
Use of Slaughterhouses as Sentinel Points for Genomic Surveillance of Foot-and-Mouth Disease Virus in Southern Vietnam

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
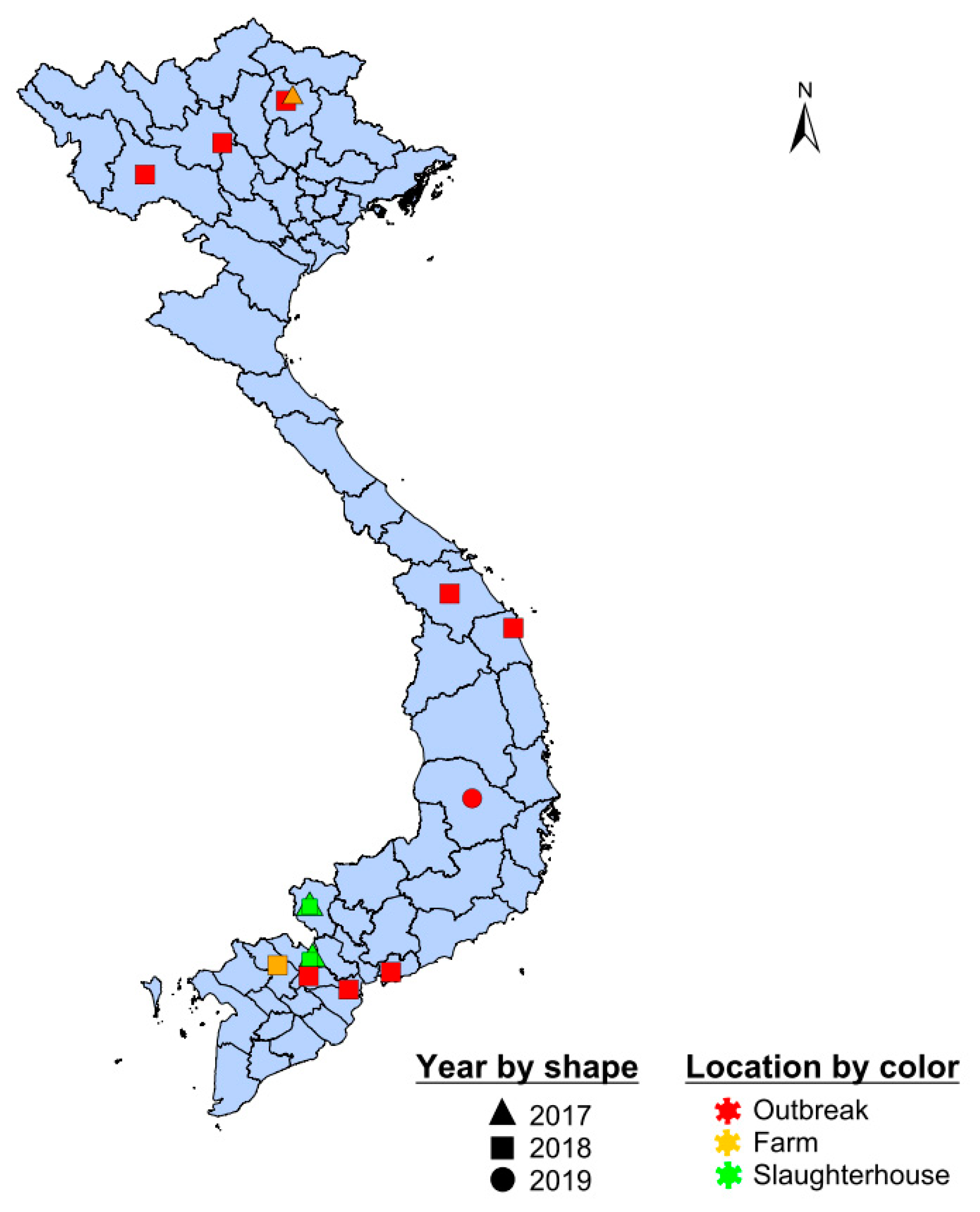
2.1. Study Populations and Sampling Design
2.1.1. Farm-Based Sampling
2.1.2. Slaughterhouse-Based Sampling
2.2. Outbreak Virus Sequences
2.3. Laboratory Analysis
2.4. Analysis of Diagnostic Data
2.5. Phylogenetic Analysis
2.5.1. Identification of Circulating Clusters
2.5.2. Time-Scaled Phylogenies
3. Results
3.1. Descriptive Data (Sample Screening)
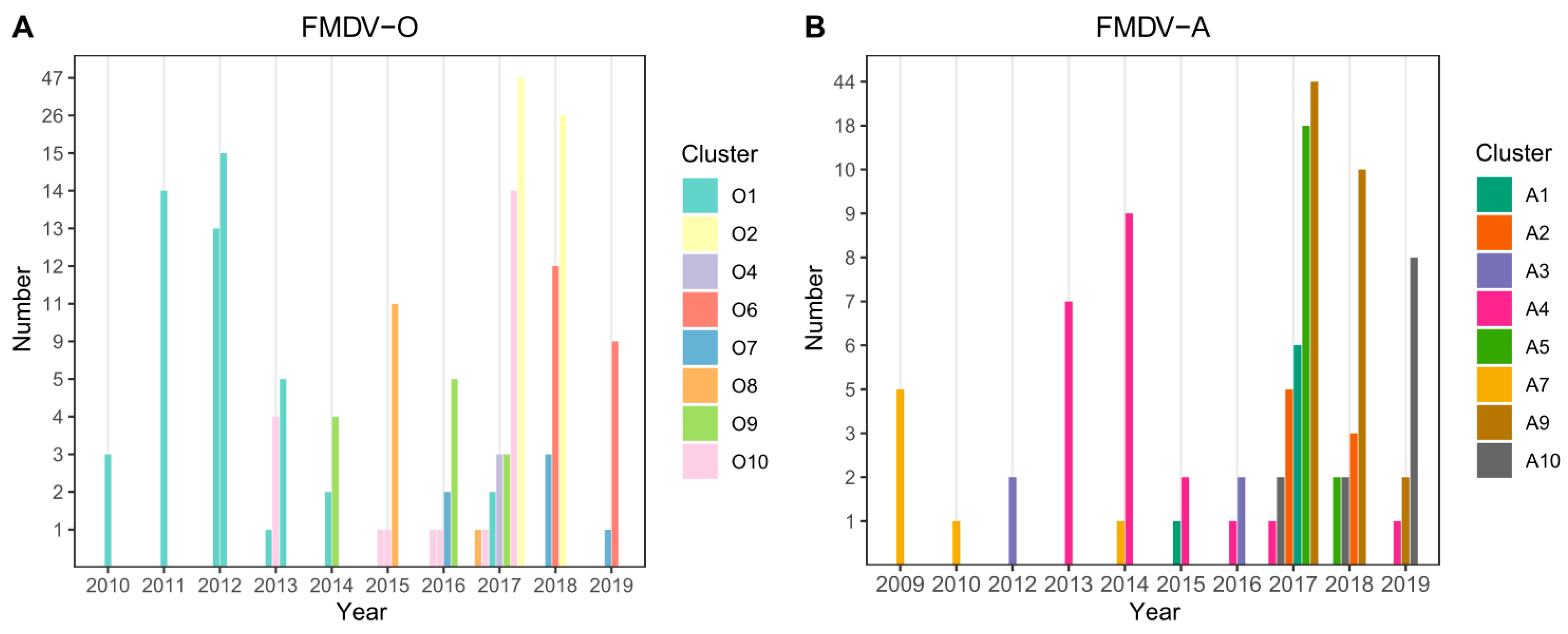
3.2. Cluster Analysis
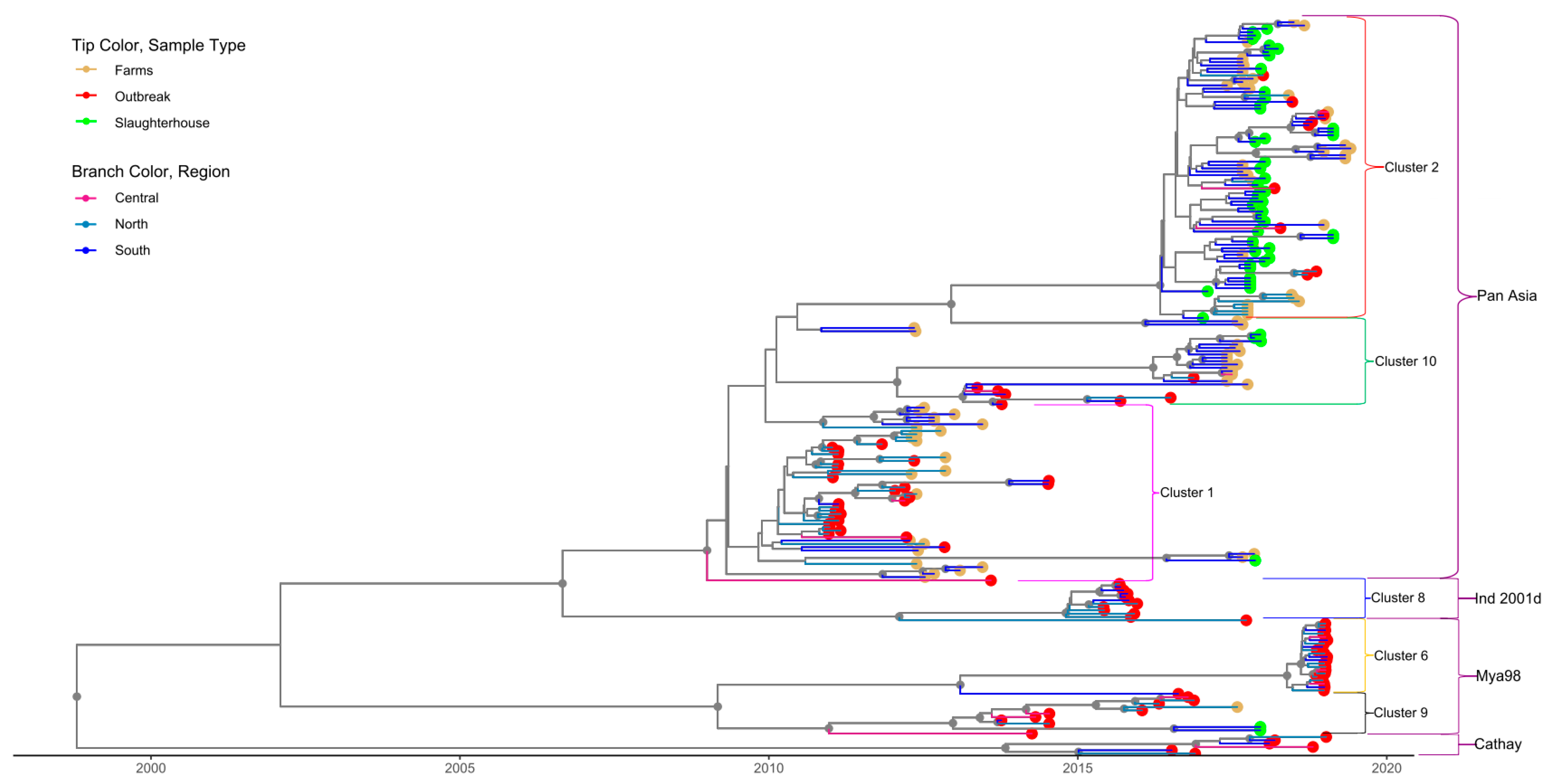
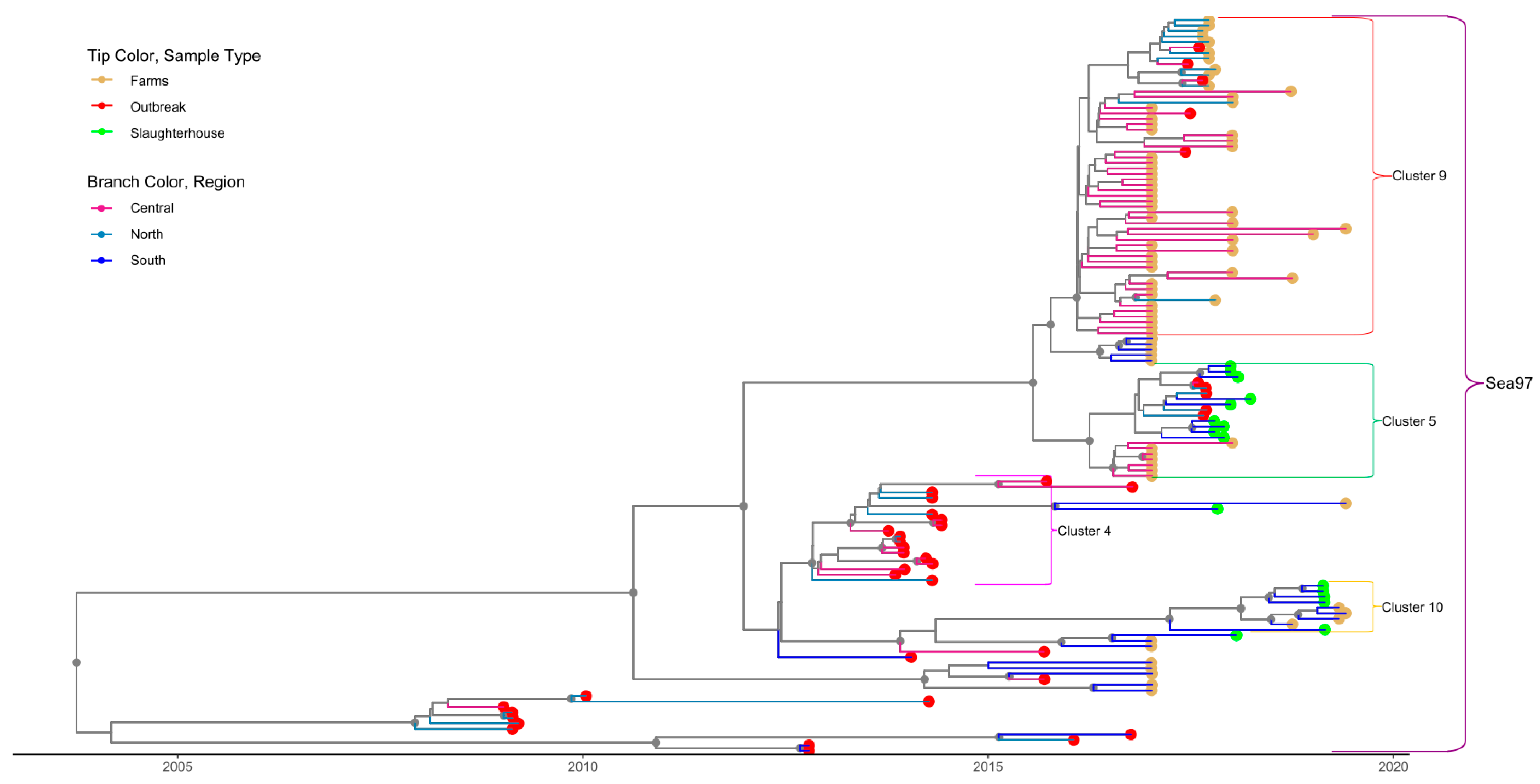
3.3. Phylogenetic Data Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stenfeldt, C.; Arzt, J. The Carrier Conundrum; A Review of Recent Advances and Persistent Gaps Regarding the Carrier State of Foot-and-Mouth Disease Virus. Pathogens 2020, 9, 167. [Google Scholar] [CrossRef] [Green Version]
- Cameron, A.R.; Meyer, A.; Faverjon, C.; Mackenzie, C. Quantification of the Sensitivity of Early Detection Surveillance. Transbound. Emerg. Dis. 2020, 67, 2532–2543. [Google Scholar] [CrossRef]
- Armson, B.; Gubbins, S.; Mioulet, V.; Qasim, I.A.; King, D.P.; Lyons, N.A. Foot-and-Mouth Disease Surveillance Using Pooled Milk on a Large-Scale Dairy Farm in an Endemic Setting. Front. Vet. Sci. 2020, 7, 264. [Google Scholar] [CrossRef] [PubMed]
- Thumbi, S.M.; Njenga, M.K.; Otiang, E.; Otieno, L.; Munyua, P.; Eichler, S.; Widdowson, M.-A.; McElwain, T.F.; Palmer, G.H. Mobile phone-based surveillance for animal disease in rural communities: Implications for detection of zoonoses spillover. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20190020. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, H.C.D.C.; Pauszek, S.J.; Ludi, A.; Huston, C.L.; Pacheco, J.M.; Le, V.T.; Nguyen, P.T.; Bui, H.H.; Nguyen, T.D.; Ngo, L.T.; et al. An Integrative Analysis of Foot-and-Mouth Disease Virus Carriers in Vietnam Achieved Through Targeted Surveillance and Molecular Epidemiology. Transbound. Emerg. Dis. 2015, 64, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Bertram, M.R.; Yadav, S.; Stenfeldt, C.; Delgado, A.; Arzt, J. Extinction Dynamics of the Foot-and-Mouth Disease Virus Carrier State Under Natural Conditions. Front. Vet. Sci. 2020, 7, 276. [Google Scholar] [CrossRef]
- Omondi, G.P.; Gakuya, F.; Arzt, J.; Sangula, A.; Hartwig, E.; Pauszek, S.; Smoliga, G.; Brito, B.; Perez, A.; Obanda, V.; et al. The role of African buffalo in the epidemiology of foot-and-mouth disease in sympatric cattle and buffalo populations in Kenya. Transbound. Emerg. Dis. 2020, 67, 2206–2221. [Google Scholar] [CrossRef]
- Farooq, U.; Ahmed, Z.; Naeem, K.; Bertram, M.; Brito, B.; Stenfeldt, C.; Pauszek, S.J.; LaRocco, M.; Rodriguez, L.; Arzt, J. Characterization of naturally occurring, new and persistent subclinical foot-and-mouth disease virus infection in vaccinated Asian buffalo in Islamabad Capital Territory, Pakistan. Transbound. Emerg. Dis. 2018, 65, 1836–1850. [Google Scholar] [CrossRef]
- Innocent, G.T.; Gilbert, L.; Jones, E.O.; McLeod, J.E.; Gunn, G.; McKendrick, I.J.; Albon, S. Combining Slaughterhouse Surveillance Data with Cattle Tracing Scheme and Environmental Data to Quantify Environmental Risk Factors for Liver Fluke in Cattle. Front. Vet. Sci. 2017, 4, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willeberg, P.W.; McAloon, C.; Houtsma, E.; Higgins, I.; Clegg, T.A.; More, S. The Herd-Level Sensitivity of Abattoir Surveillance for Bovine Tuberculosis: Simulating the Effects of Current and Potentially Modified Meat Inspection Procedures in Irish Cattle. Front. Vet. Sci. 2018, 5, 82. [Google Scholar] [CrossRef] [Green Version]
- Arguello, H.; Alvarez-Ordóñez, A.; Carvajal, A.; Rubio, P.; Prieto, M. Role of Slaughtering in Salmonella Spreading and Control in Pork Production. J. Food Prot. 2013, 76, 899–911. [Google Scholar] [CrossRef]
- Kao, S.-Y.Z.; VanderWaal, K.; Enns, E.A.; Craft, M.E.; Alvarez, J.; Picasso-Risso, C.; Wells, S.J. Modeling cost-effectiveness of risk-based bovine tuberculosis surveillance in Minnesota. Prev. Vet. Med. 2018, 159, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Peterson, E.; Remmenga, M.; Hagerman, A.; Akkina, J.E. Use of Temperature, Humidity, and Slaughter Condemnation Data to Predict Increases in Transport Losses in Three Classes of Swine and Resulting Foregone Revenue. Front. Vet. Sci. 2017, 4, 67. [Google Scholar] [CrossRef] [Green Version]
- Savin, M.; Bierbaum, G.; Hammerl, J.A.; Heinemann, C.; Parcina, M.; Sib, E.; Voigt, A.; Kreyenschmidt, J. Antibiotic-resistant bacteria and antimicrobial residues in wastewater and process water from German pig slaughterhouses and their receiving municipal wastewater treatment plants. Sci. Total Environ. 2020, 727, 138788. [Google Scholar] [CrossRef] [PubMed]
- Fertner, M.; Denwood, M.; Birkegård, A.C.; Stege, H.; Boklund, A. Associations between Antibacterial Treatment and the Prevalence of Tail-Biting-Related Sequelae in Danish Finishers at Slaughter. Front. Vet. Sci. 2017, 4, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonardi, S. Salmonellain the pork production chain and its impact on human health in the European Union. Epidemiol. Infect. 2017, 145, 1513–1526. [Google Scholar] [CrossRef] [Green Version]
- Le, V.P.; Vu, T.T.H.; Duong, H.-Q.; Than, V.T.; Song, D. Evolutionary phylodynamics of foot-and-mouth disease virus serotypes O and A circulating in Vietnam. BMC Vet. Res. 2016, 12, 269. [Google Scholar] [CrossRef] [Green Version]
- Vu, L.T.; Long, N.T.; Brito, B.; Stenfeldt, C.; Phuong, N.T.; Hoang, B.H.; Pauszek, S.J.; Hartwig, E.J.; Smoliga, G.R.; Vu, P.P.; et al. First detection of foot-and-mouth disease virus O/Ind-2001d in Vietnam. PLoS ONE 2017, 12, e0177361. [Google Scholar] [CrossRef] [Green Version]
- Van Diep, N.; Ngoc, T.T.B.; Hoa, L.Q.; Nga, B.T.T.; Kang, B.; Oh, J.; Lan, N.T.; Le, V.P. O/SEA/Mya-98 lineage foot-and-mouth disease virus was responsible for an extensive epidemic that occurred in late 2018 in Vietnam. Arch. Virol. 2020, 165, 2487–2493. [Google Scholar] [CrossRef] [PubMed]
- Le, V.P.; Nguyen, T.; Lee, K.-N.; Ko, Y.-J.; Lee, H.-S.; Nguyen, V.C.; Mai, T.D.; Do, T.H.; Kim, S.-M.; Cho, I.-S.; et al. Molecular characterization of serotype A foot-and-mouth disease viruses circulating in Vietnam in 2009. Vet. Microbiol. 2010, 144, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Pham, T.L.; Wieland, B. Temporal patterns and space-time cluster analysis of foot-and-mouth disease (FMD) cases from 2007 to 2017 in Vietnam. Transbound. Emerg. Dis. 2019, 67, 584–591. [Google Scholar] [CrossRef]
- Bertram, M.R.; De Rueda, C.B.; Garabed, R.; Jumbo, S.D.; Moritz, M.; Pauszek, S.; Abdoulkadiri, S.; Rodriguez, L.L.; Arzt, J. Molecular Epidemiology of Foot-and-Mouth Disease Virus in the Context of Transboundary Animal Movement in the Far North Region of Cameroon. Front. Vet. Sci. 2018, 5, 320. [Google Scholar] [CrossRef]
- Brito, B.; Pauszek, S.J.; Eschbaumer, M.; Stenfeldt, C.; Ferreira, H.C.D.C.; Vu, L.T.; Phuong, N.T.; Hoang, B.H.; Tho, N.D.; Dong, P.V.; et al. Phylodynamics of foot-and-mouth disease virus O/PanAsia in Vietnam 2010–2014. Vet. Res. 2017, 48, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenfeldt, C.; Pacheco, J.; Smoliga, G.R.; Bishop, E.; Pauszek, S.J.; Hartwig, E.J.; Rodriguez, L.L.; Arzt, J. Detection of Foot-and-mouth Disease Virus RNA and Capsid Protein in Lymphoid Tissues of Convalescent Pigs Does Not Indicate Existence of a Carrier State. Transbound. Emerg. Dis. 2014, 63, 152–164. [Google Scholar] [CrossRef]
- Pacheco, J.M.; Arzt, J.; Rodriguez, L.L. Early events in the pathogenesis of foot-and-mouth disease in cattle after controlled aerosol exposure. Vet. J. 2010, 183, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Bertram, M.R.; Palinski, R.M.; Ranjan, R.; Biswal, J.K.; Pauszek, S.J.; Hartwig, E.J.; Smoliga, G.R.; Fish, I.H.; Vierra, D.; Subramanaim, S.; et al. Genome Sequences of 18 Foot-and-Mouth Disease Virus Outbreak Strains of Serotype O Sublineage Ind2001d from India, 2013 to 2014. Microbiol. Resour. Announc. 2019, 8, e00776-19. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Hurtle, W.; Rowland, J.M.; Casteran, K.A.; Bucko, S.M.; Grau, F.R.; Valdazo-González, B.; Knowles, N.J.; King, D.; Beckham, T.R.; et al. Development of a universal RT-PCR for amplifying and sequencing the leader and capsid-coding region of foot-and-mouth disease virus. J. Virol. Methods 2013, 189, 70–76. [Google Scholar] [CrossRef]
- Palinski, R.M.; Bertram, M.R.; Vu, L.T.; Pauszek, S.J.; Hartwig, E.J.; Smoliga, G.R.; Stenfeldt, C.; Fish, I.H.; Hoang, B.H.; Phuong, N.T.; et al. First Genome Sequence of Foot-and-Mouth Disease Virus Serotype O Sublineage Ind2001e from Southern Vietnam. Microbiol. Resour. Announc. 2019, 8, e01424-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Baele, G.; Lemey, P.; Suchard, M.A. Genealogical Working Distributions for Bayesian Model Testing with Phylogenetic Uncertainty. Syst. Biol. 2015, 65, 250–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A. Tracer Version 1.7.1, Computer Program and Documentation Distributed by the Author. 2007. Available online: http://tree.bio.ed.ac.uk/software/tracer/ (accessed on 21 December 2020).
- Rambault, A. Fig Tree Tree Figure Drawing Tool, Version 1.4.4. 2006–2009. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 21 December 2020).
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2016, 8, 28–36. [Google Scholar] [CrossRef]
- Bertram, M.R.; Brito, B.; Palinski, R.M.; Fish, I.H.; Pauszek, S.J.; Hartwig, E.J.; Smoliga, G.R.; Vu, L.T.; Hoang, B.H.; Phuong, N.T.; et al. Novel Recombinant Foot-and-Mouth Disease Virus Circulating in Vietnam. Microbiol. Resour. Announc. 2021, 10, e01263-20. [Google Scholar] [CrossRef]
- Bertram, M.; Vu, L.T.; Pauszek, S.J.; Brito, B.P.; Hartwig, E.J.; Smoliga, G.R.; Hoang, B.H.; Phuong, N.T.; Stenfeldt, C.; Fish, I.H.; et al. Lack of Transmission of Foot-and-Mouth Disease Virus From Persistently Infected Cattle to Naïve Cattle Under Field Conditions in Vietnam. Front. Vet. Sci. 2018, 5, 174. [Google Scholar] [CrossRef] [Green Version]
- Seeyo, K.B.; Nishi, T.; Kawaguchi, R.; Ungvanijban, S.; Udon, R.; Fukai, K.; Yamakawa, M.; Rukkwamsuk, T. Evolution of antigenic and genetic characteristics of foot-and-mouth disease virus serotype A circulating in Thailand, 2007–2019. Virus Res. 2020, 290, 198166. [Google Scholar] [CrossRef] [PubMed]
- Bronsvoort, B.M.D.; Handel, I.G.; Nfon, C.K.; Sørensen, K.-J.; Malirat, V.; Bergmann, I.; Tanya, V.N.; Morgan, K.L. Redefining the “carrier” state for foot-and-mouth disease from the dynamics of virus persistence in endemically affected cattle populations. Sci. Rep. 2016, 6, 29059. [Google Scholar] [CrossRef] [Green Version]
- Hayer, S.S.; VanderWaal, K.; Ranjan, R.; Biswal, J.K.; Subramaniam, S.; Mohapatra, J.K.; Sharma, G.K.; Rout, M.; Dash, B.B.; Das, B.; et al. Foot-and-mouth disease virus transmission dynamics and persistence in a herd of vaccinated dairy cattle in India. Transbound. Emerg. Dis. 2017, 65, e404–e415. [Google Scholar] [CrossRef] [PubMed]
- Fournié, G.; Waret-Szkuta, A.; Camacho, A.; Yigezu, L.M.; Pfeiffer, D.; Roger, F. A dynamic model of transmission and elimination of peste des petits ruminants in Ethiopia. Proc. Natl. Acad. Sci. USA 2018, 115, 8454–8459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapshak, P.; Sinnott, J.; Somboonwit, C.; Kuhn, J.; Feldblyum, T.; Segal, D. Seasonal and Pandemic Influenza Surveillance and Disease Severity. Glob. Virol. I Identifying Investig. Viral Dis. 2015, 761–789. [Google Scholar] [CrossRef]
- Henritzi, D.; Petric, P.P.; Lewis, N.S.; Graaf, A.; Pessia, A.; Starick, E.; Breithaupt, A.; Strebelow, G.; Luttermann, C.; Parker, L.M.K.; et al. Surveillance of European Domestic Pig Populations Identifies an Emerging Reservoir of Potentially Zoonotic Swine Influenza A Viruses. Cell Host Microbe 2020, 28, 614–627.e6. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Province | Sampling Dates | No. of Farms | NSP Serology (Positive/Total); Percent Positive | RNA Detection in OPF Samples (Positive/Total); Percent Positive | No. VP1 Sequences Obtained | |
|---|---|---|---|---|---|---|
| Southern Provinces | Ninh Thuan | October 2016 June–September 2017 June–September 2018 January–February 2019 | 69 | (1010/1290); 78.3% | (72/1003); 7.2% | 18 |
| Dong Thap | August 2015 October 2016 June, September–November 2017 June–August 2018 January–February 2019 | 135 | (888/1965); 45.2% | (197/882); 22.3% | 30 | |
| Dak Lak | August 2015 August 2017 June–October 2018 January–February 2019 | 212 | (1233/2173); 56.7% | (97/1230); 7.8% | 48 | |
| Binh Phuoc | September 2015 | 160 | (84/514); 16.3% | (2/80); 2.5% | 0 | |
| Northern Provinces | Lang Son | 2015 2016 June–September 2017 May–August 2018 | 227 | (208/1387); 15% | (3/223); 1.3% | 1 |
| Phu Tho | 2015 2016 August–November 2017 June–September 2018 January–February 2019 | 442 | (269/1256); 21.4% | (2/274); 0.8% | 0 | |
| Bak Kan | October 2016 August–November 2017 June–September 2018 January–February 2019 | 303 | (1264/2790); 45.3% | (73/1241); 5.8% | 18 | |
| Ha Tinh | August 2015 | 274 | (86/500); 17.2% | (0/112); 0% | 0 |
| Province | Sampling Dates | NSP Serology (Positive/Total); Percent Positive | RNA Detection in OPF Samples (Positive/Total); Percent Positive | No.VP1 Sequences Obtained |
|---|---|---|---|---|
| Long An | October 2017–May 2018 January–February 2019 | (179/480); 37.3% | (51/480); 10.6% | 30 |
| Tay Ninh | October 2017–June 2018 January–February 2019 | (277/480); 57.7% | (71/480); 14.8% | 34 |
| Serotype/Cluster ID (Lineage) | Source | Number of Sequences per Source | Total Number of Sequences | Region of First Detection | Earliest Date Detected | t MRCA |
|---|---|---|---|---|---|---|
| O-1 | OB | 26 | 54 | 2008.6 | ||
| (PanAsia) | FA | 28 | North (FA) | 2010-12-22 | (1998.7, 2020) | |
| O-2 † | OB | 9 | 90 | South (SH) | 2017-01-10 | 2015.9 |
| (PanAsia) | FA | 22 | (2013.4, 2019.6) | |||
| SH | 42 | |||||
| O-6 † | OB | 21 | 21 | South (OB) | 2018-02-07 | 2017.9 |
| (Mya-98) | (2017.5, 2019.1) | |||||
| O-8 | OB | 12 | 12 | North (OB) | 2015-06-02 | 2011.7 |
| (Ind2001d) | (2006.6, 2020.9) | |||||
| O-9 † | OB | 10 | 13 | Central (OB) | 2013-10-07 | 2013.1 |
| (Mya-98) | FA | 2 | (2007.2, 2019.2) | |||
| SH | 1 | |||||
| O-10 † | OB | 2 | 22 | South (OB) | 2013-05-17 | 2009.7 |
| (PanAsia) | FA | 3 | (2002.9, 2018.8) | |||
| SH | 9 | |||||
| A-4 † | OB | 19 | 21 | Central (OB) | 2013-10-09 | 2012.4 |
| (Sea/97) | FA | 1 | (2006.4, 2019.4) | |||
| SH | 1 | |||||
| A-5 † | FA | 6 | 20 | Central (FA) | 2017-01-08 | 2015.8 |
| (Sea/97) | OB | 5 | ||||
| SH | 9 | (2013.1, 2019.5) | ||||
| A-9 | FA | 5 | 56 | Central (FA) | 2017-01-08 | 2015.3 |
| (Sea/97) | OB | 50 | (2012.2, 2019.5) | |||
| A-10 † | FA | 6 | 12 | South (FA) | 2018-10-03 | 2016.8 |
| (Sea/97) | SH | 6 | (2015.5, 2020.1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gunasekara, U.; Bertram, M.R.; Dung, D.H.; Hoang, B.H.; Phuong, N.T.; Hung, V.V.; Long, N.V.; Minh, P.Q.; Vu, L.T.; Dong, P.V.; et al. Use of Slaughterhouses as Sentinel Points for Genomic Surveillance of Foot-and-Mouth Disease Virus in Southern Vietnam. Viruses 2021, 13, 2203. https://doi.org/10.3390/v13112203
Gunasekara U, Bertram MR, Dung DH, Hoang BH, Phuong NT, Hung VV, Long NV, Minh PQ, Vu LT, Dong PV, et al. Use of Slaughterhouses as Sentinel Points for Genomic Surveillance of Foot-and-Mouth Disease Virus in Southern Vietnam. Viruses. 2021; 13(11):2203. https://doi.org/10.3390/v13112203
Chicago/Turabian StyleGunasekara, Umanga, Miranda R. Bertram, Do H. Dung, Bui H. Hoang, Nguyen T. Phuong, Vo V. Hung, Nguyen V. Long, Phan Q. Minh, Le T. Vu, Pham V. Dong, and et al. 2021. "Use of Slaughterhouses as Sentinel Points for Genomic Surveillance of Foot-and-Mouth Disease Virus in Southern Vietnam" Viruses 13, no. 11: 2203. https://doi.org/10.3390/v13112203
APA StyleGunasekara, U., Bertram, M. R., Dung, D. H., Hoang, B. H., Phuong, N. T., Hung, V. V., Long, N. V., Minh, P. Q., Vu, L. T., Dong, P. V., Perez, A., VanderWaal, K., & Arzt, J. (2021). Use of Slaughterhouses as Sentinel Points for Genomic Surveillance of Foot-and-Mouth Disease Virus in Southern Vietnam. Viruses, 13(11), 2203. https://doi.org/10.3390/v13112203