Current Knowledge



 , ,
, ,  ,
, 
 and
and
Abstract
1. Introduction
2. HPV Epidemiology and Association to Different Cancers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Average Number of Cases # | Cancer Attributable to HR HPVs * | ||||||
|---|---|---|---|---|---|---|---|
| Anatomical Site of Cancer (ICD-10 Code) ** | In Women | In Men | Total | HR HPVs | HPV-16/18 | Estimated Total Number | References |
| Cervix (C53) | 604,127 | / | 604,127 | 89.5% | 70.8% | 540,694 | [32,33] |
| Vagina (C52) | 17,908 | / | 17,908 | 85.3% | 63.7% | 15,276 | [33] |
| Vulva (C51) | 45,240 | / | 45,240 | 87.1% | 72.6% | 39,404 | [33] |
| Penis (C60) | / | 36,068 | 36,068 | 84.6% | 70.2% | 30,514 | [33] |
| Anus (C21) | 29,159 | 21,706 | 50,865 | 95.9% | 87.0% | 48,780 | [33] |
| Oral cavity and lip (C00-06) | 113,502 | 264,211 | 377,713 | 7.4% | 68.8% | 27,951 | [34] |
| Nasopharynx (C11) | 36,983 | 96,371 | 133,354 | 7.9% | 75% | 10,535 | [34] |
| Oropharynx (C09-C10) | 19,367 | 79,045 | 98,412 | 24.9% | 83% | 24,505 | [34] |
| Hypopharynx (C12-C13) | 14,000 | 70,254 | 84,254 | 3.9% | 80 | 3286 | [34] |
| Larynx (C32) | 24,350 | 160,265 | 184,615 | 5.7% | 50.8% | 10,523 | [34] |
| Total | 904,636 | 727,920 | 1,548,302 | / | / | 751,468 | / |
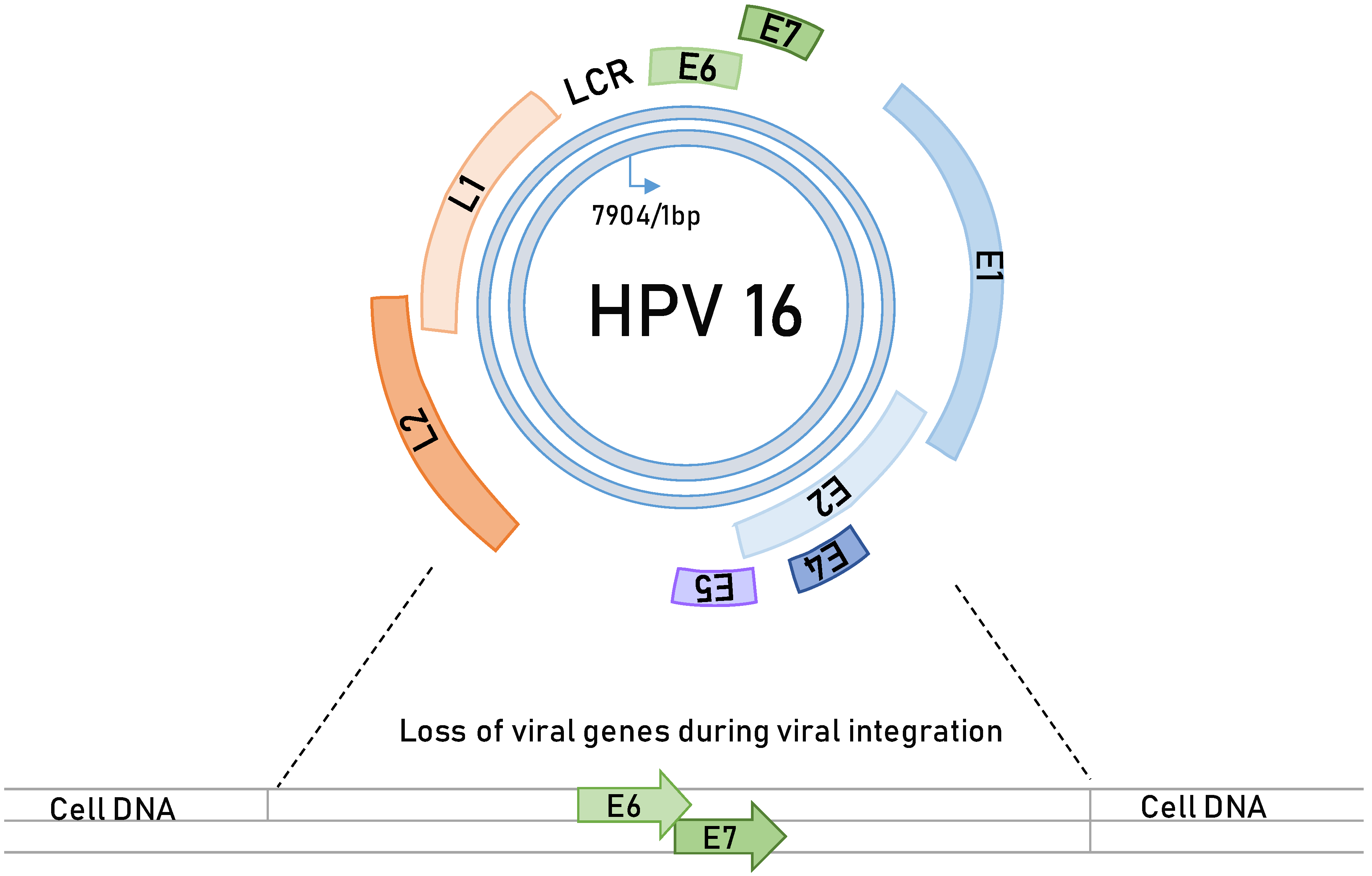
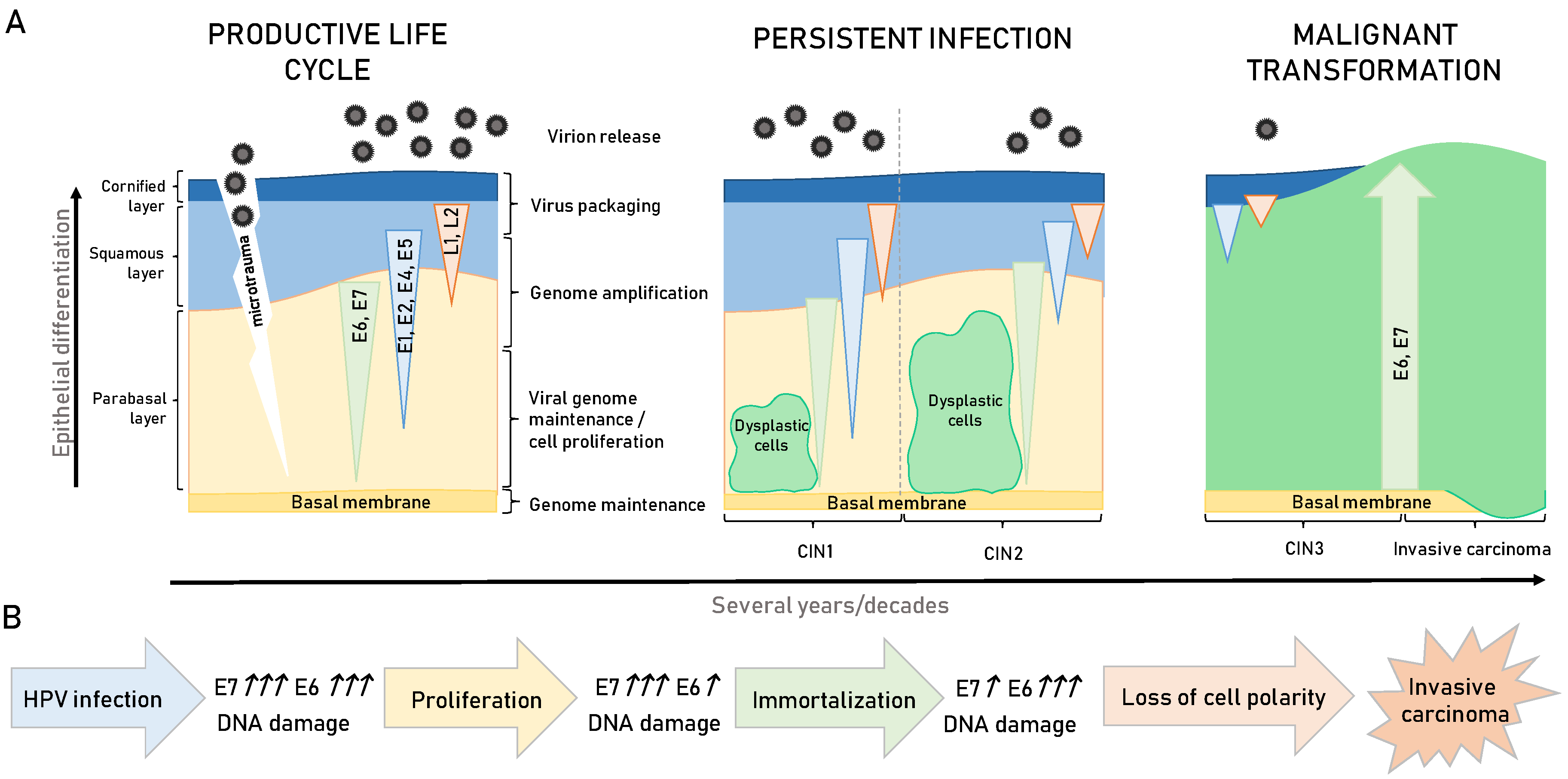
3. HPV-Induced Carcinogenesis
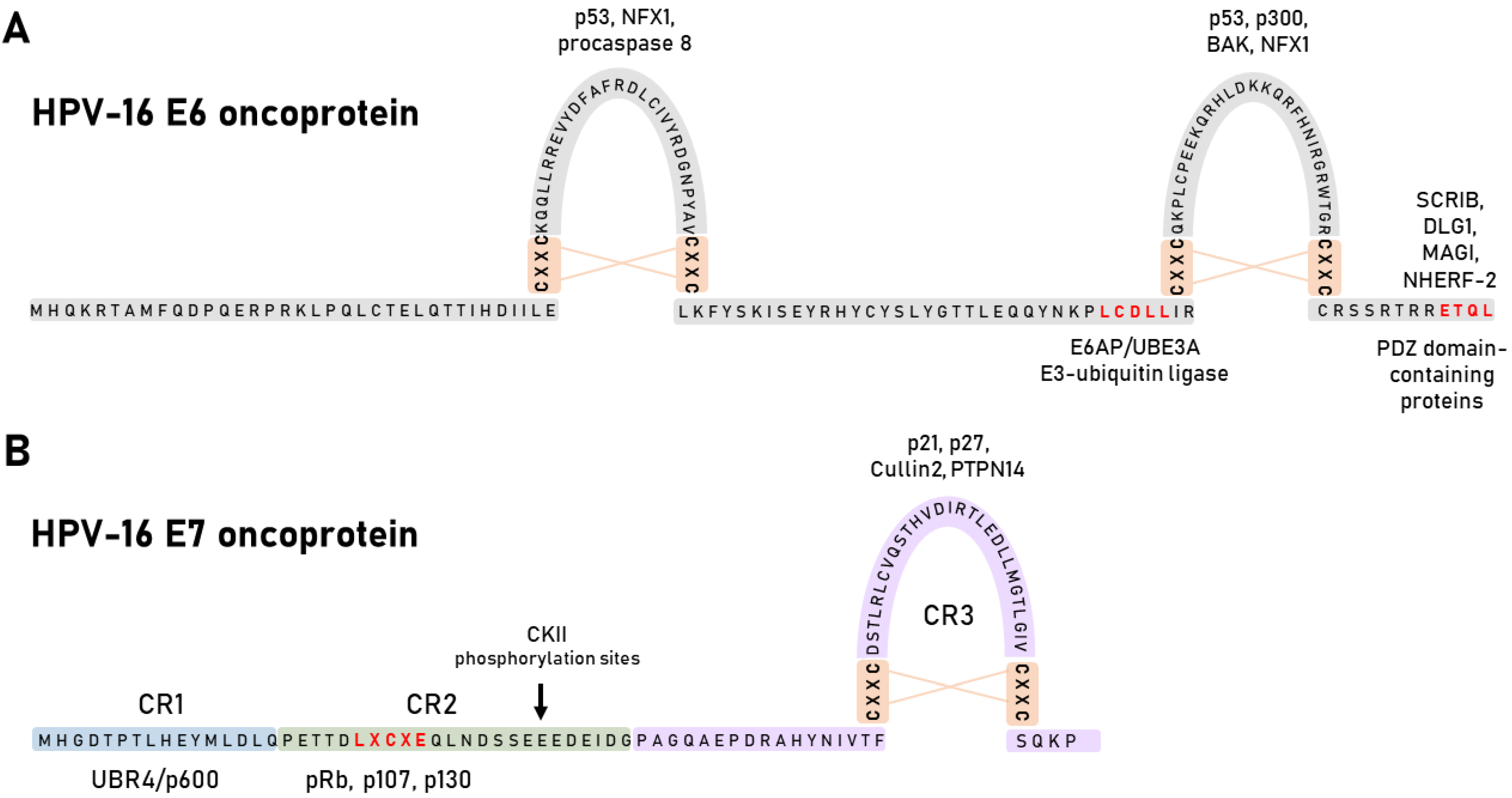
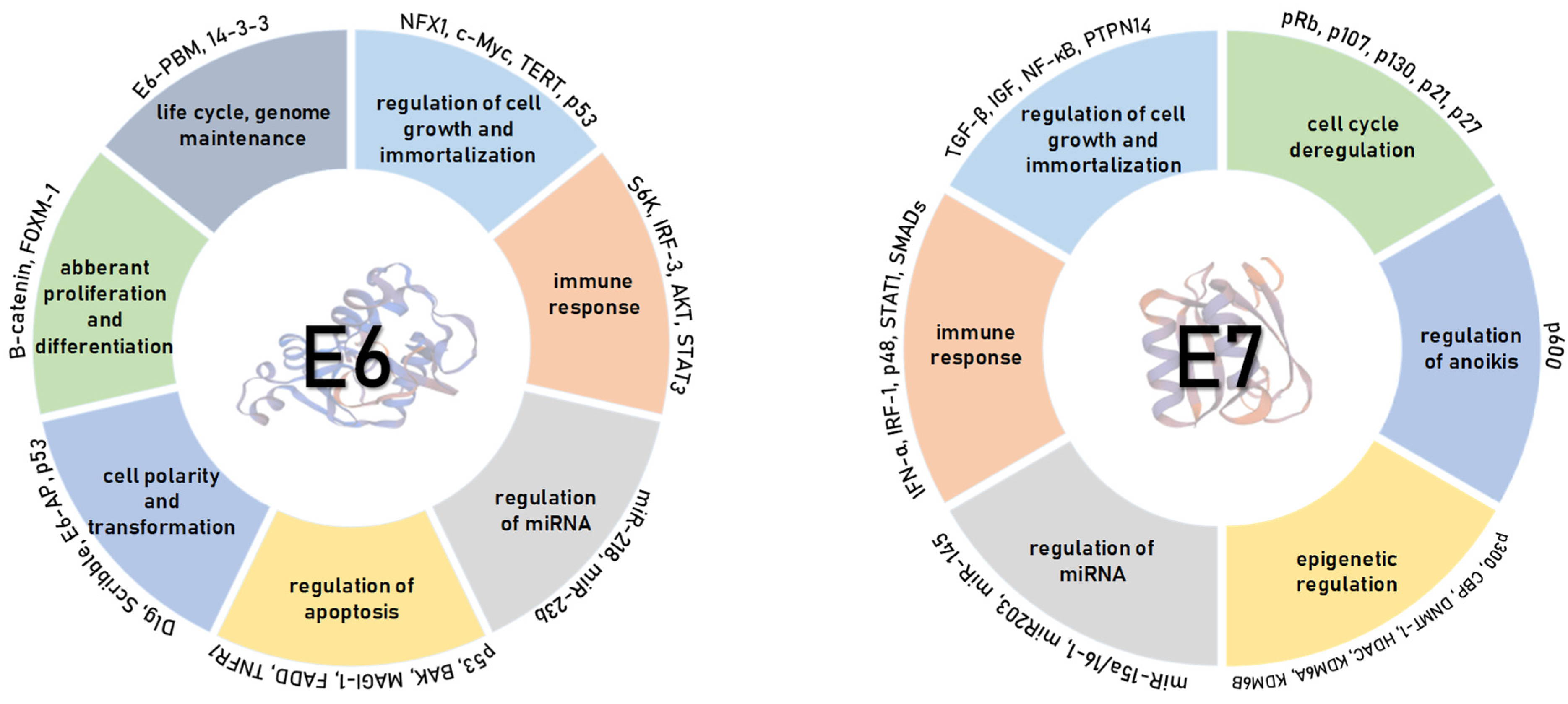
4. HPV Oncoproteins
4.1. HPV E6 Oncoprotein
4.2. HPV E7 Oncoprotein
5. HPV Biomarkers in Clinical Practice
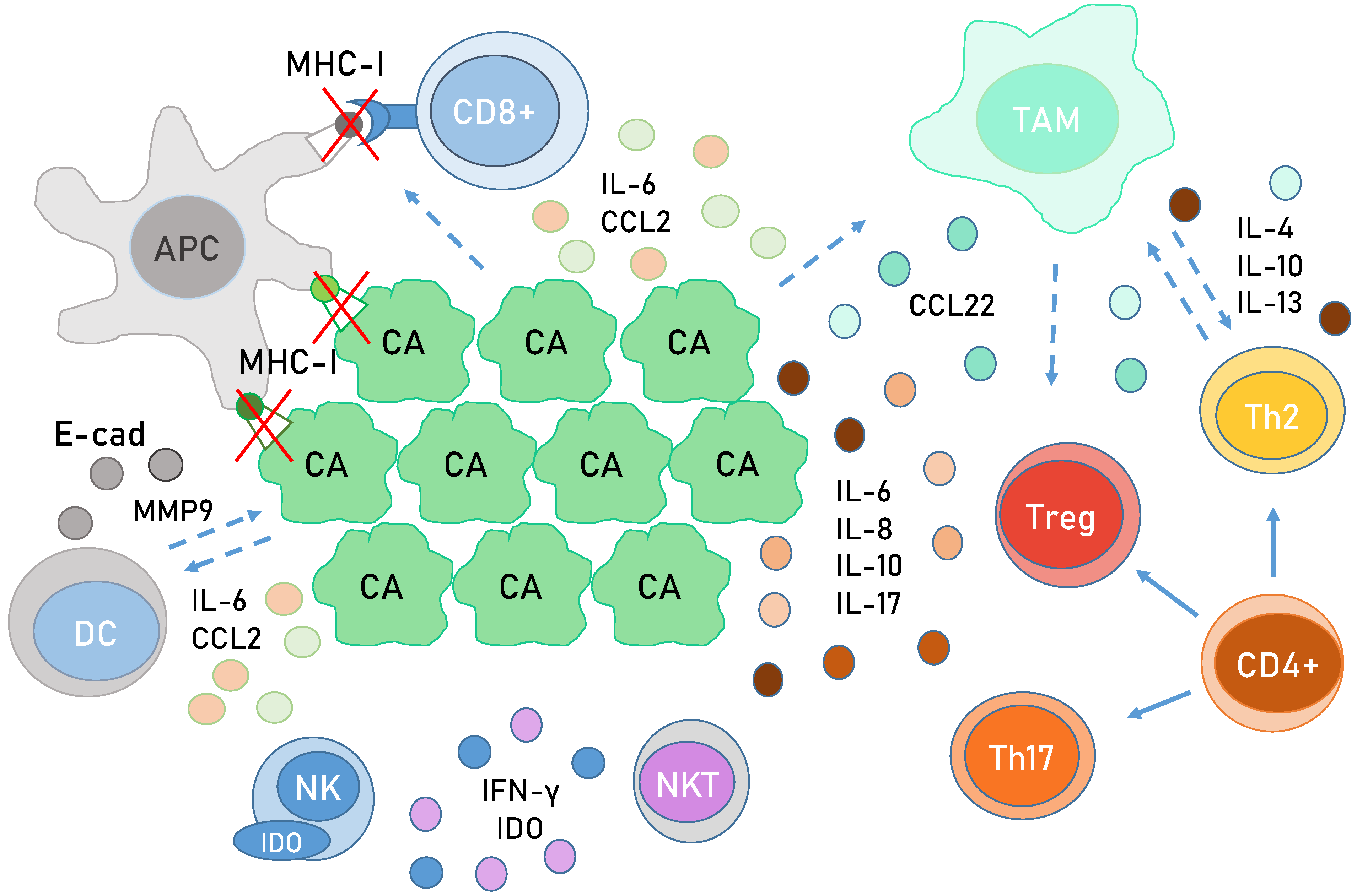
6. Immunology in HPV-Related Cancer
7. Epigenetic Changes in HPV-Associated Cancers
7.1. DNA Acetylation in HPV-Associated Cancers
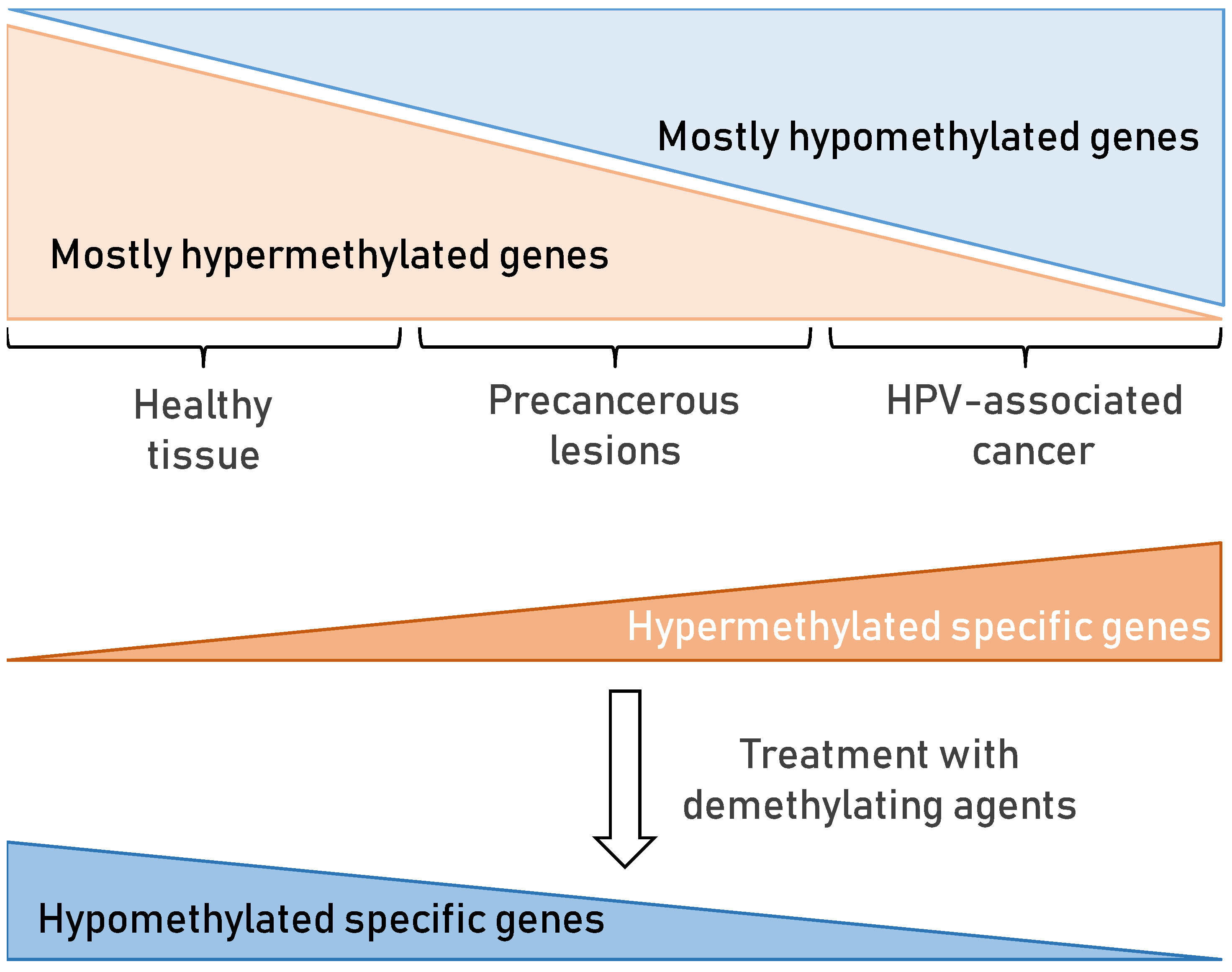
7.2. DNA Methylation in HPV-Associated Cancer
7.3. miRNA in HPV-Associated Cancer
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CC | cervical cancer |
| E | early |
| HNC | head and neck cancer |
| HNSCC | head and neck squamous cell carcinoma |
| HPV | human papillomavirus |
| HR | high-risk |
| LR | low-risk |
| SCC | squamous cell carcinoma |
| L | late |
| LCR | long control region |
References
- zur Hausen, H. Infections Causing Human Cancer; Wiley-VCH verlag GmbH &, Co. KGaA, Weinheim: Württemberg, Germany, 2006; ISBN 3-527-31056-8. [Google Scholar]
- Olson, C.; Cook, R.H. Cutaneous Sarcoma-like Lesions of the Horse Caused by the Agent of Bovine Papilloma. Proc. Soc. Exp. Biol. Med. 1951, 77, 281–284. [Google Scholar] [CrossRef]
- Jablonska, S.; Dabrowski, J.; Jakubowicz, K. Epidermodysplasia Verruciformis as a Model in Studies on the Role of Papovaviruses in Oncogenesis. Cancer Res. 1972, 32, 583–589. [Google Scholar]
- Orth, G.; Favre, M.; Jablonska, S.; Brylak, K.; Croissant, O. Viral Sequences Related to a Human Skin Papillomavirus in Genital Warts. Nature 1978, 275, 334–336. [Google Scholar] [CrossRef]
- zur Hausen, H. Oncogenic Herpes Viruses. Biochim. Et Biophys. Acta (Bba) -Rev. Cancer 1975, 417, 25–53. [Google Scholar] [CrossRef]
- zur Hausen, H. Papillomaviruses in Anogenital Cancer as a Model to Understand the Role of Viruses in Human Cancers. Cancer Res. 1989, 49, 4677–4681. [Google Scholar]
- Mühr, L.S.A.; Eklund, C.; Dillner, J. Towards Quality and Order in Human Papillomavirus Research. Virology 2018, 519, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.T.; Nardelli-Haefliger, D. Chapter 17: Second Generation HPV Vaccines to Prevent Cervical Cancer. Vaccine 2006, 24, S147–S153. [Google Scholar] [CrossRef]
- Technical Guidance on the Introduction of HPV Vaccines in European Union Countries—An Update. Available online: https://www.ecdc.europa.eu/en/publications-data/technical-guidance-introduction-hpv-vaccines-european-union-countries-update (accessed on 25 October 2021).
- Martínez-Gómez, X.; Curran, A.; Campins, M.; Alemany, L.; Rodrigo-Pendás, J.Á.; Borruel, N.; Castellsagué, X.; Díaz-de-Heredia, C.; Moraga-Llop, F.A.; del Pino, M.; et al. Multidisciplinary, Evidence-Based Consensus Guidelines for Human Papillomavirus (HPV) Vaccination in High-Risk Populations, Spain, 2016. Eurosurveillance 2019, 24, 1700857. [Google Scholar] [CrossRef]
- Brdar, B.; Grce, M. Human papillomavirus and assocition with genital tumours. In Onkogene and Growth Factors; Ikić, D., Pavelić, K., Spaventi, R., Eds.; JAZU Globus, Zagreb: Zagreb, Croatia, 1989; pp. 185–192. ISBN 86-343-0586-4. [Google Scholar]
- Grce, M.; Krusic, J.; Magdic, L.; Brdar, B. Detection of Human Papilloma Viruses Related to Carcinoma, Condyloma Acuminatum and Dysplasia of the Uterine Cervix. Period. Biol. 1991, 93, 165–168. [Google Scholar]
- Sabol, I.; Milutin Gašperov, N.; Matovina, M.; Božinović, K.; Grubišić, G.; Fistonić, I.; Belci, D.; Alemany, L.; Džebro, S.; Dominis, M.; et al. Cervical HPV Type-Specific Pre-Vaccination Prevalence and Age Distribution in Croatia. PLoS ONE 2017, 12, e0180480. [Google Scholar] [CrossRef] [PubMed]
- Matovina, M.; Husnjak, K.; Milutin, N.; Ciglar, S.; Grce, M. Possible Role of Bacterial and Viral Infections in Miscarriages. Fertil. Steril. 2004, 81, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Sabol, I.; Salakova, M.; Smahelova, J.; Pawlita, M.; Schmitt, M.; Milutin Gasperov, N.; Grce, M.; Tachezy, R. Evaluation of Different Techniques for Identification of Human Papillomavirus Types of Low Prevalence. J. Clin. Microbiol. 2008, 46, 1606–1613. [Google Scholar] [CrossRef][Green Version]
- Matovina, M.; Sabol, I.; Grubisić, G.; Gasperov, N.M.; Grce, M. Identification of Human Papillomavirus Type 16 Integration Sites in High-Grade Precancerous Cervical Lesions. Gynecol. Oncol 2009, 113, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Sabol, I.; Čretnik, M.; Hadžisejdić, I.; Si-Mohamed, A.; Matovina, M.; Grahovac, B.; Levanat, S.; Grce, M. A New Approach for the Evaluation of the Human Papillomavirus Type 16 Variability with High Resolution Melting Analysis. J. Virol. Methods 2009, 162, 142–147. [Google Scholar] [CrossRef][Green Version]
- Sabol, I.; Matovina, M.; Milutin Gasperov, N.; Grce, M. Identification of a Novel Human Papillomavirus Type 16 E1 Gene Variant with Potentially Reduced Oncogenicity. J. Med. Virol. 2008, 80, 2134–2140. [Google Scholar] [CrossRef]
- Sabol, I.; Matovina, M.; Si-Mohamed, A.; Grce, M. Characterization and Whole Genome Analysis of Human Papillomavirus Type 16 E1-1374∧63nt Variants. PLoS ONE 2012, 7, e41045. [Google Scholar] [CrossRef]
- Grce, M.; Husnjak, K.; Skerlev, M.; Lipozencić, J.; Pavelić, K. Detection and Typing of Human Papillomaviruses by Means of Polymerase Chain Reaction and Fragment Length Polymorphism in Male Genital Lesions. Anticancer Res. 2000, 20, 2097–2102. [Google Scholar]
- Mravak-Stipetić, M.; Sabol, I.; Kranjčić, J.; Knežević, M.; Grce, M. Human Papillomavirus in the Lesions of the Oral Mucosa According to Topography. PLoS ONE 2013, 8, e69736. [Google Scholar] [CrossRef] [PubMed]
- Sabol, I.; Smahelova, J.; Klozar, J.; Mravak-Stipetic, M.; Gheit, T.; Tommasino, M.; Grce, M.; Tachezy, R. Beta-HPV Types in Patients with Head and Neck Pathology and in Healthy Subjects. J. Clin. Virol. 2016, 82, 159–165. [Google Scholar] [CrossRef]
- Milutin Gašperov, N.; Sabol, I.; Planinić, P.; Grubišić, G.; Fistonić, I.; Ćorušić, A.; Grce, M. Methylated Host Cell Gene Promoters and Human Papillomavirus Type 16 and 18 Predicting Cervical Lesions and Cancer. PLoS ONE 2015, 10, e0129452. [Google Scholar] [CrossRef] [PubMed]
- Božinović, K.; Sabol, I.; Dediol, E.; Gašperov, N.M.; Manojlović, S.; Vojtechova, Z.; Tachezy, R.; Grce, M. Genome-Wide MiRNA Profiling Reinforces the Importance of MiR-9 in Human Papillomavirus Associated Oral and Oropharyngeal Head and Neck Cancer. Sci. Rep. 2019, 9, 2306. [Google Scholar] [CrossRef] [PubMed]
- Milutin Gašperov, N.; Sabol, I.; Božinović, K.; Dediol, E.; Mravak-Stipetić, M.; Licastro, D.; Dal Monego, S.; Grce, M. DNA Methylome Distinguishes Head and Neck Cancer from Potentially Malignant Oral Lesions and Healthy Oral Mucosa. Int. J. Mol. Sci. 2020, 21, 6853. [Google Scholar] [CrossRef] [PubMed]
- Saidu, N.E.B.; Filić, V.; Thomas, M.; Sarabia-Vega, V.; Đukić, A.; Miljković, F.; Banks, L.; Tomaić, V. PDZ Domain-Containing Protein NHERF-2 Is a Novel Target of Human Papillomavirus 16 (HPV-16) and HPV-18. J. Virol. 2019, 94, e00663-19. [Google Scholar] [CrossRef] [PubMed]
- Đukić, A.; Lulić, L.; Thomas, M.; Skelin, J.; Saidu, N.E.B.; Grce, M.; Banks, L.; Tomaić, V. HPV Oncoproteins and the Ubiquitin Proteasome System: A Signature of Malignancy? Pathogens 2020, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- IARC, W. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans Volume 90 Human Papillomaviruses; Iarc Press: Lyon, France, 2007; Volume 90, ISBN 978-92-832-1290-4. [Google Scholar]
- Božinović, K.; Sabol, I.; Rakušić, Z.; Jakovčević, A.; Šekerija, M.; Lukinović, J.; Prgomet, D.; Grce, M. HPV-Driven Oropharyngeal Squamous Cell Cancer in Croatia—Demography and Survival. PLoS ONE 2019, 14. [Google Scholar] [CrossRef]
- de Sanjosé, S.; Diaz, M.; Castellsagué, X.; Clifford, G.; Bruni, L.; Muñoz, N.; Bosch, F.X. Worldwide Prevalence and Genotype Distribution of Cervical Human Papillomavirus DNA in Women with Normal Cytology: A Meta-Analysis. Lancet Infect. Dis. 2007, 7, 453–459. [Google Scholar] [CrossRef]
- Global Strategy to Accelerate the Elimination of Cervical Cancer as a Public Health Problem. Available online: https://www.who.int/publications-detail-redirect/9789240014107 (accessed on 13 August 2021).
- de Sanjose, S.; Quint, W.G.; Alemany, L.; Geraets, D.T.; Klaustermeier, J.E.; Lloveras, B.; Tous, S.; Felix, A.; Bravo, L.E.; Shin, H.-R.; et al. Human Papillomavirus Genotype Attribution in Invasive Cervical Cancer: A Retrospective Cross-Sectional Worldwide Study. Lancet Oncol. 2010, 11, 1048–1056. [Google Scholar] [CrossRef]
- de Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide Burden of Cancer Attributable to HPV by Site, Country and HPV Type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef]
- Castellsagué, X.; Alemany, L.; Quer, M.; Halec, G.; Quirós, B.; Tous, S.; Clavero, O.; Alòs, L.; Biegner, T.; Szafarowski, T.; et al. HPV Involvement in Head and Neck Cancers: Comprehensive Assessment of Biomarkers in 3680 Patients. JNCI J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Tomaić, V. Functional Roles of E6 and E7 Oncoproteins in HPV-Induced Malignancies at Diverse Anatomical Sites. Cancers 2016, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- PaVE: Papilloma Virus Genome Database. Available online: https://pave.niaid.nih.gov/ (accessed on 25 October 2021).
- Combes, J.-D.; Chen, A.A.; Franceschi, S. Prevalence of Human Papillomavirus in Cancer of the Oropharynx by Gender. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2954–2958. [Google Scholar] [CrossRef]
- de Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of Papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef]
- Murakami, I.; Egawa, N.; Griffin, H.; Yin, W.; Kranjec, C.; Nakahara, T.; Kiyono, T.; Doorbar, J. Roles for E1-Independent Replication and E6-Mediated P53 Degradation during Low-Risk and High-Risk Human Papillomavirus Genome Maintenance. PLoS Pathog. 2019, 15, e1007755. [Google Scholar] [CrossRef]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The Biology and Life-Cycle of Human Papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Vats, A.; Trejo-Cerro, O.; Thomas, M.; Banks, L. Human Papillomavirus E6 and E7: What Remains? Tumour Virus Res. 2021, 11, 200213. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Bergant, M.; Boon, S.S.; Ganti, K.; Kranjec, C.; Massimi, P.; Subbaiah, V.K.; Thomas, M.; Tomaić, V.; Banks, L. Human Papillomaviruses and the Specificity of PDZ Domain Targeting. Febs J. 2012, 279, 3530–3537. [Google Scholar] [CrossRef] [PubMed]
- Manzo-Merino, J.; Thomas, M.; Fuentes-Gonzalez, A.M.; Lizano, M.; Banks, L. HPV E6 Oncoprotein as a Potential Therapeutic Target in HPV Related Cancers. Expert Opin. Ther. Targets 2013, 17, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Jabbar, S.F.; Park, S.; Schweizer, J.; Berard-Bergery, M.; Pitot, H.C.; Lee, D.; Lambert, P.F. Cervical Cancers Require the Continuous Expression of the Human Papillomavirus Type 16 E7 Oncoprotein Even in the Presence of the Viral E6 Oncoprotein. Cancer Res. 2012, 72, 4008–4016. [Google Scholar] [CrossRef]
- Hawley-Nelson, P.; Vousden, K.H.; Hubbert, N.L.; Lowy, D.R.; Schiller, J.T. HPV16 E6 and E7 Proteins Cooperate to Immortalize Human Foreskin Keratinocytes. Embo J. 1989, 8, 3905–3910. [Google Scholar] [CrossRef]
- Riley, R.R.; Duensing, S.; Brake, T.; Munger, K.; Lambert, P.F.; Arbeit, J.M. Dissection of Human Papillomavirus E6 and E7 Function in Transgenic Mouse Models of Cervical Carcinogenesis. Cancer Res. 2003, 63, 4862. [Google Scholar] [PubMed]
- Ganti, K.; Broniarczyk, J.; Manoubi, W.; Massimi, P.; Mittal, S.; Pim, D.; Szalmas, A.; Thatte, J.; Thomas, M.; Tomaić, V.; et al. The Human Papillomavirus E6 PDZ Binding Motif: From Life Cycle to Malignancy. Viruses 2015, 7, 3530–3551. [Google Scholar] [CrossRef]
- Kranjec, C.; Tomaić, V.; Massimi, P.; Nicolaides, L.; Doorbar, J.; Banks, L. The High-Risk HPV E6 Target Scribble (HScrib) Is Required for HPV E6 Expression in Cervical Tumour-Derived Cell Lines. Papillomavirus Res. 2016, 2, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Strati, K.; Lambert, P.F. Role of Rb-Dependent and Rb-Independent Functions of Papillomavirus E7 Oncogene in Head and Neck Cancer. Cancer Res. 2007, 67, 11585–11593. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.S.; Wettstein, F.O. Transcription of the Cottontail Rabbit Papillomavirus Early Region and Identification of Two E6 Polypeptides in COS-7 Cells. J. Virol. 1987, 61, 2938–2942. [Google Scholar] [CrossRef]
- Kanda, T.; Watanabe, S.; Zanma, S.; Sato, H.; Furuno, A.; Yoshiike, K. Human Papillomavirus Type 16 E6 Proteins with Glycine Substitution for Cysteine in the Metal-Binding Motif. Virology 1991, 185, 536–543. [Google Scholar] [CrossRef]
- Brimer, N.; Drews, C.M.; Vande Pol, S.B. Association of Papillomavirus E6 Proteins with Either MAML1 or E6AP Clusters E6 Proteins by Structure, Function, and Evolutionary Relatedness. PloS Pathog. 2017, 13, e1006781. [Google Scholar] [CrossRef]
- Thomas, M.; Narayan, N.; Pim, D.; Tomaić, V.; Massimi, P.; Nagasaka, K.; Kranjec, C.; Gammoh, N.; Banks, L. Human Papillomaviruses, Cervical Cancer and Cell Polarity. Oncogene 2008, 27, 7018–7030. [Google Scholar] [CrossRef] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. Localization of the E6-AP Regions That Direct Human Papillomavirus E6 Binding, Association with P53, and Ubiquitination of Associated Proteins. Mol. Cell. Biol. 1993, 13, 4918–4927. [Google Scholar] [CrossRef]
- Howie, H.L.; Katzenellenbogen, R.A.; Galloway, D.A. Papillomavirus E6 Proteins. Virology 2009, 384, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Brimer, N.; Lyons, C.; Vande Pol, S.B. Association of E6AP (UBE3A) with Human Papillomavirus Type 11 E6 Protein. Virology 2007, 358, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.L.; Keiger, K.E.; Lee, C.J.; Huibregtse, J.M. The Global Transcriptional Effects of the Human Papillomavirus E6 Protein in Cervical Carcinoma Cell Lines Are Mediated by the E6AP Ubiquitin Ligase. J. Virol. 2005, 79, 3737–3747. [Google Scholar] [CrossRef] [PubMed]
- Tomaić, V.; Pim, D.; Banks, L. The Stability of the Human Papillomavirus E6 Oncoprotein Is E6AP Dependent. Virology 2009, 393, 7–10. [Google Scholar] [CrossRef]
- Tomaic, V.; Pim, D.; Thomas, M.; Massimi, P.; Myers, M.P.; Banks, L. Regulation of the Human Papillomavirus Type 18 E6/E6AP Ubiquitin Ligase Complex by the HECT Domain-Containing Protein EDD. J. Virol. 2011, 85, 3120–3127. [Google Scholar] [CrossRef]
- Martínez-Noël, G.; Galligan, J.T.; Sowa, M.E.; Arndt, V.; Overton, T.M.; Harper, J.W.; Howley, P.M. Identification and Proteomic Analysis of Distinct UBE3A/E6AP Protein Complexes. Mol. Cell Biol. 2012, 32, 3095–3106. [Google Scholar] [CrossRef]
- Tomaić, V.; Ganti, K.; Pim, D.; Bauer, C.; Blattner, C.; Banks, L. Interaction of HPV E6 Oncoproteins with Specific Proteasomal Subunits. Virology 2013, 446, 389–396. [Google Scholar] [CrossRef]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP Complex Functions as a Ubiquitin-Protein Ligase in the Ubiquitination of P53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 Oncoprotein Encoded by Human Papillomavirus Types 16 and 18 Promotes the Degradation of P53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human Papillomavirus Oncoproteins: Pathways to Transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Lechner, M.S.; Laimins, L.A. Inhibition of P53 DNA Binding by Human Papillomavirus E6 Proteins. J. Virol. 1994, 68, 4262–4273. [Google Scholar] [CrossRef]
- Kumar, A.; Zhao, Y.; Meng, G.; Zeng, M.; Srinivasan, S.; Delmolino, L.M.; Gao, Q.; Dimri, G.; Weber, G.F.; Wazer, D.E.; et al. Human Papillomavirus Oncoprotein E6 Inactivates the Transcriptional Coactivator Human ADA3. Mol. Cell Biol. 2002, 22, 5801–5812. [Google Scholar] [CrossRef]
- Thomas, M.; Banks, L. Inhibition of Bak-Induced Apoptosis by HPV-18 E6. Oncogene 1998, 17, 2943–2954. [Google Scholar] [CrossRef] [PubMed]
- Snijders, P.J.; van Duin, M.; Walboomers, J.M.; Steenbergen, R.D.; Risse, E.K.; Helmerhorst, T.J.; Verheijen, R.H.; Meijer, C.J. Telomerase Activity Exclusively in Cervical Carcinomas and a Subset of Cervical Intraepithelial Neoplasia Grade III Lesions: Strong Association with Elevated Messenger RNA Levels of Its Catalytic Subunit and High-Risk Human Papillomavirus DNA. Cancer Res. 1998, 58, 3812–3818. [Google Scholar]
- Veldman, T.; Horikawa, I.; Barrett, J.C.; Schlegel, R. Transcriptional Activation of the Telomerase HTERT Gene by Human Papillomavirus Type 16 E6 Oncoprotein. J. Virol. 2001, 75, 4467–4472. [Google Scholar] [CrossRef] [PubMed]
- Boon, S.S.; Tomaić, V.; Thomas, M.; Roberts, S.; Banks, L. Cancer-causing human papillomavirus E6 proteins display major differences in the phospho-regulation of their PDZ interactions. J. Virol. 2015, 89, 1579–1586. [Google Scholar] [CrossRef]
- Boon, S.S.; Banks, L. High-Risk Human Papillomavirus E6 Oncoproteins Interact with 14-3-3ζ in a PDZ Binding Motif-Dependent Manner. J. Virol. 2013, 87, 1586–1595. [Google Scholar] [CrossRef]
- Muslin, A.J.; Tanner, J.W.; Allen, P.M.; Shaw, A.S. Interaction of 14-3-3 with Signaling Proteins Is Mediated by the Recognition of Phosphoserine. Cell 1996, 84, 889–897. [Google Scholar] [CrossRef]
- zur Hausen, H. Papillomaviruses and Cancer: From Basic Studies to Clinical Application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.; Munger, K. The Papillomavirus E7 Proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef]
- Phelps, W.C.; Munger, K.; Yee, C.L.; Barnes, J.A.; Howley, P.M. Structure-Function Analysis of the Human Papillomavirus Type 16 E7 Oncoprotein. J. Virol. 1992, 66, 2418–2427. [Google Scholar] [CrossRef] [PubMed]
- Clemens, K.E.; Brent, R.; Gyuris, J.; Munger, K. Dimerization of the Human Papillomavirus E7 Oncoprotein in Vivo. Virology 1995, 214, 289–293. [Google Scholar] [CrossRef][Green Version]
- Oh, S.T.; Longworth, M.S.; Laimins, L.A. Roles of the E6 and E7 Proteins in the Life Cycle of Low-Risk Human Papillomavirus Type 11. J. Virol. 2004, 78, 2620–2626. [Google Scholar] [CrossRef] [PubMed]
- Huh, K.; Zhou, X.; Hayakawa, H.; Cho, J.-Y.; Libermann, T.A.; Jin, J.; Wade Harper, J.; Munger, K. Human Papillomavirus Type 16 E7 Oncoprotein Associates with the Cullin 2 Ubiquitin Ligase Complex, Which Contributes to Degradation of the Retinoblastoma Tumor Suppressor. J. Virol. 2007, 81, 9737–9747. [Google Scholar] [CrossRef] [PubMed]
- Yeo-Teh, N.S.L.; Ito, Y.; Jha, S. High-Risk Human Papillomaviral Oncogenes E6 and E7 Target Key Cellular Pathways to Achieve Oncogenesis. Int. J. Mol. Sci. 2018, 19, 1706. [Google Scholar] [CrossRef] [PubMed]
- Durzynska, J.; Lesniewicz, K.; Poreba, E. Human Papillomaviruses in Epigenetic Regulations. Mutat. Res.-Rev. Mutat. Res. 2017, 772, 36–50. [Google Scholar] [CrossRef]
- Szalmás, A.; Tomaic, V.; Basukala, O.; Massimi, P.; Mittal, S.; Kónya, J.; Banks, L. The PTPN14 Tumor Suppressor Is a Degradation Target of Human. J. Virol. 2017, 91, e00057-17. [Google Scholar]
- Hatterschide, J.; Bohidar, A.E.; Grace, M.; Nulton, T.J.; Kim, H.W.; Windle, B.; Morgan, I.M.; Munger, K.; White, E.A. PTPN14 Degradation by High-Risk Human Papillomavirus E7 Limits Keratinocyte Differentiation and Contributes to HPV-Mediated Oncogenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 7033–7042. [Google Scholar] [CrossRef] [PubMed]
- Huh, K.-W.; DeMasi, J.; Ogawa, H.; Nakatani, Y.; Howley, P.M.; Munger, K. Association of the Human Papillomavirus Type 16 E7 Oncoprotein with the 600-KDa Retinoblastoma Protein-Associated Factor, P600. Proc. Natl. Acad. Sci. USA 2005, 102, 11492–11497. [Google Scholar] [CrossRef]
- Dang, F.; Nie, L.; Wei, W. Ubiquitin Signaling in Cell Cycle Control and Tumorigenesis. Cell Death Differ. 2021, 28, 427–438. [Google Scholar] [CrossRef]
- Jozefiak, A.; Larska, M.; Pomorska-Mol, M.; Ruszkowski, J.J. The IGF-1 Signaling Pathway in Viral Infections. Viruses 2021, 13, 1488. [Google Scholar] [CrossRef]
- Gammoh, N.; Grm, H.S.; Massimi, P.; Banks, L. Regulation of Human Papillomavirus Type 16 E7 Activity through Direct Protein Interaction with the E2 Transcriptional Activator. J. Virol. 2006, 80, 1787–1797. [Google Scholar] [CrossRef][Green Version]
- Songock, W.K.; Kim, S.; Bodily, J.M.; State, L. The Human Papillomavirus E7 Oncoprotein as a Regulator of Transcription. Virus Res. 2017, 231, 56–75. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Wang, S. E3 Ubiquitin Ligases and Human Papillomavirus-Induced Carcinogenesis. J. Int. Med. Res. 2014, 42, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Habig, M.; Smola, H.; Dole, V.S.; Derynck, R.; Pfister, H.; Smola-Hess, S. E7 Proteins from High- and Low-Risk Human Papillomaviruses Bind to TGF-β-Regulated Smad Proteins and Inhibit Their Transcriptional Activity. Arch. Virol. 2006, 151, 1961–1972. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M.; Münger, K.; Werness, B.A.; Phelps, W.C.; Schlegel, R. Molecular Mechanisms of Transformation by the Human Papillomaviruses. Princess Takamatsu Symp. 1989, 20, 199–206. [Google Scholar]
- Arbyn, M.; Sasieni, P.; Meijer, C.J.L.M.; Clavel, C.; Koliopoulos, G.; Dillner, J. Chapter 9: Clinical Applications of HPV Testing: A Summary of Meta-Analyses. Vaccine 2006, 24 (Suppl. 3), S78–S89. [Google Scholar] [CrossRef]
- Poljak, M.; Kocjan, B.J.; Oštrbenk, A.; Seme, K. Commercially Available Molecular Tests for Human Papillomaviruses (HPV): 2015 Update. J. Clin. Virol. 2016, 76, S3–S13. [Google Scholar] [CrossRef]
- Grce, M. Primary and Secondary Prevention of Cervical Cancer. Expert Rev. Mol. Diagn. 2009, 9, 851–857. [Google Scholar] [CrossRef]
- Cuzick, J.; Arbyn, M.; Sankaranarayanan, R.; Tsu, V.; Ronco, G.; Mayrand, M.-H.; Dillner, J.; Meijer, C.J.L.M. Overview of Human Papillomavirus-Based and Other Novel Options for Cervical Cancer Screening in Developed and Developing Countries. Vaccine 2008, 26 (Suppl. 10), K29–K41. [Google Scholar] [CrossRef]
- Grce, M.; Davies, P. Human Papillomavirus Testing for Primary Cervical Cancer Screening. Expert Rev. Mol. Diagn. 2008, 8, 599–605. [Google Scholar] [CrossRef]
- Ying, H.; Jing, F.; Fanghui, Z.; Youlin, Q.; Yali, H. High-Risk HPV Nucleic Acid Detection Kit–the Care HPV Test—A New Detection Method for Screening. Sci. Rep. 2014, 4, 4704. [Google Scholar] [CrossRef]
- Wang, M.B.; Liu, I.Y.; Gornbein, J.A.; Nguyen, C.T. HPV-Positive Oropharyngeal Carcinoma: A Systematic Review of Treatment and Prognosis. Otolaryngol. Head Neck Surg. 2015, 153, 758–769. [Google Scholar] [CrossRef]
- Burger, E.A.; Kornør, H.; Klemp, M.; Lauvrak, V.; Kristiansen, I.S. HPV MRNA Tests for the Detection of Cervical Intraepithelial Neoplasia: A Systematic Review. Gynecol. Oncol. 2011, 120, 430–438. [Google Scholar] [CrossRef]
- Macedo, A.C.L.; Gonçalves, J.C.N.; Bavaresco, D.V.; Grande, A.J.; Chiaramonte Silva, N.; Rosa, M.I. Accuracy of MRNA HPV Tests for Triage of Precursor Lesions and Cervical Cancer: A Systematic Review and Meta-Analysis. J. Oncol. 2019, 2019, 1–14. [Google Scholar] [CrossRef]
- Luhn, P.; Wentzensen, N. HPV-Based Tests for Cervical Cancer Screening and Management of Cervical Disease. Curr. Obs. Gynecol. Rep. 2013, 2, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Ferrera, A.; Valladares, W.; Cabrera, Y.; de la Luz Hernandez, M.; Darragh, T.; Baena, A.; Almonte, M.; Herrero, R. Performance of an HPV 16/18 E6 Oncoprotein Test for Detection of Cervical Precancer and Cancer. Int. J. Cancer 2019, 145, 2042–2050. [Google Scholar] [CrossRef] [PubMed]
- Chernesky, M.; Jang, D.; Schweizer, J.; Arias, M.; Doerwald-Munoz, L.; Gupta, M.; Jackson, B.; Archibald, S.; Young, J.; Lytwyn, A.; et al. HPV E6 Oncoproteins and Nucleic Acids in Neck Lymph Node Fine Needle Aspirates and Oral Samples from Patients with Oropharyngeal Squamous Cell Carcinoma. Papillomavirus Res. 2018, 6, 1–5. [Google Scholar] [CrossRef]
- Brown, C.A.; Bogers, J.; Sahebali, S.; Depuydt, C.E.; De Prins, F.; Malinowski, D.P. Role of Protein Biomarkers in the Detection of High-Grade Disease in Cervical Cancer Screening Programs. J. Oncol. 2012, 2012, e289315. [Google Scholar] [CrossRef] [PubMed]
- de Freitas, A.C.; Coimbra, E.C.; Leitão, M. da C.G. Molecular Targets of HPV Oncoproteins: Potential Biomarkers for Cervical Carcinogenesis. Biochim. Et Biophys. Acta (BBA) -Rev. Cancer 2014, 1845, 91–103. [Google Scholar] [CrossRef]
- Wentzensen, N.; Clarke, M.A.; Bremer, R.; Poitras, N.; Tokugawa, D.; Goldhoff, P.E.; Castle, P.E.; Schiffman, M.; Kingery, J.D.; Grewal, K.K.; et al. Clinical Evaluation of Human Papillomavirus Screening With P16/Ki-67 Dual Stain Triage in a Large Organized Cervical Cancer Screening Program. JAMA Intern. Med. 2019, 179, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.A.; Wentzensen, N.; Mirabello, L.; Ghosh, A.; Wacholder, S.; Harari, A.; Lorincz, A.; Schiffman, M.; Burk, R.D. Human Papillomavirus DNA Methylation as a Potential Biomarker for Cervical Cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 2125–2137. [Google Scholar] [CrossRef]
- Prigge, E.-S.; Toth, C.; Dyckhoff, G.; Wagner, S.; Müller, F.; Wittekindt, C.; Freier, K.; Plinkert, P.; Hoffmann, J.; Vinokurova, S.; et al. P16INK4a/Ki-67 Co-Expression Specifically Identifies Transformed Cells in the Head and Neck Region. Int. J. Cancer 2015, 136, 1589–1599. [Google Scholar] [CrossRef] [PubMed]
- Jenson, E.G.; Baker, M.; Paydarfar, J.A.; Gosselin, B.J.; Li, Z.; Black, C.C. MCM2/TOP2A (ProExC) Immunohistochemistry as a Predictive Marker in Head and Neck Mucosal Biopsies. Pathol. -Res. Pract. 2014, 210, 346–350. [Google Scholar] [CrossRef] [PubMed]
- IARC. IARC Handbooks of Cancer Prevention: Cervix Cancer Screening; IARC Press, International Agency for Research on Cancer: Lyon, France; World Health Organization: Lyon, France, 2005; ISBN 978-92-832-3010-6. [Google Scholar]
- Del Moral-Hernández, O.; Hernández-Sotelo, D.; del Carmen Alarcón-Romero, L.; Mendoza-Catalán, M.A.; Flores-Alfaro, E.; Castro-Coronel, Y.; Ortiz-Ortiz, J.; Leyva-Vázquez, M.A.; Ortuño-Pineda, C.; Castro-Mora, W.; et al. TOP2A/MCM2, P16INK4a, and Cyclin E1 Expression in Liquid-Based Cytology: A Biomarkers Panel for Progression Risk of Cervical Premalignant Lesions. BMC Cancer 2021, 21, 39. [Google Scholar] [CrossRef]
- Litjens, R.J.; Hopman, A.H.; van de Vijver, K.K.; Ramaekers, F.C.; Kruitwagen, R.F.; Kruse, A.-J. Molecular Biomarkers in Cervical Cancer Diagnosis: A Critical Appraisal. Expert Opin. Med. Diagn. 2013, 7, 365–377. [Google Scholar] [CrossRef]
- Iida, M.; Banno, K.; Yanokura, M.; Nakamura, K.; Adachi, M.; Nogami, Y.; Umene, K.; Masuda, K.; Kisu, I.; Iwata, T.; et al. Candidate Biomarkers for Cervical Cancer Treatment: Potential for Clinical Practice (Review). Mol. Clin. Oncol. 2014, 2, 647–655. [Google Scholar] [CrossRef]
- Balachandra, S.; Kusin, S.B.; Lee, R.; Blackwell, J.; Tiro, J.A.; Cowell, L.G.; Chiang, C.; Wu, S.; Varma, S.; Rivera, E.L.; et al. Blood-based Biomarkers of Human Papillomavirus–Associated Cancers: A Systematic Review and Meta-analysis. Cancer 2021, 127, 850–864. [Google Scholar] [CrossRef]
- Koslabova, E.; Hamsikova, E.; Salakova, M.; Klozar, J.; Foltynova, E.; Salkova, E.; Rotnaglova, E.; Ludvikova, V.; Tachezy, R. Markers of HPV Infection and Survival in Patients with Head and Neck Tumors: Markers of HPV Infection. Int. J. Cancer 2013, 133, 1832–1839. [Google Scholar] [CrossRef]
- Stanley, M.A. Epithelial Cell Responses to Infection with Human Papillomavirus. Clin. Microbiol. Rev. 2012, 25, 215–222. [Google Scholar] [CrossRef]
- Zhou, C.; Tuong, Z.K.; Frazer, I.H. Papillomavirus Immune Evasion Strategies Target the Infected Cell and the Local Immune System. Front. Oncol. 2019, 9, 682. [Google Scholar] [CrossRef]
- Ferreira, A.R.; Ramalho, A.C.; Marques, M.; Ribeiro, D. The Interplay between Antiviral Signalling and Carcinogenesis in Human Papillomavirus Infections. Cancers 2020, 12, 646. [Google Scholar] [CrossRef]
- Zhang, B.; Li, P.; Wang, E.; Brahmi, Z.; Dunn, K.W.; Blum, J.S.; Roman, A. The E5 Protein of Human Papillomavirus Type 16 Perturbs MHC Class II Antigen Maturation in Human Foreskin Keratinocytes Treated with Interferon-Gamma. Virology 2003, 310, 100–108. [Google Scholar] [CrossRef]
- Ashrafi, G.H.; Haghshenas, M.R.; Marchetti, B.; O’Brien, P.M.; Campo, M.S. E5 Protein of Human Papillomavirus Type 16 Selectively Downregulates Surface HLA Class I. Int. J. Cancer 2005, 113, 276–283. [Google Scholar] [CrossRef]
- Georgopoulos, N.T.; Proffitt, J.L.; Blair, G.E. Transcriptional Regulation of the Major Histocompatibility Complex (MHC) Class I Heavy Chain, TAP1 and LMP2 Genes by the Human Papillomavirus (HPV) Type 6b, 16 and 18 E7 Oncoproteins. Oncogene 2000, 19, 4930–4935. [Google Scholar] [CrossRef]
- Chandra, J.; Miao, Y.; Romoff, N.; Frazer, I.H. Epithelium Expressing the E7 Oncoprotein of HPV16 Attracts Immune-Modulatory Dendritic Cells to the Skin and Suppresses Their Antigen-Processing Capacity. PLoS ONE 2016, 11, e0152886. [Google Scholar] [CrossRef] [PubMed]
- Hasan, U.A.; Zannetti, C.; Parroche, P.; Goutagny, N.; Malfroy, M.; Roblot, G.; Carreira, C.; Hussain, I.; Müller, M.; Taylor-Papadimitriou, J.; et al. The Human Papillomavirus Type 16 E7 Oncoprotein Induces a Transcriptional Repressor Complex on the Toll-like Receptor 9 Promoter. J. Exp. Med. 2013, 210, 1369–1387. [Google Scholar] [CrossRef]
- Hopfner, K.P.; Hornung, V. Molecular Mechanisms and Cellular Functions of CGAS–STING Signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding Type I and III Interferon Signalling during Viral Infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, E.J.; Kwon, H.J.; Hwang, E.S.; Namkoong, S.E.; Um, S.J. Inactivation of Interferon Regulatory Factor-1 Tumor Suppressor Protein by HPV E7 Oncoprotein. Implication for the E7-Mediated Immune Evasion Mechanism in Cervical Carcinogenesis. J. Biol. Chem. 2000, 275, 6764–6769. [Google Scholar] [CrossRef]
- Um, S.-J.; Rhyu, J.-W.; Kim, E.-J.; Jeon, K.-C.; Hwang, E.-S.; Park, J.-S. Abrogation of IRF-1 Response by High-Risk HPV E7 Protein in Vivo. Cancer Lett. 2002, 179, 205–212. [Google Scholar] [CrossRef]
- Ronco, L.V.; Karpova, A.Y.; Vidal, M.; Howley, P.M. Human Papillomavirus 16 E6 Oncoprotein Binds to Interferon Regulatory Factor-3 and Inhibits Its Transcriptional Activity. Genes Dev. 1998, 12, 2061–2072. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Wu, C.; Zhang, Y.J. Interplay between Janus Kinase/Signal Transducer and Activator of Transcription Signaling Activated by Type I Interferons and Viral Antagonism. Front. Immunol. 2017, 8, 1–17. [Google Scholar] [CrossRef]
- Hong, S.; Mehta, K.P.; Laimins, L.A. Suppression of STAT-1 Expression by Human Papillomaviruses Is Necessary for Differentiation-Dependent Genome Amplification and Plasmid Maintenance. J. Virol. 2011, 85, 9486–9494. [Google Scholar] [CrossRef]
- Yi, Y.; Fang, Y.; Wu, K.; Liu, Y.; Zhang, W. Comprehensive Gene and Pathway Analysis of Cervical Cancer Progression. Oncol. Lett. 2020, 19, 3316–3332. [Google Scholar] [CrossRef]
- Sobti, R.C.; Singh, N.; Hussain, S.; Suri, V.; Bharadwaj, M.; Das, B.C. Deregulation of STAT-5 Isoforms in the Development of HPV-Mediated Cervical Carcinogenesis. J. Recept. Signal Transduct. 2010, 30, 178–188. [Google Scholar] [CrossRef]
- Morgan, E.L.; Macdonald, A. Autocrine STAT3 Activation in HPV Positive Cervical Cancer through a Virus-Driven Rac1—NFκB—IL-6 Signalling Axis. PloS Pathog. 2019, 15, e1007835. [Google Scholar] [CrossRef]
- Hong, S.; Laimins, L.A. The JAK-STAT Transcriptional Regulator, STAT-5, Activates the ATM DNA Damage Pathway to Induce HPV 31 Genome Amplification upon Epithelial Differentiation. PloS Pathog. 2013, 9, e1003295. [Google Scholar] [CrossRef]
- Morgan, E.L.; Wasson, C.W.; Hanson, L.; Kealy, D.; Pentland, I.; McGuire, V.; Scarpini, C.; Coleman, N.; Arthur, J.S.C.; Parish, J.L.; et al. STAT3 Activation by E6 Is Essential for the Differentiation-Dependent HPV18 Life Cycle. PloS Pathog. 2018, 14, e1006975. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Shishodia, G.; Mahata, S.; Hedau, S.; Pandey, A.; Bhambhani, S.; Batra, S.; Basir, S.F.; Das, B.C.; Bharti, A.C. Aberrant Expression and Constitutive Activation of STAT3 in Cervical Carcinogenesis: Implications in High-Risk Human Papillomavirus Infection. Mol. Cancer 2010, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Gaykalova, D.A.; Manola, J.B.; Ozawa, H.; Zizkova, V.; Morton, K.; Bishop, J.A.; Sharma, R.; Zhang, C.; Michailidi, C.; Considine, M.; et al. NF-ΚB and Stat3 Transcription Factor Signatures Differentiate HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinoma. Int. J. Cancer 2015, 137, 1879–1889. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; He, S.; Minassian, A.; Li, J.; Feng, P. Recent Advances on Viral Manipulation of NF-ΚB Signaling Pathway. Curr. Opin. Virol. 2015, 15, 103–111. [Google Scholar] [CrossRef]
- Nakahara, T.; Tanaka, K.; Ohno, S.; Egawa, N.; Yugawa, T.; Kiyono, T. Activation of NF-ΚB by Human Papillomavirus 16 E1 Limits E1-Dependent Viral Replication through Degradation of E1. J. Virol. 2015, 89, 5040–5059. [Google Scholar] [CrossRef]
- Karim, R.; Tummers, B.; Meyers, C.; Biryukov, J.L.; Alam, S.; Backendorf, C.; Jha, V.; Offringa, R.; van Ommen, G.-J.B.; Melief, C.J.M.; et al. Human Papillomavirus (HPV) Upregulates the Cellular Deubiquitinase UCHL1 to Suppress the Keratinocyte’s Innate Immune Response. PloS Pathog. 2013, 9, e1003384. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Degenkolbe, R.; Bernard, H.U.; O’Connor, M.J. The Human Papillomavirus Type 16 E6 Oncoprotein Can Down-Regulate P53 Activity by Targeting the Transcriptional Coactivator CBP/P300. J. Virol. 1999, 73, 6209–6219. [Google Scholar] [CrossRef]
- Da Costa, R.; Bastidos, M.; Medeiros, R.; Oliveira, P.A. The NFκB Signaling Pathway in Papillomavirus-Induced Lesions: Friend or Foe? Anticancer Res. 2016, 36, 2073–2083. [Google Scholar] [PubMed]
- Monisha, J.; Roy, N.K.; Bordoloi, D.; Kumar, A.; Golla, R.; Kotoky, J.; Padmavathi, G.; Kunnumakkara, A.B. Nuclear Factor Kappa B: A Potential Target to Persecute Head and Neck Cancer. Curr. Drug Targets 2017, 18, 232–253. [Google Scholar] [CrossRef]
- Branca, M.; Giorgi, C.; Ciotti, M.; Santini, D.; Di Bonito, L.; Costa, S.; Benedetto, A.; Bonifacio, D.; Di Bonito, P.; Paba, P.; et al. Upregulation of Nuclear Factor-ΚB (NF-ΚB) Is Related to the Grade of Cervical Intraepithelial Neoplasia, but Is Not an Independent Predictor of High-Risk Human Papillomavirus or Disease Outcome in Cervical Cancer. Diagn. Cytopathol. 2006, 34, 555–563. [Google Scholar] [CrossRef]
- Guess, J.C.; McCance, D.J. Decreased Migration of Langerhans Precursor-like Cells in Response to Human Keratinocytes Expressing Human Papillomavirus Type 16 E6/E7 Is Related to Reduced Macrophage Inflammatory Protein-3α Production. J. Virol. 2005, 79, 14852–14862. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, Z.J.; Jolly, C.; Androphy, E.J.; Mercer, A.; Matthews, C.M.; Hibma, M.H. Transcriptional Repression of E-Cadherin by Human Papillomavirus Type 16 E6. PLoS ONE 2012, 7, e48954. [Google Scholar] [CrossRef]
- Bashaw, A.A.; Leggatt, G.R.; Chandra, J.; Tuong, Z.K.; Frazer, I.H. Modulation of Antigen Presenting Cell Functions during Chronic HPV Infection. Papillomavirus Res. 2017, 4, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Artaza-Irigaray, C.; Molina-Pineda, A.; Aguilar-Lemarroy, A.; Ortiz-Lazareno, P.; Limón-Toledo, L.P.; Pereira-Suárez, A.L.; Rojo-Contreras, W.; Jave-Suárez, L.F. E6/E7 and E6* From HPV16 and HPV18 Upregulate IL-6 Expression Independently of P53 in Keratinocytes. Front. Immunol. 2019, 10, 1676. [Google Scholar] [CrossRef] [PubMed]
- Pahne-Zeppenfeld, J.; Schröer, N.; Walch-Rückheim, B.; Oldak, M.; Gorter, A.; Hegde, S.; Smola, S. Cervical Cancer Cell-Derived Interleukin-6 Impairs CCR7-Dependent Migration of MMP-9-Expressing Dendritic Cells. Int. J. Cancer 2014, 134, 2061–2073. [Google Scholar] [CrossRef]
- Walch-Rückheim, B.; Pahne-Zeppenfeld, J.; Fischbach, J.; Wickenhauser, C.; Horn, L.C.; Tharun, L.; Büttner, R.; Mallmann, P.; Stern, P.; Kim, Y.-J.; et al. STAT3/IRF1 Pathway Activation Sensitizes Cervical Cancer Cells to Chemotherapeutic Drugs. Cancer Res. 2016, 76, 3872–3883. [Google Scholar] [CrossRef] [PubMed]
- Litwin, T.R.; Irvin, S.R.; Chornock, R.L.; Sahasrabuddhe, V.V.; Stanley, M.; Wentzensen, N. Infiltrating T-Cell Markers in Cervical Carcinogenesis: A Systematic Review and Meta-Analysis. Br. J. Cancer 2021, 124, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Jung, A.C.; Guihard, S.; Krugell, S.; Ledrappier, S.; Brochot, A.; Dalstein, V.; Job, S.; de Reynies, A.; Noël, G.; Wasylyk, B.; et al. CD8-Alpha T-Cell Infiltration in Human Papillomavirus-Related Oropharyngeal Carcinoma Correlates with Improved Patient Prognosis. Int. J. Cancer 2013, 132, E26–E36. [Google Scholar] [CrossRef]
- Xue, J.; Wang, Y.; Chen, C.; Zhu, X.; Zhu, H.; Hu, Y. Effects of Th17 Cells and IL-17 in the Progression of Cervical Carcinogenesis with High-Risk Human Papillomavirus Infection. Cancer Med. 2018, 7, 297–306. [Google Scholar] [CrossRef]
- Dasgupta, S.; Bhattacharya-Chatterjee, M.; O’Malley, B.W.; Chatterjee, S.K. Inhibition of NK Cell Activity through TGF-Β1 by Down-Regulation of NKG2D in a Murine Model of Head and Neck Cancer. J. Immunol. 2005, 175, 5541–5550. [Google Scholar] [CrossRef]
- Garcia-Iglesias, T.; Del Toro-Arreola, A.; Albarran-Somoza, B.; Del Toro-Arreola, S.; Sanchez-Hernandez, P.E.; Ramirez-Dueñas, M.G.; Balderas-Peña, L.M.A.; Bravo-Cuellar, A.; Ortiz-Lazareno, P.C.; Daneri-Navarro, A. Low NKp30, NKp46 and NKG2D Expression and Reduced Cytotoxic Activity on NK Cells in Cervical Cancer and Precursor Lesions. BMC Cancer 2009, 9, 186. [Google Scholar] [CrossRef] [PubMed]
- Koromilas, A.E.; Li, S.; Matlashewski, G. Control of Interferon Signaling in Human Papillomavirus Infection. Cytokine Growth Factor Rev. 2001, 12, 157–170. [Google Scholar] [CrossRef]
- Sato, N.; Saga, Y.; Mizukami, H.; Wang, D.; Takahashi, S.; Nonaka, H.; Fujiwara, H.; Takei, Y.; Machida, S.; Takikawa, O.; et al. Downregulation of Indoleamine-2,3-Dioxygenase in Cervical Cancer Cells Suppresses Tumor Growth by Promoting Natural Killer Cell Accumulation. Oncol. Rep. 2012, 28, 1574–1578. [Google Scholar] [CrossRef]
- de Freitas, A.C.; de Oliveira, T.H.A.; Barros, M.R.J.; Venuti, A. HrHPV E5 Oncoprotein: Immune Evasion and Related Immunotherapies. J. Exp. Clin. Cancer Res. Cr 2017, 36, 71. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Yang, P.; Zhu, H.; Chen, X.; Xie, X.; Yang, M.; Liu, S.; Wang, H. Accumulation of Invariant NKT Cells with Increased IFN-γ Production in Persistent High-Risk HPV-Infected High-Grade Cervical Intraepithelial Neoplasia. Diagn. Pathol. 2015, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yung, M.M.H.; Ngan, H.Y.S.; Chan, K.K.L.; Chan, D.W. The Impact of the Tumor Microenvironment on Macrophage Polarization in Cancer Metastatic Progression. Int. J. Mol. Sci. 2021, 22, 6560. [Google Scholar] [CrossRef]
- Seminerio, I.; Kindt, N.; Descamps, G.; Bellier, J.; Lechien, J.R.; Mat, Q.; Pottier, C.; Journé, F.; Saussez, S. High Infiltration of CD68+ Macrophages Is Associated with Poor Prognoses of Head and Neck Squamous Cell Carcinoma Patients and Is Influenced by Human Papillomavirus. Oncotarget 2018, 9, 11046–11059. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, X.; Tan, H.; Lu, Y.; Tao, D.; Liu, Y.; Ma, Y. PIWIL2 Suppresses Siah2-Mediated Degradation of HDAC3 and Facilitates CK2α-Mediated HDAC3 Phosphorylation. Cell Death Dis. 2018, 9, 423. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Grunstein, M. Functions of Site-Specific Histone Acetylation and Deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef]
- Liu, S.; Chang, W.; Jin, Y.; Feng, C.; Wu, S.; He, J.; Xu, T. The Function of Histone Acetylation in Cervical Cancer Development. Biosci. Rep. 2019, 39, BSR20190527. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, N.S.; Moore, D.W.; Broker, T.R.; Chow, L.T. Vorinostat, a Pan-HDAC Inhibitor, Abrogates Productive HPV-18 DNA Amplification. Proc. Natl. Acad. Sci. USA 2018, 115, E11138–E11147. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Piao, L.; Bullock, B.N.; Smith, A.; Su, T.; Zhang, M.; Teknos, T.N.; Arora, P.S.; Pan, Q. Targeting HPV16 E6-P300 Interaction Reactivates P53 and Inhibits the Tumorigenicity of HPV-Positive Head and Neck Squamous Cell Carcinoma. Oncogene 2014, 33, 1037–1046. [Google Scholar] [CrossRef]
- Hong, S.; Dutta, A.; Laimins, L.A. The Acetyltransferase Tip60 Is a Critical Regulator of the Differentiation-Dependent Amplification of Human Papillomaviruses. J. Virol. 2015, 89, 4668–4675. [Google Scholar] [CrossRef] [PubMed]
- Holliday, R.; Pugh, J.E. DNA Modification Mechanisms and Gene Activity during Development. Science 1975, 187, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Sova, P.; Feng, Q.; Geiss, G.; Wood, T.; Strauss, R.; Rudolf, V.; Lieber, A.; Kiviat, N. Discovery of Novel Methylation Biomarkers in Cervical Carcinoma by Global Demethylation and Microarray Analysis. Cancer Epidemiol. Biomark. Prev. 2006, 15, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, A.T. Virtues and Weaknesses of DNA Methylation as a Test for Cervical Cancer Prevention. Acta Cytol. 2016, 60, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Verlaat, W.; Snijders, P.J.F.; Novianti, P.W.; Wilting, S.M.; De Strooper, L.M.A.; Trooskens, G.; Vandersmissen, J.; Van Criekinge, W.; Wisman, G.B.A.; Meijer, C.J.L.M.; et al. Genome-Wide DNA Methylation Profiling Reveals Methylation Markers Associated with 3q Gain for Detection of Cervical Precancer and Cancer. Clin. Cancer Res. 2017, 23, 3813–3822. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Ganguly, P.; Ganguly, N. Modulation of DNA Methylation by Human Papillomavirus E6 and E7 Oncoproteins in Cervical Cancer. Oncol. Lett. 2018, 15, 11–22. [Google Scholar] [CrossRef]
- Leonard, S.M.; Wei, W.; Collins, S.I.; Pereira, M.; Diyaf, A.; Constandinou-Williams, C.; Young, L.S.; Roberts, S.; Woodman, C.B. Oncogenic Human Papillomavirus Imposes an Instructive Pattern of DNA Methylation Changes Which Parallel the Natural History of Cervical HPV Infection in Young Women. Carcinogenesis 2012, 33, 1286–1293. [Google Scholar] [CrossRef]
- Yin, F.; Wang, N.; Wang, S.; Yu, F.; Sun, X.; Yu, X.; Luo, B.; Zhao, C.; Wang, Y. HPV16 Oncogenes E6 or/and E7 May Influence the Methylation Status of RASSFIA Gene Promoter Region in Cervical Cancer Cell Line HT-3. Oncol. Rep. 2017, 37, 2324–2334. [Google Scholar] [CrossRef]
- Jiao, X.; Zhang, S.; Jiao, J.; Zhang, T.; Qu, W.; Muloye, G.M.; Kong, B.; Zhang, Q.; Cui, B. Promoter Methylation of SEPT9 as a Potential Biomarker for Early Detection of Cervical Cancer and Its Overexpression Predicts Radioresistance. Clin. Epigenetics 2019, 11, 120. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, C.; Long, J.; Shen, D.; Zhou, W.; Zhou, Q.; Yang, J.; Jiang, M. E6 and E7 Gene Silencing Results in Decreased Methylation of Tumor Suppressor Genes and Induces Phenotype Transformation of Human Cervical Carcinoma Cell Lines. Oncotarget 2015, 6, 23930–23943. [Google Scholar] [CrossRef][Green Version]
- Farkas, S.A.; Milutin-Gašperov, N.; Grce, M.; Nilsson, T.K. Genome-Wide DNA Methylation Assay Reveals Novel Candidate Biomarker Genes in Cervical Cancer. Epigenetics 2013, 8, 1213–1225. [Google Scholar] [CrossRef]
- Milutin Gašperov, N.; Farkas, S.A.; Nilsson, T.K.; Grce, M. Epigenetic Activation of Immune Genes in Cervical Cancer. Immunol. Lett. 2014, 162, 256–257. [Google Scholar] [CrossRef]
- Ekanayake Weeramange, C.; Tang, K.D.; Vasani, S.; Langton-Lockton, J.; Kenny, L.; Punyadeera, C. DNA Methylation Changes in Human Papillomavirus-Driven Head and Neck Cancers. Cells 2020, 9, 1359. [Google Scholar] [CrossRef]
- Liu, G. CDH1 Promoter Methylation in Patients with Cervical Carcinoma: A Systematic Meta-Analysis with Trial Sequential Analysis. Future Oncol. 2017, 14, 51–63. [Google Scholar] [CrossRef]
- Strzelczyk, J.K.; Krakowczyk, Ł.; Owczarek, A.J. Aberrant DNA Methylation of the P16, APC, MGMT, TIMP3 and CDH1 Gene Promoters in Tumours and the Surgical Margins of Patients with Oral Cavity Cancer. J. Cancer 2018, 9, 1896–1904. [Google Scholar] [CrossRef]
- Wen, G.; Wang, H.; Zhong, Z. Associations of RASSF1A, RARβ, and CDH1 Promoter Hypermethylation with Oral Cancer Risk: A PRISMA-Compliant Meta-Analysis. Medicine 2018, 97, e9971. [Google Scholar] [CrossRef]
- Dong, S.M.; Sun, D.-I.; Benoit, N.E.; Kuzmin, I.; Lerman, M.I.; Sidransky, D. Epigenetic Inactivation of RASSF1A in Head and Neck Cancer. Clin. Cancer Res. 2003, 9, 3635–3640. [Google Scholar] [PubMed]
- Sartor, M.A.; Dolinoy, D.C.; Jones, T.R.; Colacino, J.A.; Prince, M.E.P.; Carey, T.E.; Rozek, L.S. Genome-Wide Methylation and Expression Differences in HPV(+) and HPV(-) Squamous Cell Carcinoma Cell Lines Are Consistent with Divergent Mechanisms of Carcinogenesis. Epigenetics 2011, 6, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Hansel, A.; Steinbach, D.; Greinke, C.; Schmitz, M.; Eiselt, J.; Scheungraber, C.; Gajda, M.; Hoyer, H.; Runnebaum, I.B.; Dürst, M. A Promising DNA Methylation Signature for the Triage of High-Risk Human Papillomavirus DNA-Positive Women. PLoS ONE 2014, 9, e91905. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Fenton, T.; West, J.; Wilson, G.; Feber, A.; Henderson, S.; Thirlwell, C.; Dibra, H.K.; Jay, A.; Butcher, L.; et al. Identification and Functional Validation of HPV-Mediated Hypermethylation in Head and Neck Squamous Cell Carcinoma. Genome Med. 2013, 5, 15. [Google Scholar] [CrossRef]
- Widschwendter, A.; Ivarsson, L.; Blassnig, A.; Müller, H.M.; Fiegl, H.; Wiedemair, A.; Müller-Holzner, E.; Goebel, G.; Marth, C.; Widschwendter, M. CDH1 and CDH13 Methylation in Serum Is an Independent Prognostic Marker in Cervical Cancer Patients. Int. J. Cancer 2004, 109, 163–166. [Google Scholar] [CrossRef]
- Hilska, M.; Roberts, P.J.; Collan, Y.U.; Laine, V.J.O.; Kössi, J.; Hirsimäki, P.; Rahkonen, O.; Laato, M. Prognostic Significance of Matrix Metalloproteinases-1, -2, -7 and -13 and Tissue Inhibitors of Metalloproteinases-1, -2, -3 and -4 in Colorectal Cancer. Int. J. Cancer 2007, 121, 714–723. [Google Scholar] [CrossRef]
- Dueñas-González, A.; Lizano, M.; Candelaria, M.; Cetina, L.; Arce, C.; Cervera, E. Epigenetics of Cervical Cancer. An Overview and Therapeutic Perspectives. Mol. Cancer 2005, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. Elegans Heterochronic Gene Lin-4 Encodes Small RNAs with Antisense Complementarity to Lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From MicroRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of MicroRNA Biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Gebert, L.F.R.; MacRae, I.J. Regulation of MicroRNA Function in Animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—MicroRNAs with a Role in Cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA Expression Profiles Classify Human Cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Waldman, S.A.; Terzic, A. Translating MicroRNA Discovery Into Clinical Biomarkers in Cancer. JAMA 2007, 297, 1923–1925. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.R. Introduction to Predictive Biomarkers: Definitions and Characteristics. In Predictive Biomarkers in Oncology: Applications in Precision Medicine; Badve, S., Kumar, G.L., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 3–18. ISBN 978-3-319-95228-4. [Google Scholar]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating MicroRNAs as Stable Blood-Based Markers for Cancer Detection. PNAS 2008, 105, 10513–10518. [Google Scholar] [CrossRef]
- Mall, C.; Rocke, D.M.; Durbin-Johnson, B.; Weiss, R.H. Stability of MiRNA in Human Urine Supports Its Biomarker Potential. Biomark Med. 2013, 7, 623–631. [Google Scholar] [CrossRef]
- Park, N.J.; Zhou, H.; Elashoff, D.; Henson, B.S.; Kastratovic, D.A.; Abemayor, E.; Wong, D.T. Salivary MicroRNA: Discovery, Characterization, and Clinical Utility for Oral Cancer Detection. Clin. Cancer Res. 2009, 15, 5473–5477. [Google Scholar] [CrossRef]
- Teplyuk, N.M.; Mollenhauer, B.; Gabriely, G.; Giese, A.; Kim, E.; Smolsky, M.; Kim, R.Y.; Saria, M.G.; Pastorino, S.; Kesari, S.; et al. MicroRNAs in Cerebrospinal Fluid Identify Glioblastoma and Metastatic Brain Cancers and Reflect Disease Activity. Neuro-oncology 2012, 14, 689–700. [Google Scholar] [CrossRef]
- Hall, J.S.; Taylor, J.; Valentine, H.R.; Irlam, J.J.; Eustace, A.; Hoskin, P.J.; Miller, C.J.; West, C.M.L. Enhanced Stability of MicroRNA Expression Facilitates Classification of FFPE Tumour Samples Exhibiting near Total MRNA Degradation. Br. J. Cancer 2012, 107, 684–694. [Google Scholar] [CrossRef]
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. MiRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells 2020, 9, 276. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, R.; Suleiman, S.; Pentimalli, F.; O’Toole, S.A.; O’Leary, J.J.; Ward, M.P.; Conlon, N.T.; Sabol, M.; Ozretić, P.; Erson-Bensan, A.E.; et al. Could MicroRNAs Be Useful Tools to Improve the Diagnosis and Treatment of Rare Gynecological Cancers? A Brief Overview. Int. J. Mol. Sci. 2021, 22, 3822. [Google Scholar] [CrossRef]
- Kang, J.-W.; Eun, Y.-G.; Lee, Y.-C. Diagnostic Value of Salivary MiRNA in Head and Neck Squamous Cell Cancer: Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2021, 22, 7026. [Google Scholar] [CrossRef]
- Stojanovic, J.; Tognetto, A.; Tiziano, D.F.; Leoncini, E.; Posteraro, B.; Pastorino, R.; Boccia, S. MicroRNAs Expression Profiles as Diagnostic Biomarkers of Gastric Cancer: A Systematic Literature Review. Biomarkers 2019, 24, 110–119. [Google Scholar] [CrossRef]
- Xue, J.; Jia, E.; Ren, N.; Lindsay, A.; Yu, H. Circulating MicroRNAs as Promising Diagnostic Biomarkers for Pancreatic Cancer: A Systematic Review. Onco. Targets 2019, 12, 6665–6684. [Google Scholar] [CrossRef]
- Zhong, S.; Golpon, H.; Zardo, P.; Borlak, J. MiRNAs in Lung Cancer. A Systematic Review Identifies Predictive and Prognostic MiRNA Candidates for Precision Medicine in Lung Cancer. Transl. Res. 2021, 230, 164–196. [Google Scholar] [CrossRef]
- Reshmi, G.; Pillai, M.R. Beyond HPV: Oncomirs as New Players in Cervical Cancer. Febs Lett. 2008, 582, 4113–4116. [Google Scholar] [CrossRef]
- Lui, W.-O.; Pourmand, N.; Patterson, B.K.; Fire, A. Patterns of Known and Novel Small RNAs in Human Cervical Cancer. Cancer Res. 2007, 67, 6031–6043. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.; McLean, T.; Zhang, X.; Zhao, C.J.; Thomson, J.M.; O’Brien, C.; Rose, B. MicroRNA Expression Profiles in Head and Neck Cancer Cell Lines. Biochem. Biophys. Res. Commun. 2007, 358, 12–17. [Google Scholar] [CrossRef]
- Jiang, Y.; Hu, Z.; Zuo, Z.; Li, Y.; Pu, F.; Wang, B.; Tang, Y.; Guo, Y.; Tao, H. Identification of Circulating MicroRNAs as a Promising Diagnostic Biomarker for Cervical Intraepithelial Neoplasia and Early Cancer: A Meta-Analysis. Biomed. Res. Int. 2020, 2020, e4947381. [Google Scholar] [CrossRef] [PubMed]
- Kumarasamy, C.; Madhav, M.R.; Sabarimurugan, S.; Krishnan, S.; Baxi, S.; Gupta, A.; Gothandam, K.M.; Jayaraj, R. Prognostic Value of MiRNAs in Head and Neck Cancers: A Comprehensive Systematic and Meta-Analysis. Cells 2019, 8, 772. [Google Scholar] [CrossRef]
- Binabaj, M.M.; Bahrami, A.; Khazaei, M.; Avan, A.; Ferns, G.A.; Soleimanpour, S.; Ryzhikov, M.; Hassanian, S.M. The Prognostic Value of Small Noncoding MicroRNA-21 Expression in the Survival of Cancer Patients: A Meta-Analysis. Crit Rev. Eukaryot Gene Expr. 2020, 30, 207–221. [Google Scholar] [CrossRef]
- Jamali, Z.; Asl Aminabadi, N.; Attaran, R.; Pournagiazar, F.; Ghertasi Oskouei, S.; Ahmadpour, F. MicroRNAs as Prognostic Molecular Signatures in Human Head and Neck Squamous Cell Carcinoma: A Systematic Review and Meta-Analysis. Oral Oncol. 2015, 51, 321–331. [Google Scholar] [CrossRef]






| Methods | Indicators | Commercial Tests | Clinical Applications |
|---|---|---|---|
| HR HPV DNA detection | Present or transient HR HPV infection | Hybrid Capture® 2 (HC2) HPV DNA Test (Qiagen, Hilden, Germany) Cervista HPV HR Test (Hologic, Marlborough, MA, USA) AMPLICOR HPV Test (Roche, Basel, Switzerland) careHPV™ Test (Qiagen, Hilden, Germany) | Triage of ASC-US or LSIL Follow-up women after treatment Primary screening test |
| HR HPV DNA genotyping | Determination of HR HPV type | INNO-LiPA® HPV Genotyping (Fujirebio Diagnostics, Gothenburg, Sweden) Linear Array HPV Genotyping Test (Roche, Basel, Switzerland) | Triage of HR-HPV positive women |
| HR HPV E6/E7 mRNA detection | Active HPV infection | PreTect HPV-Proofer (NorChip, Klokkarstua, Norway) NucliSENS EasyQ HPV (bioMérieux, Marcy-l’Étoile, France) Aptima® HPV Assay (Hologic, Marlborough, MA, USA) | Triage of LSIL |
| HPV proteins detection | Active HR HPV infection and progressive lesions | OncoE6™ (Arbor Vitae, Fremont, CA, USA) | Triage of HPV-positive women |
| Productive HPV infection | Cytoactive® HPV L1 screening set (Cytoimmun Diagnostics, Pirmasens, Germany) | Detecting CIN3 | |
| Detection of cellular proteins | Increased p16 and Ki-67 indicates cell proliferation | CINtec® PLUS cytology, p16/Ki-67 dual staining (Roche, Basel, Switzerland) | Screening tool for patients younger than 30 years of age; reduces unnecessary colposcopy, cervical biopsies, and treatment |
| Increased MCM2 and TOP2A indicates proliferative capacity | ProEx™ C, MCM2/TOP2a dual staining (BD Diagnostics, Franklin Lakes, NJ, USA) | Identification of cervical lesions that are more likely to progress Detection of CIN2+in ASC-US Distinguish true dysplasia from reactive/reparative changes, immature squamous metaplasia, and atrophy | |
| Increased CDC6, MCM5, and PCNA indicates abnormal cell proliferation beyond basal cell layers | No commercial test available, research use only | Discrimination between LSIL and HSIL Progression to cervical cancer | |
| Increased cyclin E indicates passage from G1 to S phase through phosphorylation of pRb and other targets | Discrimination between LSIL and HSIL Progression to cervical cancer | ||
| Decreased p53 indicates degradation through E6 mediated ubiquitination | Presence of active HR HPV infection | ||
| Overexpression of C-myc | Advanced cancer and poor survival | ||
| Increased telomerase indicates cell immortalization | Telomerase activity is correlated with the severity of the cervical abnormalities (biopsies and smears) |
| miRNA | CC | HNC | CC/HNC Similarity | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total Studies | Up/Down Regulated | Diagnostic Utility * | Prognostic Utility ** | Total Studies | Up/Down | ||||||
| N/ AUC/Sensitivity | AUC Max | Sensitivity AUC Max/Median | Specificity AUC Max/Median | N/Survival/Cox HR | Cox HR Min/Max | ||||||
| miR-21 | 38 | 38/0 | 7/6/2 | 0.97 | 88/85 | 98/85 | 6/4/0 | 34 | 34/0 | +/+ | |
| miR-145 | 18 | 2/16 | 3/2/2 | 0.85 | 89.1/85.0 | 91.7/77.4 | 3/2/2 | 1.6/2.6 | 15 | 1/13 | −/− |
| miR-143 | 17 | 2/15 | 2/1/1 | 0.94 | 72.4 | 80 | 3/0/0 | 13 | 2/11 | −/− | |
| miR-125b | 15 | 1/14 | 3/3/2 | 0.8 | 88/80 | 69/58 | 2/1/0 | 21 | 4/17 | −/− | |
| miR-195 | 14 | 1/13 | 3/2/1 | 0.86 | 88.9 | 78 | 0 | 11 | 2/9 | −/− | |
| miR-20a | 14 | 14/0 | 6/4/4 | 0.83 | 77.8/72.6 | 97.3/82.8 | 3/1/0 | 9 | 7/2 | +/+ | |
| miR-203a | 14 | 3/11 | 1/1/1 | 0.65 | 65 | 62.5 | 2/1/0 | 9 | 3/6 | −/− | |
| miR-34a | 14 | 5/9 | 4/3/2 | 0.95 | 83.3/80.6 | 87.5/80.1 | 2/2/2 | 0.2/0.2 | 10 | 6/4 | −/+ |
| miR-155 | 14 | 13/1 | 3/3/3 | 0.98 | 95/86.7 | 96/91.7 | 2/2/1 | 2.3 | 18 | 15/3 | +/+ |
| miR-218 | 13 | 0/13 | 2/2/1 | 0.84 | 67.7 | 60 | 2/2/1 | 0.2 | 8 | 0/8 | −/− |
| miR-9 | 13 | 12/1 | 3/2/2 | 0.8 | 67/62.5 | 94/87 | 2/2/1 | 2.7 | 15 | 14/1 | +/+ |
| miR-100 | 12 | 1/11 | 1/1/0 | 0.88 | 2/1/0 | 20 | 3/17 | −/− | |||
| miR-205 | 12 | 11/1 | 4/4/4 | 0.84 | 76.5/71 | 94/86.5 | 2/2/2 | 3/3.1 | 12 | 10/2 | +/+ |
| miR-200a | 11 | 9/2 | 2/2/1 | 0.66 | 60 | 62.5 | 2/2/0 | 11 | 10/1 | +/+ | |
| miR-141 | 11 | 9/2 | 1/1/1 | 0.94 | 82.8 | 91.7 | 1/1/0 | 8 | 7/1 | +/+ | |
| miR-17 | 11 | 9/2 | 0 | 0 | 11 | 8/3 | +/+ | ||||
| miR-29a | 10 | 2/8 | 3/3/1 | 0.89 | 92.6 | 80.7 | 0 | 10 | 1/8 | −/− | |
| miR-210 | 9 | 6/2 | 2/2/2 | 0.97 | 91.6/86.3 | 86.8/73.8 | 3/2/0 | 12 | 11/1 | +/+ | |
| miR-20b | 8 | 8/0 | 1/1/1 | 0.94 | 83 | 93 | 0 | 6 | 5/1 | +/+ | |
| miR-126 | 8 | 2/6 | 2/1/1 | 0.91 | 86 | 91 | 1/1/1 | 4 | 17 | 4/13 | −/− |
| miR-15b | 8 | 8/0 | 2/2/1 | 0.82 | 62 | 56 | 0 | 9 | 8/1 | +/+ | |
| miR-224 | 7 | 7/0 | 1/1/1 | 0.89 | 81 | 93 | 3/2/2 | 4.4/6.8 | 11 | 8/3 | +/+ |
| miR-92a | 7 | 7/0 | 3/3/3 | 0.94 | 94/69 | 87/80 | 1/1/0 | 7 | 7/0 | +/+ | |
| miR-497 | 7 | 1/6 | 1/1/0 | 0.64 | 1/1/1 | 2 | 6 | 0/5 | −/− | ||
| miR-424 | 7 | 1/6 | 1/1/1 | 0.98 | 100 | 90.9 | 1/1/0 | 11 | 9/2 | −/+ | |
| miR-93 | 7 | 7/0 | 0 | 1/1/1 | 3.7 | 13 | 11/2 | +/+ | |||
| miR-106a | 7 | 7/0 | 0 | 1/1/1 | 4 | 5 | 3/2 | +/+ | |||
| miR-206 | 6 | 2/4 | 0 | 2/2/1 | 9.1 | 7 | 2/5 | −/− | |||
| miR-192 | 6 | 5/1 | 3/3/2 | 0.95 | 91.7/83.4 | 100/97 | 0/1/0 | 2 | 1/1 | +/0 | |
| miR-486 | 5 | 4/1 | 3/3/0 | 0.9 | 0 | 12 | 4/7 | +/− | |||
| miR-34b | 5 | 2/3 | 1/1/1 | 1 | 100 | 100/ | 1/1/0 | 8 | 8/0 | −/+ | |
| miR-127 | 5 | 3/2 | 1/1/1 | 0.82 | 75 | 83 | 0 | 8 | 1/7 | +/− | |
| miR-136 | 5 | 1/4 | 1/1/1 | 0.81 | 70.8 | 90.9 | 0/1/0 | 5 | 3/2 | −/+ | |
| miR-142 | 5 | 4/1 | 1/0/0 | 1/1/1 | 3.2 | 12 | 9/3 | +/+ | |||
| miR-204 | 5 | 0/5 | 1/1/1 | 0.88 | 80.3 | 90.9 | 0/1/0 | 9 | 1/8 | −/− | |
| miR-1246 | 5 | 4/1 | 2/1/1 | 0.88 | 86 | 75 | 1/0/0 | 2 | 2/0 | +/+ | |
| miR-335 | 4 | 1/3 | 0 | 1/1/1 | 0.3 | 3 | 3/0 | −/+ | |||
| miR-425 | 4 | 4/0 | 0 | 1/1/1 | 2.4 | 5 | 4/1 | +/+ | |||
| miR-363 | 4 | 4/0 | 0 | 1/1/1 | 0.1 | 8 | 6/2 | +/+ | |||
| miR-455 | 4 | 0/4 | 1/1/1 | 0.84 | 78 | 81.3 | 1/1/0 | 13 | 12/1 | −/+ | |
| miR-181a | 4 | 2/2 | 0 | 1/1/1 | 2.4 | 11 | 6/5 | 0/+ | |||
| miR-26b | 4 | 3/1 | 2/0/0 | 1/1/1 | 2.6 | 14 | 2/12 | +/− | |||
| miR-101 | 4 | 2/2 | 0 | 1/1/1 | 2.8 | 14 | 6/7 | 0/− | |||
| miR-196a | 4 | 4/0 | 1/0/0 | 1/1/1 | 3.5 | 14 | 11/3 | +/+ | |||
| miR-215 | 3 | 2/1 | 0 | 1/1/1 | 2 | 4 | 3/1 | +/+ | |||
| miR-638 | 3 | 0/3 | 1/1/1 | 0.73 | 85 | 46 | 2/1/1 | 2.9 | 0 | ||
| let-7d | 3 | 2/1 | 1/1/0 | 0.82 | 0 | 7 | 3/4 | +/− | |||
| miR-144 | 3 | 2/1 | 1/1/1 | 0.95 | 89 | 93 | 1/0/0 | 4 | 3/1 | +/+ | |
| miR-494 | 3 | 1/2 | 1/1/1 | 0.91 | 91.7 | 90.9 | 1/1/0 | 4 | 1/3 | −/− | |
| miR-194 | 3 | 3/0 | 2/2/1 | 0.94 | 95.8 | 81.8 | 0/1/0 | 3 | 1/2 | +/− | |
| miR-181b | 3 | 1/2 | 0 | 1/1/1 | 2.4 | 9 | 5/4 | −/+ | |||
| miR-370 | 3 | 1/2 | 1/1/0 | 0.82 | 1/0/0 | 5 | 1/4 | −/− | |||
| miR-152 | 3 | 2/1 | 1/1/0 | 0.93 | 0 | 2 | 2/0 | +/+ | |||
| miR-411 | 3 | 0/3 | 0 | 2/1/1 | 0.4 | 5 | 0/5 | −/− | |||
| miR-135a | 2 | 2/0 | 1/1/1 | 0.83 | 70.8 | 91.8 | 0/1/0 | 3 | 1/2 | +/− | |
| miR-329 | 2 | 1/1 | 0 | 1/1/1 | 2.8 | 0 | |||||
| miR-664 | 2 | 1/1 | 0 | 1/1/1 | 4.2 | 1 | 0/1 | 0/− | |||
| miR-299 | 2 | 0/2 | 1/1/1 | 0.98 | 91.6 | 90.9 | 0/1/0 | 5 | 0/5 | −/− | |
| miR-22 | 2 | 1/1 | 0 | 1/1/1 | 1.8 | 3 | 2/1 | 0/+ | |||
| miR-362 | 2 | 0/2 | 0 | 2/2/2 | 0.4/0.5 | 1 | 0/1 | −/− | |||
| miR-3162 | 1 | 1/0 | 1/1/1 | 0.87 | 79 | 50 | 0 | 0 | |||
| miR-449a | 1 | 0/1 | 0 | 1/1/1 | 2.3 | 0 | |||||
| miR-4484 | 1 | 1/0 | 1/1/1 | 0.81 | 72 | 75 | 0 | 0 | |||
| miR-2392 | 1 | 1/0 | 1/1/1 | 0.94 | 59 | 85 | 0 | 0 | |||
| miR-1254 | 1 | 0/1 | 0 | 1/1/1 | 2.9 | 0 | |||||
| miR-766 | 1 | 1/0 | 1/1/1 | 0.85 | 79.1 | 82.1 | 1/0/0 | 3 | 3/0 | +/+ | |
| miR-503 | 1 | 0/1 | 0 | 1/1/1 | 2.8 | 9 | 7/2 | −/+ | |||
| miR-451a | 1 | 1/0 | 1/1/1 | 0.96 | 91 | 95 | 0 | 10 | 2/7 | +/− | |
| miR-1297 | 1 | 0/1 | 0 | 1/1/1 | 3.8 | 0 | |||||
| miR-153 | 1 | 0/1 | 0 | 1/1/1 | 2.1 | 0 | |||||
| miR-994 | 1 | 1/0 | 0 | 1/1/1 | 4 | 0 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pešut, E.; Đukić, A.; Lulić, L.; Skelin, J.; Šimić, I.; Milutin Gašperov, N.; Tomaić, V.; Sabol, I.; Grce, M. Human Papillomaviruses-Associated Cancers: An Update of Current Knowledge. Viruses 2021, 13, 2234. https://doi.org/10.3390/v13112234
Pešut E, Đukić A, Lulić L, Skelin J, Šimić I, Milutin Gašperov N, Tomaić V, Sabol I, Grce M. Human Papillomaviruses-Associated Cancers: An Update of Current Knowledge. Viruses. 2021; 13(11):2234. https://doi.org/10.3390/v13112234
Chicago/Turabian StylePešut, Ena, Anamaria Đukić, Lucija Lulić, Josipa Skelin, Ivana Šimić, Nina Milutin Gašperov, Vjekoslav Tomaić, Ivan Sabol, and Magdalena Grce. 2021. "Human Papillomaviruses-Associated Cancers: An Update of Current Knowledge" Viruses 13, no. 11: 2234. https://doi.org/10.3390/v13112234
APA StylePešut, E., Đukić, A., Lulić, L., Skelin, J., Šimić, I., Milutin Gašperov, N., Tomaić, V., Sabol, I., & Grce, M. (2021). Human Papillomaviruses-Associated Cancers: An Update of Current Knowledge. Viruses, 13(11), 2234. https://doi.org/10.3390/v13112234