
First Genomic Evidence of Dual African Swine Fever Virus Infection: Case Report from Recent and Historical Outbreaks in Sardinia

 , ,
, ,  , , ,
, , ,  ,
,  , , and
, , and
Abstract
1. Introduction
1.1. Dual Infections, Superinfections, and Coinfections
1.2. Sardinian Epidemiological Context
2. Materials and Methods
2.1. Ethic Statement, Sampling, and Virus Isolation
2.2. DNA Extraction, Quantification, and Sequencing
2.3. Bioinformatic Analysis
2.4. Inference of Strain Frequency and Composition from Variant Frequencies
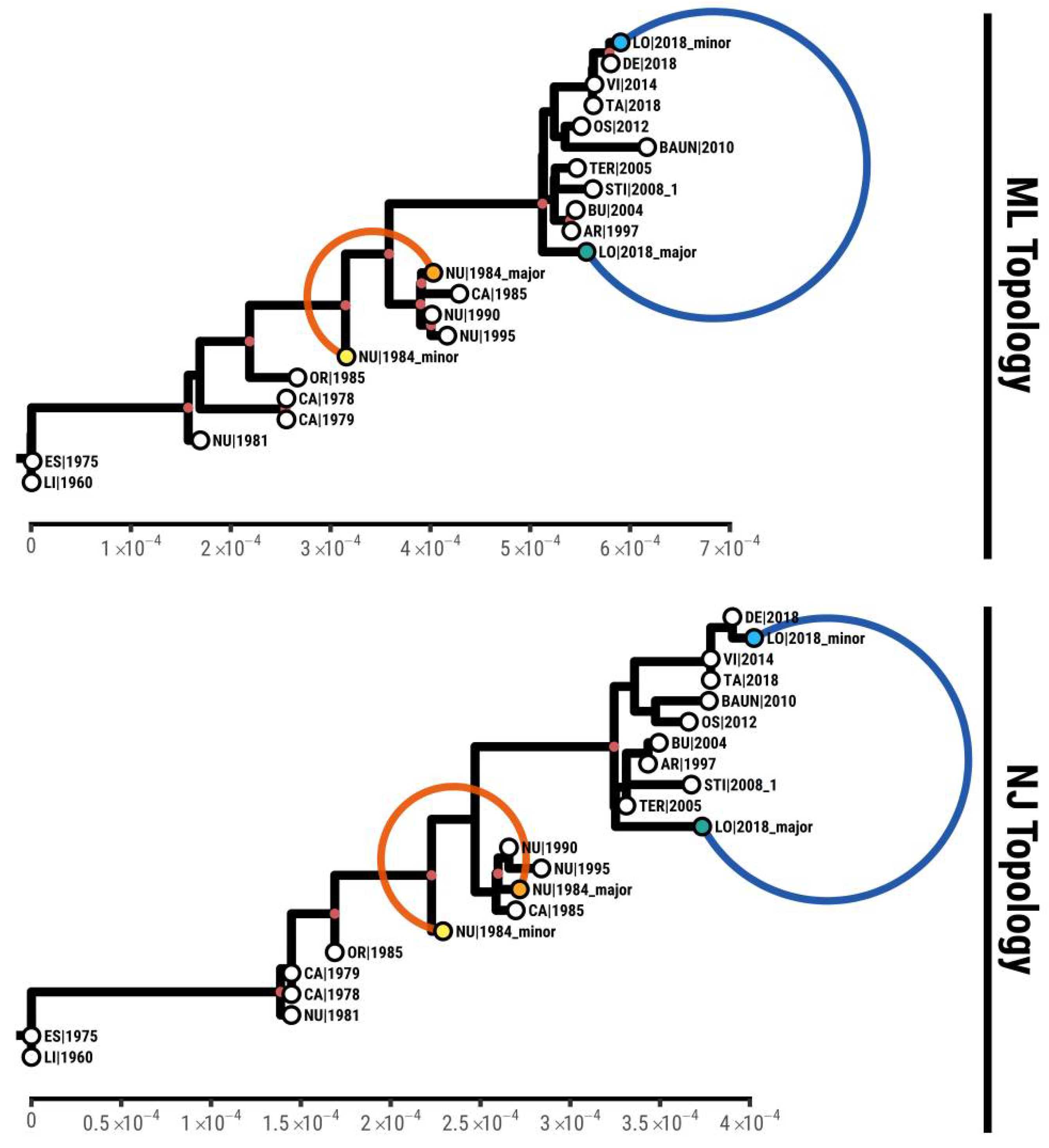
2.5. Phylogenetic Analysis
3. Results
3.1. Bioinformatic Analysis
3.2. Evidence for Dual Infection
3.3. Inference of Strain Frequency and Composition from Variant Frequencies
3.4. Phylogenetic Analysis
3.5. Genomic Differences between Coinfecting Strains
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dixon, L.K.; Chapman, D.A.; Netherton, C.L.; Upton, C. African swine fever virus replication and genomics. Virus Res. 2013, 173, 3–14. [Google Scholar] [CrossRef]
- Franzoni, G.; Graham, S.P.; Giudici, S.D.; Oggiano, A. Porcine Dendritic Cells and Viruses: An Update. Viruses 2019, 11, 445. [Google Scholar] [CrossRef]
- World Organization for Animal Health (OIE). Global Control of African Swine Fever, Resolution No. 33, 87 GS/FR; OIE Bulletine; OIE: Paris, France, 2019; Available online: https://www.oie.int/fileadmin/Home/eng/Animal_Health_in_the_World/docs/pdf/ASF/ASF_GlobalInitiative_Web.pdf (accessed on 22 October 2021).
- Iglesias, I.; Martínez, M.; Montes, F.; de la Torre, A. Velocity of ASF spread in wild boar in the European Union (2014–2017). Int. J. Infect. Dis. 2019, 79, 69. [Google Scholar] [CrossRef]
- Dixon, L.K.; Stahl, K.; Jori, F.; Vial, L.; Pfeiffer, D.U. African Swine Fever Epidemiology and Control. Annu. Rev. Anim. Biosci. 2020, 8, 221–246. [Google Scholar] [CrossRef]
- Kolbasov, D.; Titov, I.; Tsybanov, S.; Gogin, A.; Malogolovkin, A. African Swine Fever Virus, Siberia, Russia, 2017. Emerg. Infect. Dis. 2018, 24, 796–798. [Google Scholar] [CrossRef] [PubMed]
- Penrith, M.-L.; Vosloo, W.; Jori, F.; Bastos, A. African swine fever virus eradication in Africa. Virus Res. 2013, 173, 228–246. [Google Scholar] [CrossRef] [PubMed]
- Costard, S.; Wieland, B.; De Glanville, W.; Jori, F.; Rowlands, R.; Vosloo, W.; Roger, F.; Pfeiffer, D.; Dixon, L.K. African swine fever: How can global spread be prevented? Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 2683–2696. [Google Scholar] [CrossRef]
- Blome, S.; Gabriel, C.; Beer, M. Pathogenesis of African swine fever in domestic pigs and European wild boar. Virus Res. 2013, 173, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, N.; Luo, Y.; Liu, Y.; Miao, F.; Chen, T.; Zhang, S.; Cao, P.; Li, X.; Tian, K.; et al. Emergence of African Swine Fever in China. Transbound. Emerg. Dis. 2018, 65, 1482–1484. [Google Scholar] [CrossRef]
- Saade, G.; Deblanc, C.; Bougon, J.; Marois-Créhan, C.; Fablet, C.; Auray, G.; Belloc, C.; Leblanc-Maridor, M.; Gagnon, C.A.; Zhu, J.; et al. Coinfections and their molecular consequences in the porcine respiratory tract. Vet. Res. 2020, 51, 1–19. [Google Scholar] [CrossRef]
- Worobey, M.; Holmes, E.C. Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 1999, 80, 2535–2543. [Google Scholar] [CrossRef]
- Bastos, A.D.; Penrith, M.L.; Cruciere, C.; Edrich, J.L.; Hutchings, G.; Roger, F.; Couacy-Hymann, E.G.; Thomson, G.R. Genotyping field strains of African swine fever virus by partial p72 gene characterisation. Arch. Virol. 2003, 148, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Mulumba–Mfumu, L.K.; Achenbach, J.E.; Mauldin, M.R.; Dixon, L.K.; Tshilenge, C.G.; Thiry, E.; Moreno, N.; Blanco, E.; Saegerman, C.; Lamien, C.E.; et al. Genetic Assessment of African Swine Fever Isolates Involved in Outbreaks in the Democratic Republic of Congo between 2005 and 2012 Reveals Co-Circulation of p72 Genotypes I, IX and XIV, Including 19 Variants. Viruses 2017, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.K.; Wilkinson, P.J. Genetic diversity of African swine fever virus isolates from soft ticks (Ornithodoros moubata) inhabiting warthog burrows in Zambia. J. Gen. Virol. 1988, 69, 2981–2993. [Google Scholar] [CrossRef] [PubMed]
- Plowright, W.; Perry, C.; Peirce, M. Transovarial Infection with African Swine Fever Virus in the Argasid Tick, Ornithodoros moubata porcinus, Walton. Res. Vet. Sci. 1970, 11, 582–584. [Google Scholar] [CrossRef]
- Franzoni, G.; Giudici, S.D.; Loi, F.; Sanna, D.; Floris, M.; Fiori, M.; Sanna, M.L.; Madrau, P.; Scarpa, F.; Zinellu, S.; et al. African Swine Fever Circulation among Free-Ranging Pigs in Sardinia: Data from the Eradication Program. Vaccines 2020, 8, 549. [Google Scholar] [CrossRef]
- Giudici, S.D.; Franzoni, G.; Bonelli, P.; Bacciu, D.; Sanna, G.; Angioi, P.P.; Ledda, M.; Pilo, G.; Nicolussi, P.; Oggiano, A. Interaction of historical and modern Sardinian African swine fever viruses with porcine and wild-boar monocytes and monocyte-derived macrophages. Arch. Virol. 2019, 164, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Castañeda, B.; Reis, A.L.; Corteyn, A.; Parkhouse, R.M.E.; Kollnberger, S. Expression, cellular localization and antibody responses of the African swine fever virus genes B602L and K205R. Arch. Virol. 2008, 153, 2303–2306. [Google Scholar] [CrossRef]
- Giammarioli, M.; Gallardo, C.; Oggiano, A.; Iscaro, C.; Nieto, R.; Pellegrini, C.; Giudici, S.D.; Arias, M.; De Mia, G.M. Genetic characterisation of African swine fever viruses from recent and historical outbreaks in Sardinia (1978–2009). Virus Genes 2011, 42, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Sanna, G.; Dei Giudici, S.; Bacciu, D.; Angioi, P.P.; Giammarioli, M.; De Mia, G.M.; Oggiano, A. Improved strategy for molecular characteri-zation of African swine fever virus from Sardinia, based on analysis of p30, CD2V and I73R/I329L variable regions. Transbound. Emerg. Dis. 2017, 64, 1280–1286. [Google Scholar]
- Bacciu, D.; Deligios, M.; Sanna, G.; Madrau, M.P.; Sanna, M.L.; Dei Giudici, S.; Oggiano, A. Genomic analysisof sardinian 26544/OG10 isolate of African swine fever virus. Virol. Rep. 2016, 6, 81–89. [Google Scholar]
- Granberg, F.; Torresi, C.; Oggiano, A.; Malmberg, M.; Iscaro, C.; De Mia, G.M.; Belák, S. Complete Genome Sequence of an African Swine Fever Virus Isolate from Sardinia, Italy. Genome Announc. 2016, 4, e01220-16. [Google Scholar] [CrossRef] [PubMed]
- Torresi, C.; Fiori, M.S.; Bertolotti, L.; Floris, M.; Colitti, B.; Giammarioli, M.; Giudici, S.D.; Oggiano, A.; Malmberg, M.; De Mia, G.M.; et al. The evolution of African swine fever virus in Sardinia (1978 to 2014) as revealed by whole genome sequencing and comparative analysis. Transbound. Emerg. Dis. 2020, 67, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Anonymous. Bruciati Ventimila Maiali. Il Messaggero Sardo; Cooperativa Messaggero Sardo: Cagliari, Italy, 9 August 1978; p. 9. [Google Scholar]
- Fiori, M.S.S.; Sanna, D.; Scarpa, F.; Floris, M.; Nardo, A.D.D.; Ferretti, L.; Loi, F.; Cappai, S.; Sechi, A.M.M.; Angioi, P.P.P.; et al. A Deeper Insight into Evolutionary Patterns and Phylogenetic History of ASFV Epidemics in Sardinia (Italy) through Extensive Genomic Sequencing. Viruses 2021, 13, 1994. [Google Scholar] [CrossRef]
- Thiry, E.; Muylkens, B.; Meurens, F.; Gogev, S.; Thiry, J.; Vanderplasschen, A.; Schynts, F. Recombination in the alphaherpesvirus bovine herpesvirus 1. Vet. Microbiol. 2006, 113, 171–177. [Google Scholar] [CrossRef]
- Thiry, E.; Meurens, F.; Muylkens, B.; McVoy, M.; Gogev, S.; Thiry, J.; Vanderplasschen, A.; Epstein, A.; Keil, G.; Schynts, F. Recombination in alphaherpesviruses. Rev. Med. Virol. 2005, 15, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Schynts, F.; Meurens, F.; Detry, B.; Vanderplasschen, A.; Thiry, E. Rise and Survival of Bovine Herpesvirus 1 Recombinants after Primary Infection and Reactivation from Latency. J. Virol. 2003, 77, 12535–12542. [Google Scholar] [CrossRef]
- Meurens, F.; Schynts, F.; Keil, G.M.; Muylkens, B.; Vanderplasschen, A.; Gallego, P.; Thiry, E. Superinfection Prevents Recombination of the Alphaherpesvirus Bovine Herpesvirus 1. J. Virol. 2004, 78, 3872–3879. [Google Scholar] [CrossRef]
- Meurens, F.; Keil, G.M.; Muylkens, B.; Gogev, S.; Schynts, F.; Negro, S.; Wiggers, L.; Thiry, E. Interspecific Recombination between Two Ruminant Alphaherpesviruses, Bovine Herpesviruses 1 and 5. J. Virol. 2004, 78, 9828–9836. [Google Scholar] [CrossRef]
- Mannelli, A.; Sotgia, S.; Patta, C.; Oggiano, A.; Carboni, A.; Cossu, P.; Laddomada, A. Temporal and spatial patterns of African swine fever in Sardinia. Prev. Vet. Med. 1998, 35, 297–306. [Google Scholar] [CrossRef]
- Loi, F.; Laddomada, A.; Coccollone, A.; Marrocu, E.; Piseddu, T.; Masala, G.; Bandino, E.; Cappai, S.; Rolesu, S. Socio-economic factors as indicators for various animal diseases in Sardinia. PLoS ONE 2019, 14, e0217367. [Google Scholar]
- Loi, F.; Cappai, S.; Coccollone, A.; Rolesu, S. Standardized Risk Analysis Approach Aimed to Evaluate the Last African Swine Fever Eradication Program Performance, in Sardinia. Front. Vet. Sci. 2019, 6, 299. [Google Scholar] [CrossRef]
- Cappai, S.; Rolesu, S.; Coccollone, A.; Laddomada, A.; Loi, F. Evaluation of biological and socio-economic factors related to persistence of African swine fever in Sardinia. Prev. Vet. Med. 2018, 152, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mur, L.; Atzeni, M.; Martinez-Lopez, B.; Feliziani, F.; Rolesu, S.; Sánchez-Vizcaíno, J.M. Thirty-Five-Year Presence of African Swine Fever in Sardinia: History, Evolution and Risk Factors for Disease Maintenance. Transbound. Emerg. Dis. 2014, 63, e165–e177. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, P. The persistence of African swine fever in Africa and the Mediterranean. Prev. Vet. Med. 1984, 2, 71–82. [Google Scholar] [CrossRef]
- Contini, A.; Cossu, P.; Firinu, A. African swine fever in Sardinia. In African Swine Fever; EUR 8466 EN, Pro CEC/FAO Research seminar, Sardinia 1982; Wilkinson, P.J., Ed.; Commission of the European Communities: Brussels, Belgium, 1983; pp. 1–6. [Google Scholar]
- Firinu, A.; Scarano, C. La peste porcine africaine et la peste porcine classique chez le sanglier en Sardeigne. Rev. Sci. Tech. Off. Int. Epizoot. 1988, 7, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Laddomada, A.; Rolesu, S.; Loi, F.; Cappai, S.; Oggiano, A.; Madrau, M.P.; Sanna, M.L.; Pilo, G.; Bandino, E.; Brundu, D.; et al. Surveillance and control of African Swine Fever in free-ranging pigs in Sardinia. Transbound. Emerg. Dis. 2019, 66, 1114–1119. [Google Scholar] [CrossRef]
- Loi, F.; Cappai, S.; Laddomada, A.; Feliziani, F.; Oggiano, A.; Franzoni, G.; Rolesu, S.; Guberti, V. Mathematical Approach to Estimating the Main Epidemiological Parameters of African Swine Fever in Wild Boar. Vaccines 2020, 8, 521. [Google Scholar] [CrossRef]
- World Organization for Animal Health (OIE). African Swine Fever. In Manual of Diagnostic Tests and Vaccines for Terrestrial Animals, 9th ed.; Word Organisation for Animal Health: Paris, France, 2019; Chapter 3.8.1. [Google Scholar]
- Groenen, M.A.M.; Archibald, A.L.; Uenishi, H.; Tuggle, C.K.; Takeuchi, Y.; Rothschild, M.F.; Rogel-Gaillard, C.; Park, C.; Milan, D.; Megens, H.-J.; et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 2012, 491, 393–398. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Marco-Sola, S.; Sammeth, M.; Guigó, R.; Ribeca, P. The GEM mapper: Fast, accurate and versatile alignment by filtration. Nat. Methods 2012, 9, 1185–1188. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-Based Variant Detection from Short-Read Sequencing. Cornell University Press: Ithaca, NY, USA, 2012; Available online: https://arxiv.org/pdf/1207.3907.pdf (accessed on 22 October 2021).
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Tcherepanov, V.; Ehlers, A.; Upton, C. Genome Annotation Transfer Utility (GATU): Rapid annotation of viral genomes using a closely related reference genome. BMC Genom. 2006, 7, 150. [Google Scholar] [CrossRef]
- Baccam, P.; Thompson, R.J.; Fedrigo, O.; Carpenter, S.; Cornette, J.L. PAQ: Partition Analysis of Quasispecies. Bioinform 2001, 17, 16–22. [Google Scholar] [CrossRef]
- Ferretti, L.; Pérez-Martín, E.; Zhang, F.; Maree, F.; De Klerk-Lorist, L.-M.; Van Schalkwykc, L.; Juleff, N.D.; Charleston, B.; Ribeca, P. Pervasive within-host recombination and epistasis as major determinants of the molecular evolution of the foot-and-mouth disease virus capsid. PLoS Pathog. 2020, 16, e1008235. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, L.; Tennakoon, C.; Silesian, A.; Ribeca, G.F.A.; Freimanis, G.; Ribeca, P. SiNPle: Fast and Sensitive Variant Calling for Deep Sequencing Data. Genes 2019, 10, 561. [Google Scholar] [CrossRef] [PubMed]
- Wilks, S.S. The Large-Sample Distribution of the Likelihood Ratio for Testing Composite Hypotheses. Ann. Math. Stat. 1938, 9, 60–62. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Gascuel, O. BIONJ: An improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 1997, 14, 685–695. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinform 2018, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Parrish, S.; Moss, B. Characterization of a Second Vaccinia Virus mRNA-Decapping Enzyme Conserved in Poxviruses. J. Virol. 2007, 81, 12973–12978. [Google Scholar] [CrossRef]
- Cartwright, J.L.; Safrany, S.T.; Dixon, L.K.; Darzynkiewicz, E.; Stepinski, J.; Burke, R.; McLennan, A.G. The g5R (D250) Gene of African Swine Fever Virus Encodes a Nudix Hydrolase That Preferentially Degrades Diphosphoinositol Polyphosphates. J. Virol. 2002, 76, 1415–1421. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, J.; Shi, X.-J.; Sun, H.-W.; Chen, H.-J. Insights into African swine fever virus immunoevasion strategies. J. Integr. Agric. 2020, 19, 11–22. [Google Scholar] [CrossRef]
- Freitas, F.B.; Frouco, G.; Martins, C.; Ferreira, F. The QP509L and Q706L superfamily II RNA helicases of African swine fever virus are required for viral replication, having non-redundant activities. Emerg. Microbes Infect. 2019, 8, 291–302. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Sample | Position in Reference | Major Variant | Minor Variant | Major Read Count | Minor Read Count | Major Frequency | Minor Frequency |
|---|---|---|---|---|---|---|---|
| NU1984 | 12452 | T | C | 11 | 4 | 0.73 | 0.27 |
| 24115 | A | G | 13 | 2 | 0.87 | 0.13 | |
| 53275 | A | G | 52 | 42 | 0.55 | 0.45 | |
| 63153 | C | G | 144 | 76 | 0.65 | 0.35 | |
| 82709 | A | G | 57 | 9 | 0.86 | 0.14 | |
| 133600 | A | G | 28 | 17 | 0.62 | 0.38 | |
| 161784 | C | G | 70 | 22 | 0.76 | 0.24 | |
| LO2018 | 6930 | T | G | 200 | 88 | 0.69 | 0.31 |
| 9187 | C | T | 257 | 84 | 0.75 | 0.25 | |
| 23900 | C | T | 275 | 101 | 0.73 | 0.27 | |
| 25014 | T | C | 228 | 104 | 0.69 | 0.31 | |
| 27680 | G | A | 259 | 98 | 0.73 | 0.27 | |
| 33008 | T | C | 227 | 89 | 0.72 | 0.28 | |
| 34564 | G | A | 228 | 90 | 0.72 | 0.28 | |
| 66032 | T | C | 233 | 102 | 0.70 | 0.30 | |
| 71406 | G | A | 220 | 96 | 0.70 | 0.30 | |
| 77775 | T | C | 263 | 97 | 0.73 | 0.27 | |
| 77903 | A | G | 244 | 102 | 0.71 | 0.29 | |
| 80076 | A | G | 243 | 87 | 0.74 | 0.26 | |
| 81605 | C | T | 217 | 87 | 0.71 | 0.29 | |
| 82112 | G | A | 239 | 85 | 0.74 | 0.26 | |
| 100721 | C | T | 270 | 83 | 0.76 | 0.24 | |
| 106379 | T | C | 240 | 124 | 0.66 | 0.34 | |
| 107474 | C | A | 230 | 85 | 0.73 | 0.27 | |
| 116165 | C | G | 203 | 89 | 0.70 | 0.30 | |
| 154851 | C | T | 294 | 118 | 0.71 | 0.29 | |
| 166904 | C | T | 262 | 103 | 0.72 | 0.28 | |
| 180038 | C | T | 102 | 16 | 0.86 | 0.14 | |
| 180061 | T | C | 110 | 26 | 0.81 | 0.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiori, M.S.; Ferretti, L.; Floris, M.; Loi, F.; Nardo, A.D.; Sechi, A.M.; Ladu, A.; Puggioni, G.; Sanna, D.; Scarpa, F.; et al. First Genomic Evidence of Dual African Swine Fever Virus Infection: Case Report from Recent and Historical Outbreaks in Sardinia. Viruses 2021, 13, 2145. https://doi.org/10.3390/v13112145
Fiori MS, Ferretti L, Floris M, Loi F, Nardo AD, Sechi AM, Ladu A, Puggioni G, Sanna D, Scarpa F, et al. First Genomic Evidence of Dual African Swine Fever Virus Infection: Case Report from Recent and Historical Outbreaks in Sardinia. Viruses. 2021; 13(11):2145. https://doi.org/10.3390/v13112145
Chicago/Turabian StyleFiori, Mariangela Stefania, Luca Ferretti, Matteo Floris, Federica Loi, Antonello Di Nardo, Anna Maria Sechi, Anna Ladu, Graziella Puggioni, Daria Sanna, Fabio Scarpa, and et al. 2021. "First Genomic Evidence of Dual African Swine Fever Virus Infection: Case Report from Recent and Historical Outbreaks in Sardinia" Viruses 13, no. 11: 2145. https://doi.org/10.3390/v13112145
APA StyleFiori, M. S., Ferretti, L., Floris, M., Loi, F., Nardo, A. D., Sechi, A. M., Ladu, A., Puggioni, G., Sanna, D., Scarpa, F., Sanna, M. L., Madrau, M. P., Torresi, C., Sirica, R., Evangelista, E., Oggiano, A., & Dei Giudici, S. (2021). First Genomic Evidence of Dual African Swine Fever Virus Infection: Case Report from Recent and Historical Outbreaks in Sardinia. Viruses, 13(11), 2145. https://doi.org/10.3390/v13112145