
A CRISPR Activation Screen Identifies an Atypical Rho GTPase That Enhances Zika Viral Entry

 , , , ,
, , , ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
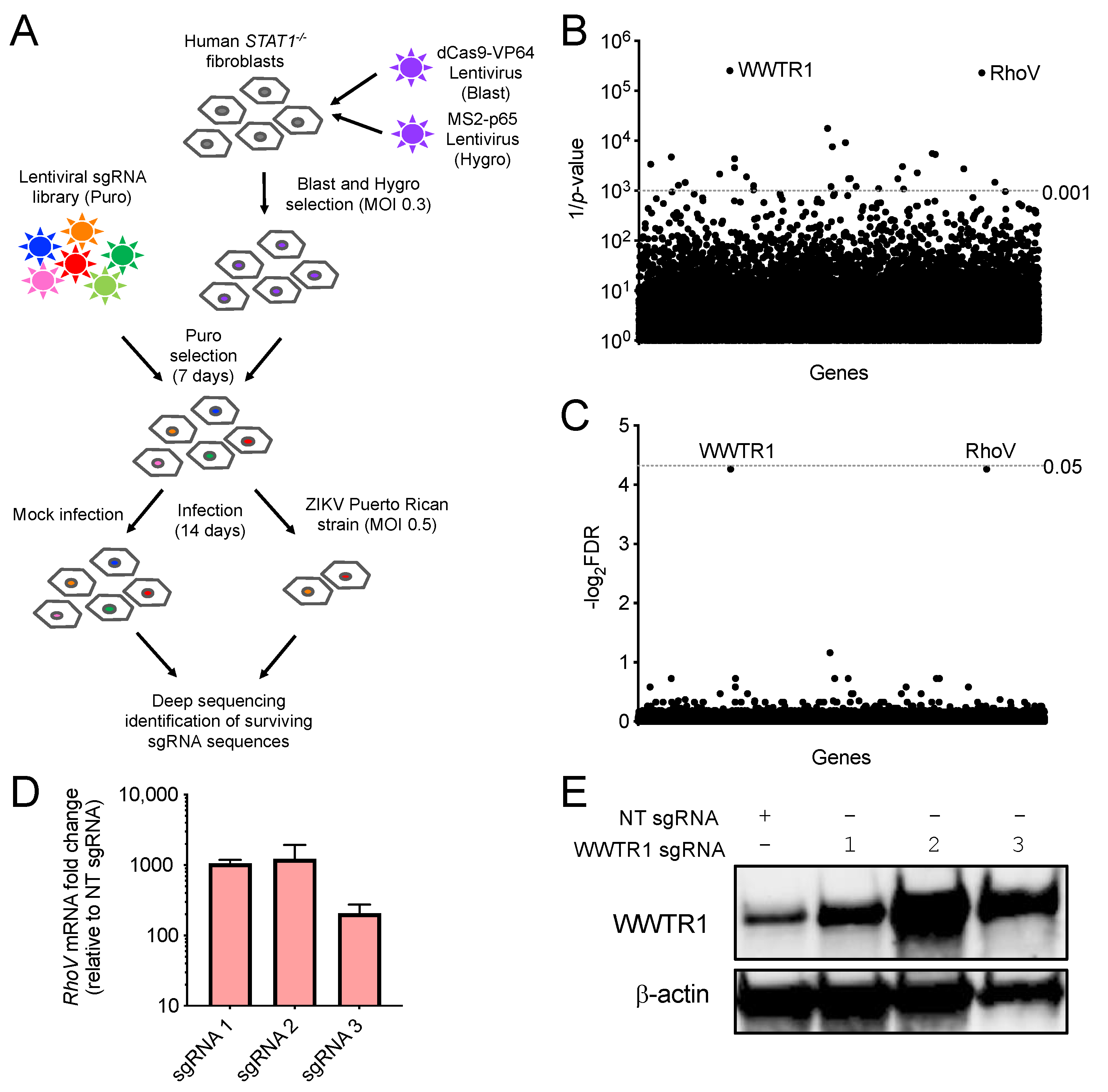
2.1. CRISPR Activation Screen and Validation of Candidate Gene Upregulation
2.2. Cells, Plasmids, Viruses, and Infections
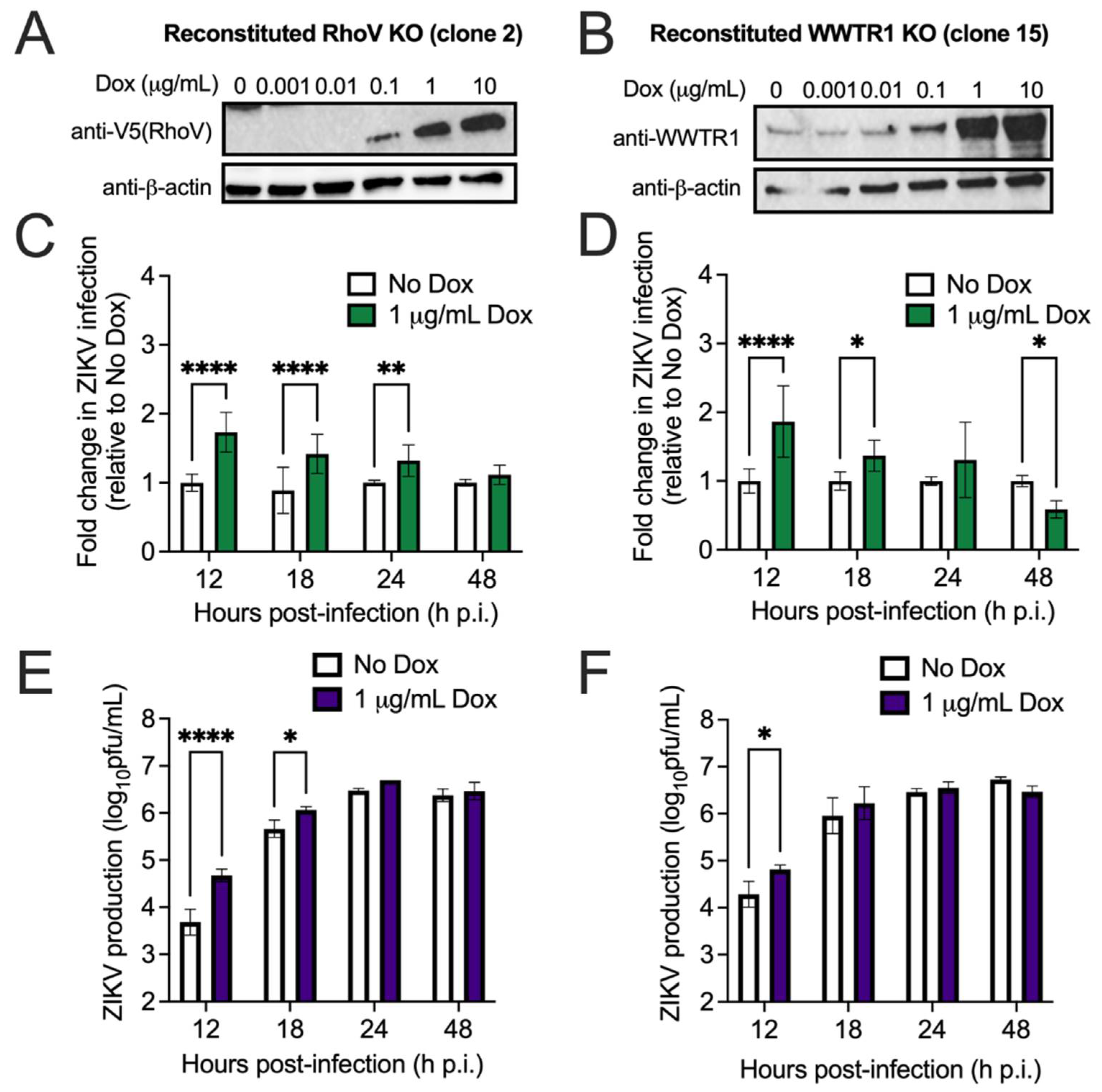
2.3. RhoV and WWTR1 Knockout (KO) by CRISPR–Cas9
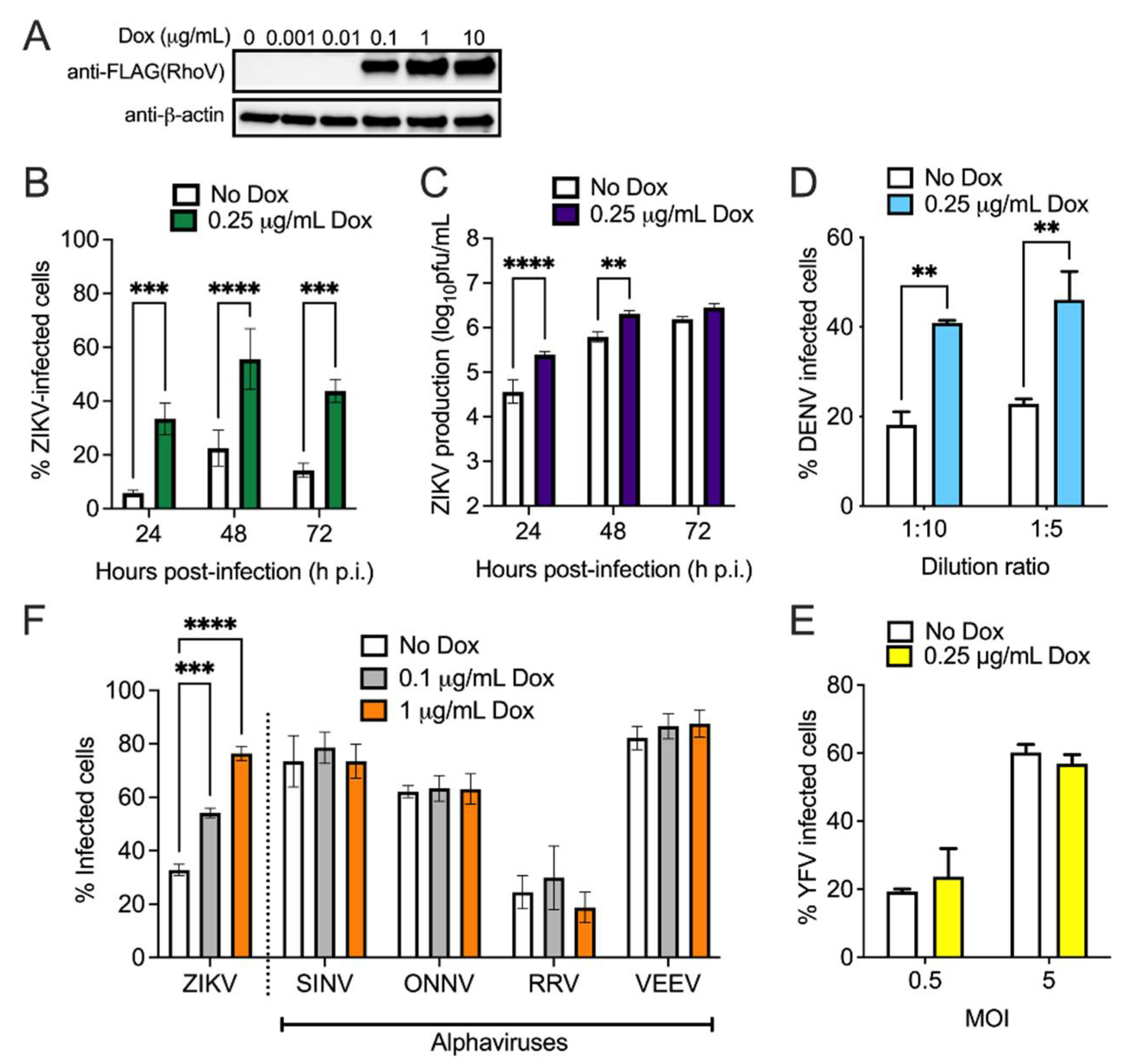
2.4. Generation and Detection of RhoV or WWTR1 Inducible Cell Lines
2.5. Staining and Analysis of ZIKV-Infected Cells
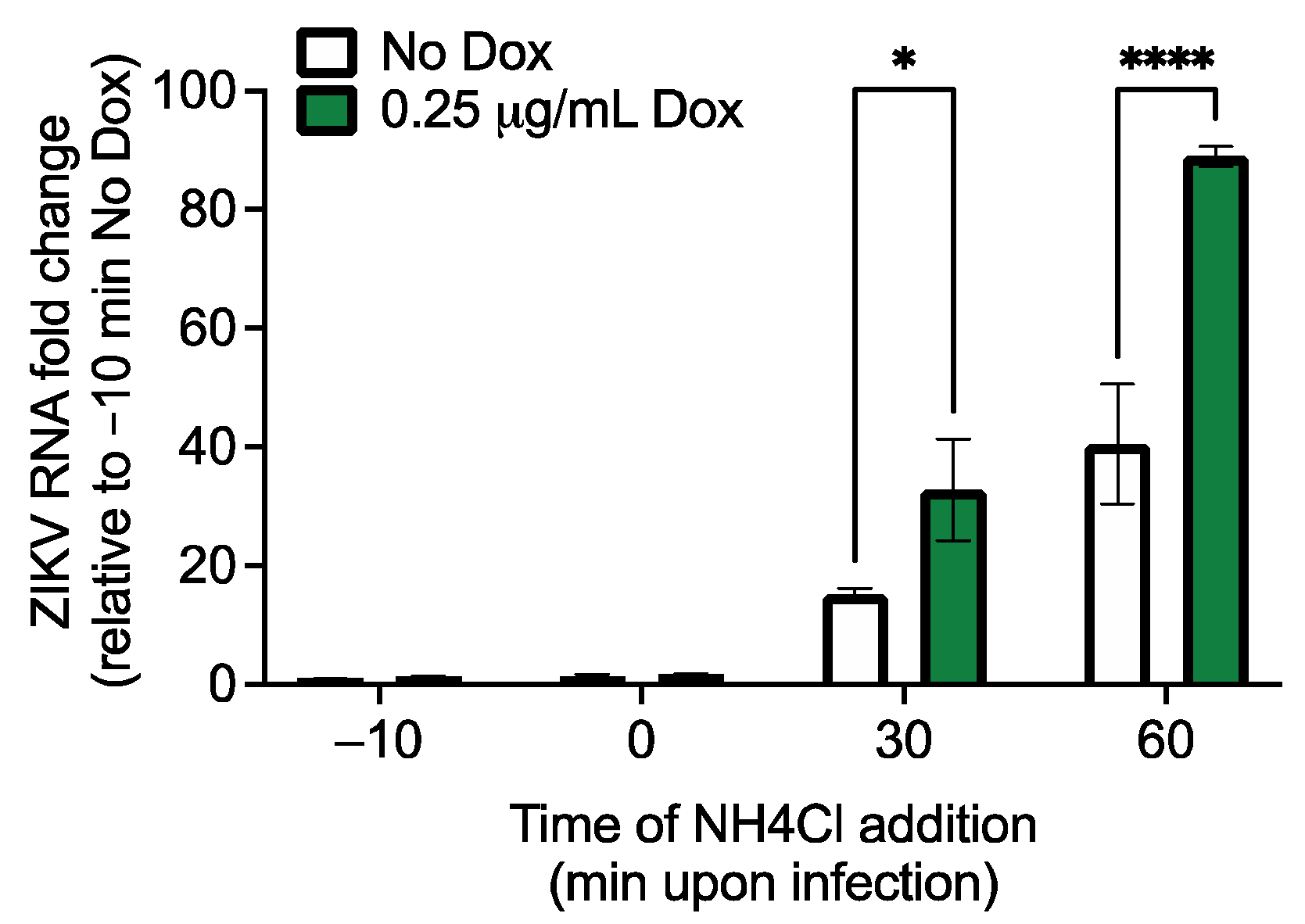
2.6. Viral Entry Assay
2.7. Immunofluorescence
2.8. siRNA Knockdown of Rho GTPases and Their Effector Proteins
2.9. Quantitative Reverse Transcription PCR (RT-qPCR)
3. Results
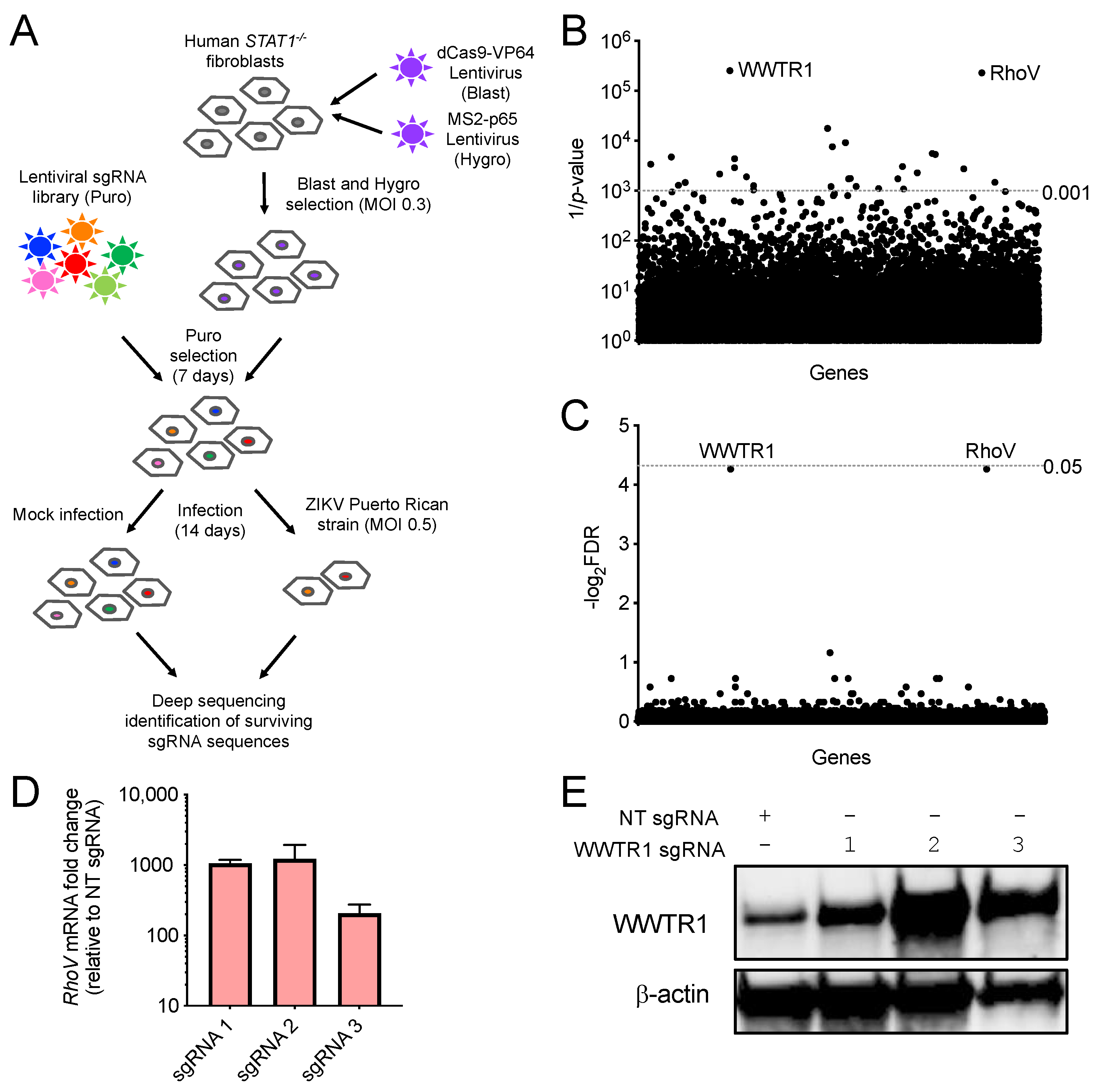
3.1. CRISPR–Cas9 Activation Screen in ZIKV-Infected Cells
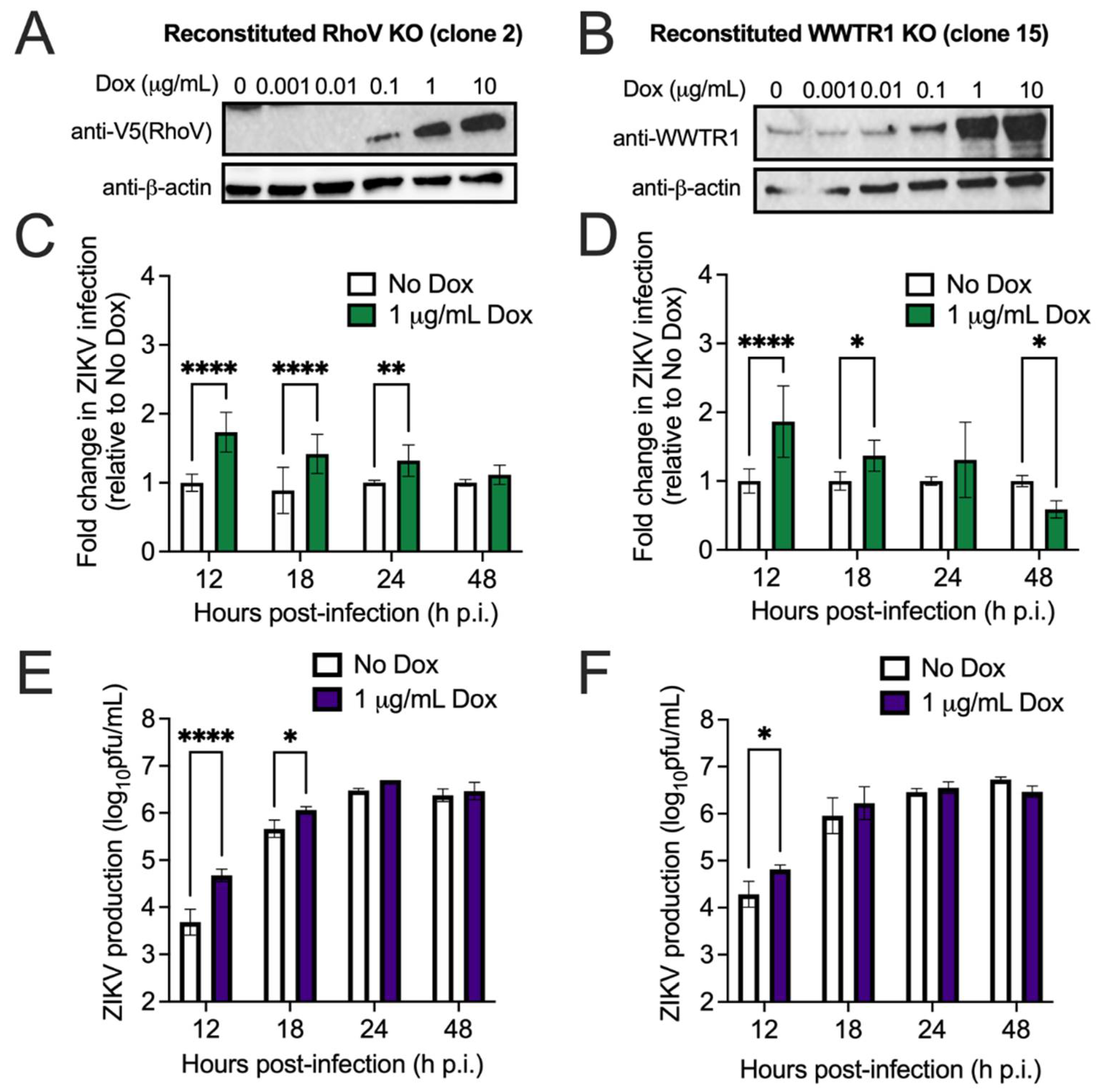
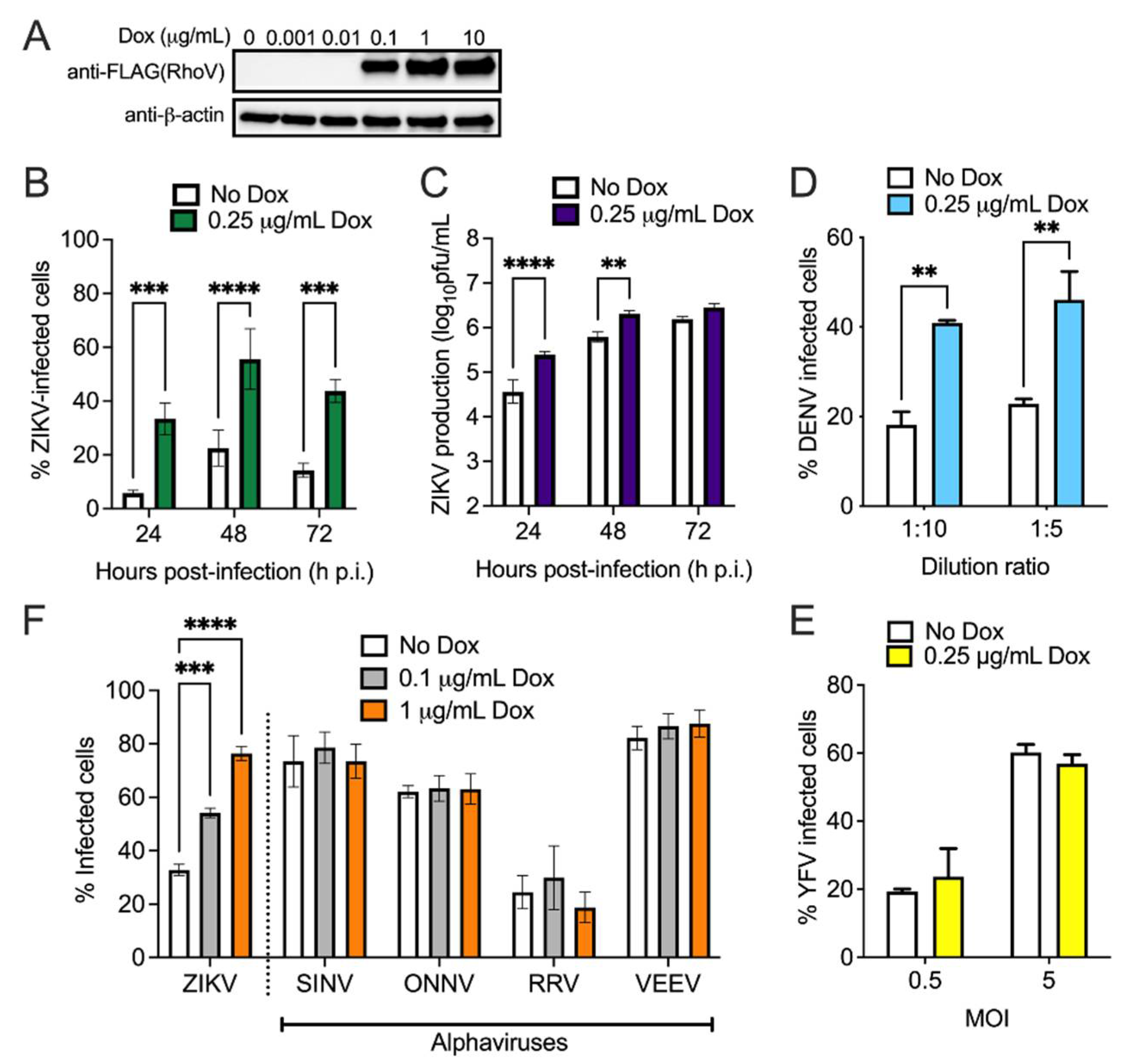
3.2. Both RhoV and WWTR1 Enhance ZIKV Infection in A549 Cells
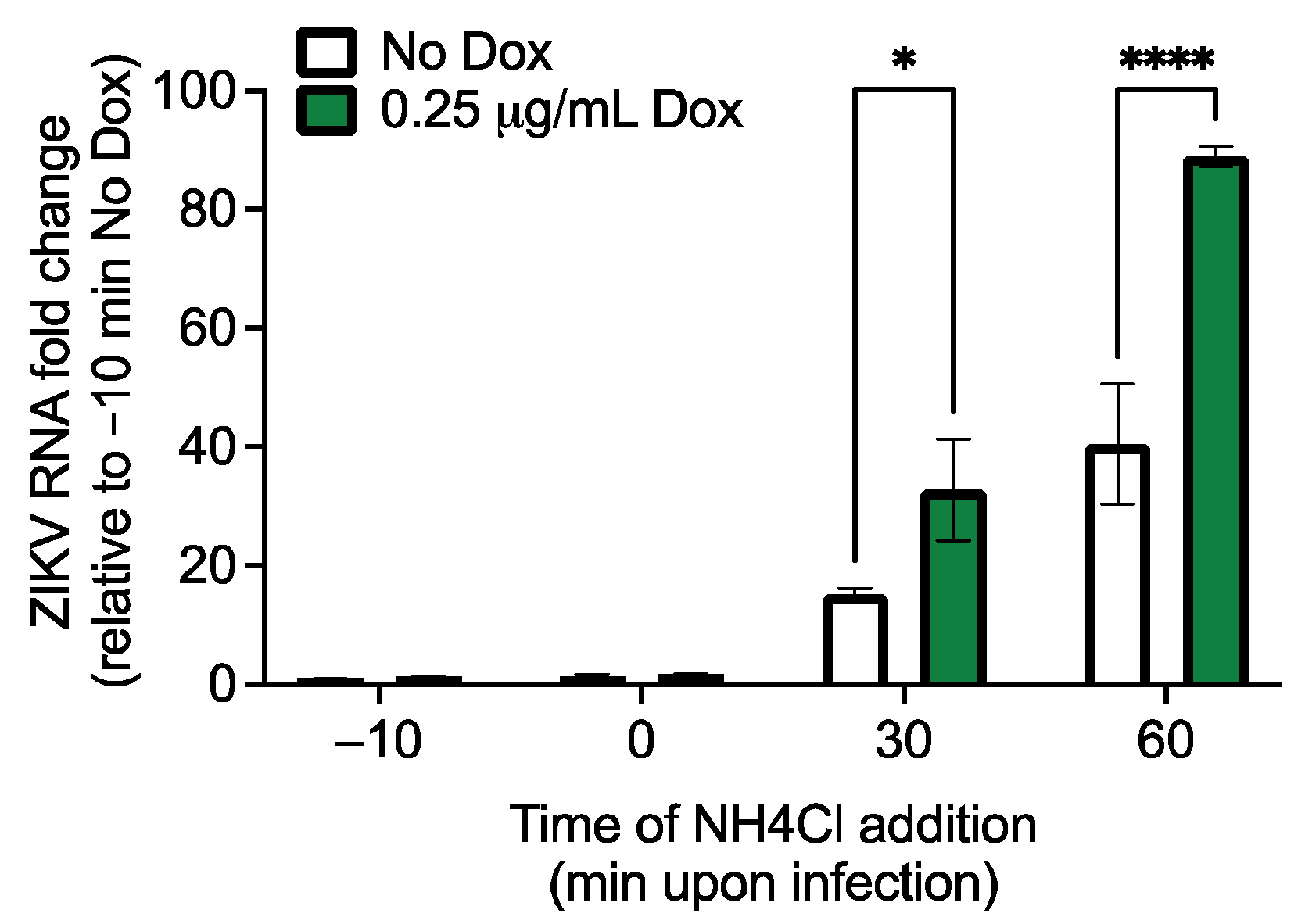
3.3. RhoV Promotes ZIKV and DENV Infection in SNB-19 Cells
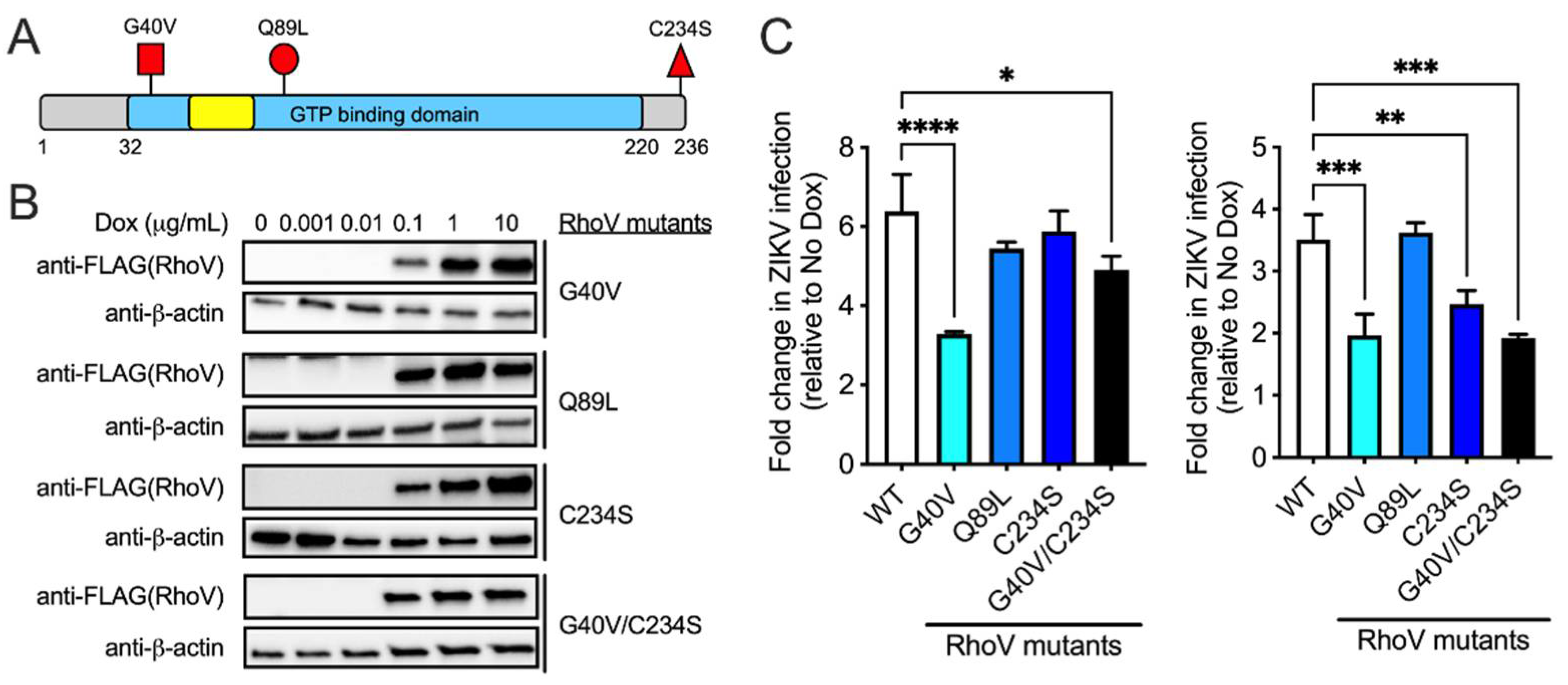
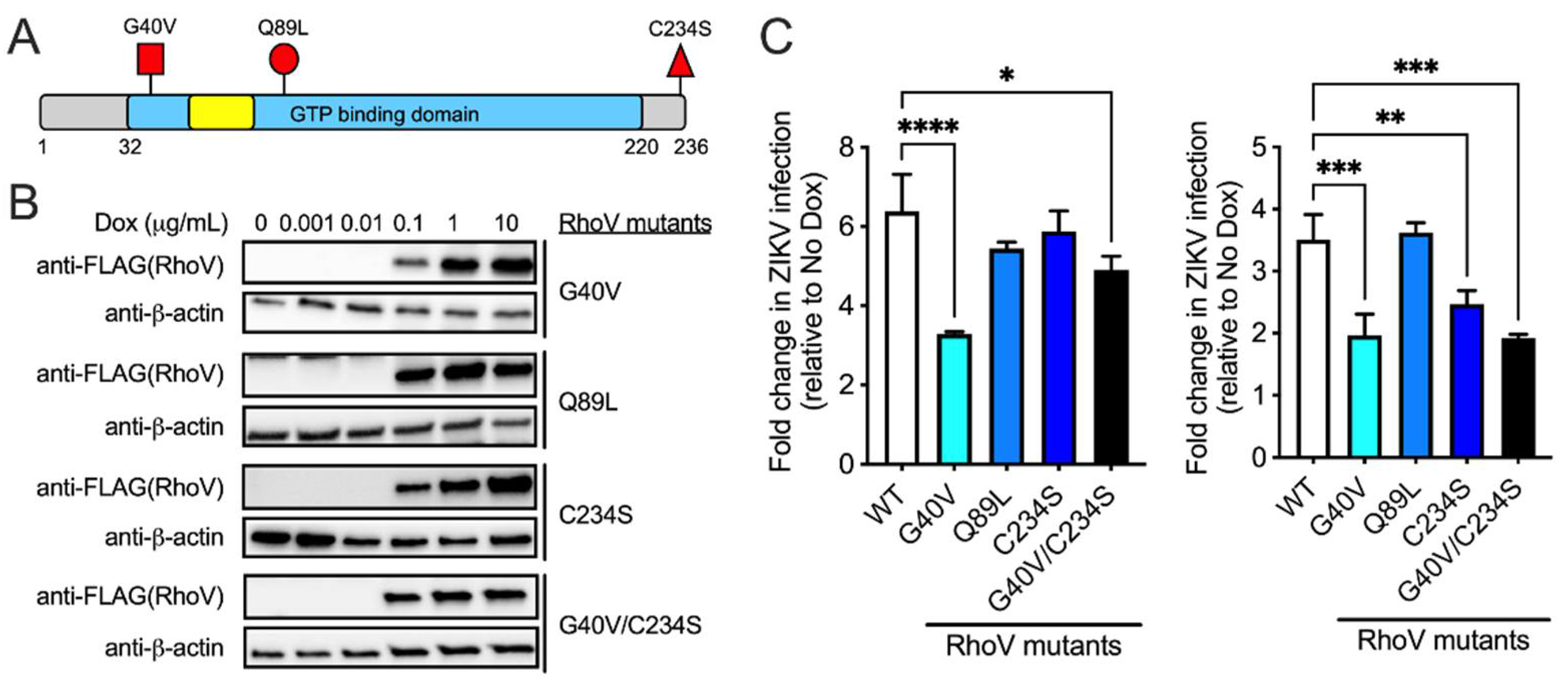
3.4. The GTPase Domain Plays an Important Role in RhoV Proviral Effects
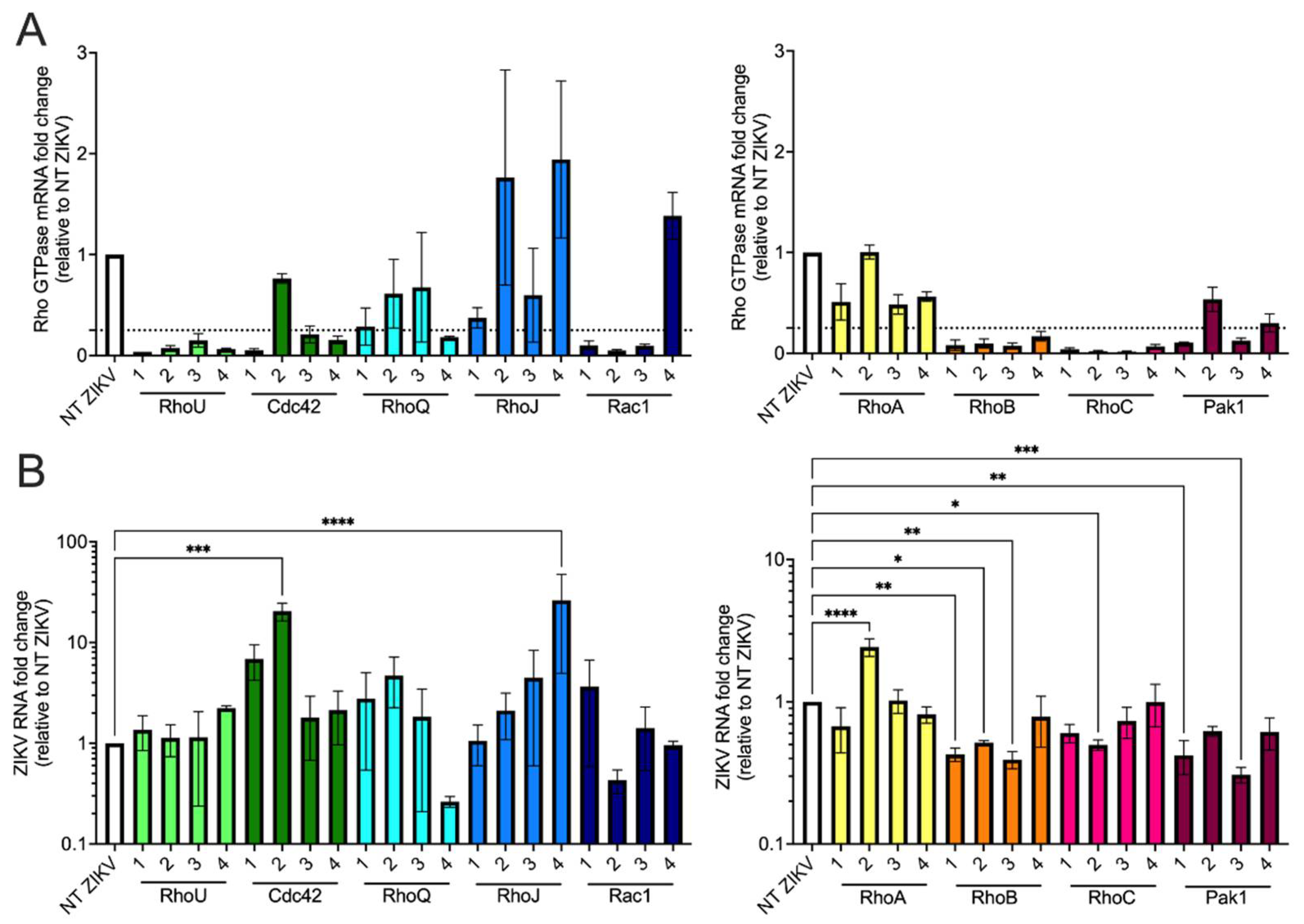
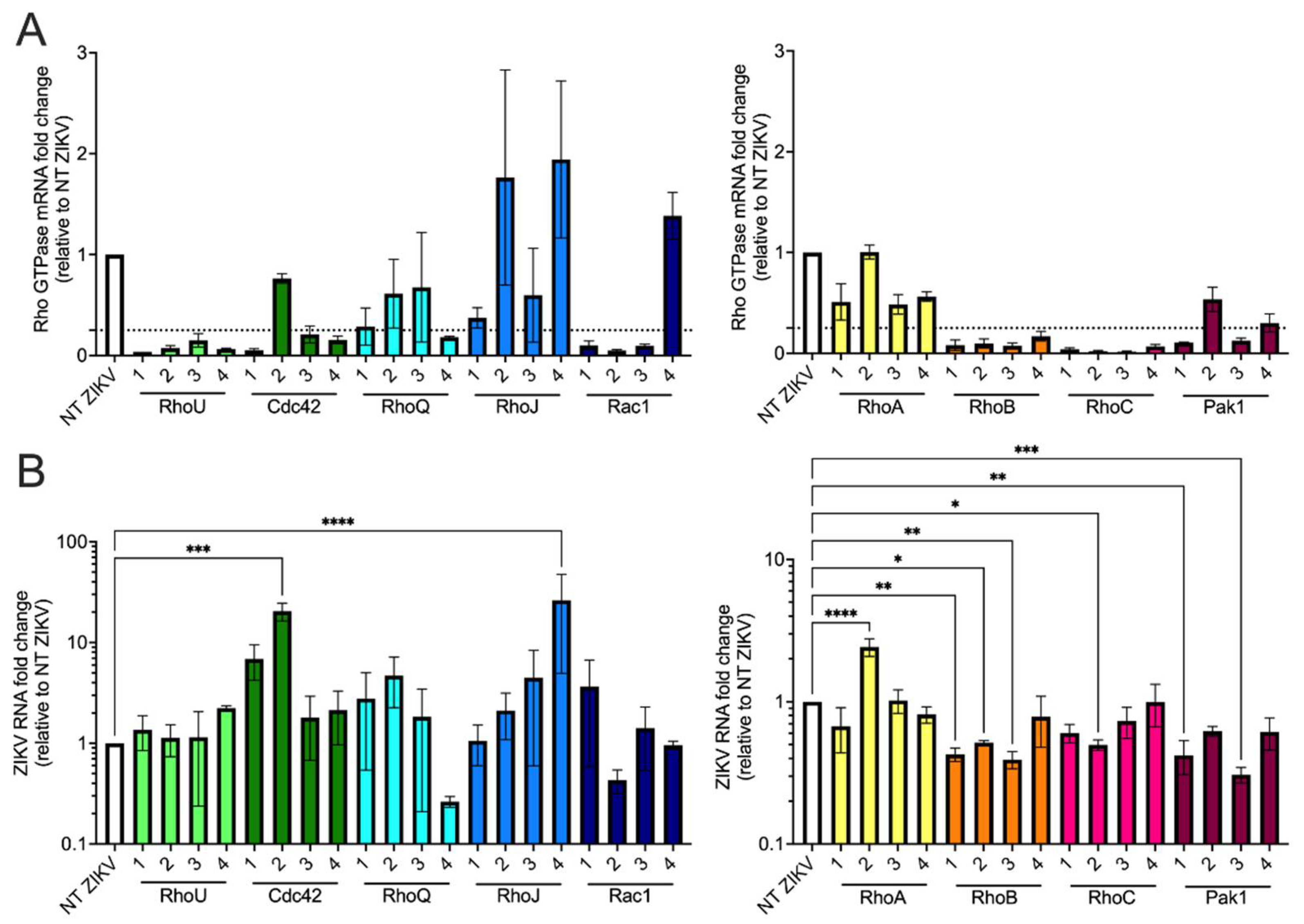
3.5. siRNA Screen of Rho GTPases and Effector Proteins Reveals That Both RhoB and Pak1 Are Proviral Factors for ZIKV
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pierson, T.C.; Diamond, M.S. The continued threat of emerging flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef]
- Chang, C.; Ortiz, K.; Ansari, A.; Gershwin, M.E. The Zika outbreak of the 21st century. J. Autoimmun. 2016, 68, 1–13. [Google Scholar] [CrossRef]
- Zorrilla, C.D.; García, I.G.; Fragoso, L.G.; De La Vega, A. Zika Virus Infection in Pregnancy: Maternal, Fetal, and Neonatal Considerations. J. Infect. Dis. 2017, 216, S891–S896. [Google Scholar] [CrossRef] [Green Version]
- Poland, G.A.; Ovsyannikova, I.G.; Kennedy, R.B. Zika Vaccine Development: Current Status. Mayo Clin. Proc. 2019, 94, 2572–2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, M.D.F.; Mazzon, M.; Jacobs, M.; Amara, A. Pathogenesis of Flavivirus Infections: Using and Abusing the Host Cell. Cell Host Microbe 2009, 5, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Marceau, C.D.; Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Brewer, S.M.; Fuchs, G.; Swaminathan, K.; Mata, M.A.; Elias, J.E.; Sarnow, P.; et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 2016, 535, 159–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savidis, G.; McDougall, W.M.; Meraner, P.; Perreira, J.M.; Portmann, J.M.; Trincucci, G.; John, S.P.; Aker, A.M.; Renzette, N.; Robbins, D.R.; et al. Identification of Zika Virus and Dengue Virus Dependency Factors using Functional Genomics. Cell Rep. 2016, 16, 232–246. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Miner, J.J.; Gorman, M.J.; Rausch, K.; Ramage, H.; White, J.P.; Zuiani, A.; Zhang, P.; Fernandez, E.; Zhang, Q.; et al. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 2016, 535, 164–168. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, H.-H.; Schneider, W.M.; Rozen-Gagnon, K.; Miles, L.A.; Schuster, F.; Razooky, B.; Jacobson, E.; Wu, X.; Yi, S.; Rudin, C.M.; et al. TMEM41B Is a Pan-flavivirus Host Factor. Cell 2021, 184, 133–148.e20. [Google Scholar] [CrossRef]
- Li, Y.; Muffat, J.; Javed, A.O.; Keys, H.R.; Lungjangwa, T.; Bosch, I.; Khan, M.; Virgilio, M.; Gehrke, L.; Sabatini, D.M.; et al. Genome-wide CRISPR screen for Zika virus resistance in human neural cells. Proc. Natl. Acad. Sci. USA 2019, 116, 9527–9532. [Google Scholar] [CrossRef] [Green Version]
- Konermann, S.; Brigham, M.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.; Habib, N.; Gootenberg, J.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2014, 517, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoggins, J.W.; Wilson, S.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Dukhovny, A.; Lamkiewicz, K.; Chen, Q.; Fricke, M.; Jabrane-Ferrat, N.; Marz, M.; Jung, J.U.; Sklan, E.H. A CRISPR Activation Screen Identifies Genes That Protect against Zika Virus Infection. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.R.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sanchez-Seco, M.P.; Evans, M.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef] [Green Version]
- Boisson-Dupuis, S.; Jouanguy, E.; Al-Hajjar, S.; Fieschi, C.; Al-Mohsen, I.Z.; Al-Jumaah, S.; Yang, K.; Chapgier, A.; Eidenschenk, C.; Eid, P.; et al. Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency. Nat. Genet. 2003, 33, 388–391. [Google Scholar] [CrossRef]
- Garcia, G.; Paul, S.; Beshara, S.; Ramanujan, V.K.; Ramaiah, A.; Nielsen-Saines, K.; Li, M.M.; French, S.W.; Morizono, K.; Kumar, A.; et al. Hippo Signaling Pathway Has a Critical Role in Zika Virus Replication and in the Pathogenesis of Neuroinflammation. Am. J. Pathol. 2020, 190, 844–861. [Google Scholar] [CrossRef]
- Hodge, R.G.; Ridley, A.J. Regulation and functions of RhoU and RhoV. Small GTPases 2017, 11, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2013, 343, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Sanjana, N.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [Green Version]
- Yau, E.H.; Rana, T.M. Next-Generation Sequencing of Genome-Wide CRISPR Screens. Next Gener. Seq. 2017, 1712, 203–216. [Google Scholar] [CrossRef]
- Li, W.; Xu, H.; Xiao, T.; Cong, L.; Love, M.I.; Zhang, F.; Irizarry, R.A.; Liu, J.S.; Brown, M.; Liu, X.S. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, A.; Berenshteyn, F.; Brivanlou, A.H. An Efficient and Reversible Transposable System for Gene Delivery and Lineage-Specific Differentiation in Human Embryonic Stem Cells. Cell Stem Cell 2009, 5, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.M.H.; Aguilar, E.G.; Michailidis, E.; Pabon, J.; Park, P.; Wu, X.; de Jong, Y.P.; Schneider, W.M.; Molina, H.; Rice, C.M.; et al. Characterization of Novel Splice Variants of Zinc Finger Antiviral Protein (ZAP). J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, D.; Zhang, T.-H.; Zhao, D.; Du, Y.; Chapa, T.J.; Shi, Y.; Wang, L.; Contreras, D.; Zeng, G.; Shi, P.-Y.; et al. High-Throughput Fitness Profiling of Zika Virus E Protein Reveals Different Roles for Glycosylation during Infection of Mammalian and Mosquito Cells. iScience 2018, 1, 97–111. [Google Scholar] [CrossRef] [Green Version]
- Blight, K.J.; McKeating, J.; Rice, C.M. Highly Permissive Cell Lines for Subgenomic and Genomic Hepatitis C Virus RNA Replication. J. Virol. 2002, 76, 13001–13014. [Google Scholar] [CrossRef] [Green Version]
- Schoggins, J.W.; Dorner, M.; Feulner, M.; Imanaka, N.; Murphy, M.Y.; Ploss, A.; Rice, C.M. Dengue reporter viruses reveal viral dynamics in interferon receptor-deficient mice and sensitivity to interferon effectors in vitro. Proc. Natl. Acad. Sci. USA 2012, 109, 14610–14615. [Google Scholar] [CrossRef] [Green Version]
- Franco, D.; Li, W.; Qing, F.; Stoyanov, C.T.; Moran, T.; Rice, C.M.; Ho, D.D. Evaluation of yellow fever virus 17D strain as a new vector for HIV-1 vaccine development. Vaccine 2010, 28, 5676–5685. [Google Scholar] [CrossRef]
- Robbiani, D.F.; Bozzacco, L.; Keeffe, J.R.; Khouri, R.; Olsen, P.; Gazumyan, A.; Schaefer-Babajew, D.; Avila-Rios, S.; Nogueira, L.; Patel, R.; et al. Recurrent Potent Human Neutralizing Antibodies to Zika Virus in Brazil and Mexico. Cell 2017, 169, 597–609.e11. [Google Scholar] [CrossRef] [Green Version]
- Bick, M.; Carroll, J.-W.N.; Gao, G.; Goff, S.P.; Rice, C.M.; MacDonald, M.R. Expression of the Zinc-Finger Antiviral Protein Inhibits Alphavirus Replication. J. Virol. 2003, 77, 11555–11562. [Google Scholar] [CrossRef] [Green Version]
- Frumence, E.; Roche, M.; Krejbich-Trotot, P.; El Kalamouni, C.; Nativel, B.; Rondeau, P.; Missé, D.; Gadea, G.; Viranaicken, W.; Desprès, P. The South Pacific epidemic strain of Zika virus replicates efficiently in human epithelial A549 cells leading to IFN-β production and apoptosis induction. Virology 2016, 493, 217–226. [Google Scholar] [CrossRef]
- Turpin, J.; Frumence, E.; Desprès, P.; Viranaicken, W.; Krejbich-Trotot, P. The ZIKA Virus Delays Cell Death through the Anti-Apoptotic Bcl-2 Family Proteins. Cells 2019, 8, 1338. [Google Scholar] [CrossRef] [Green Version]
- Hammack, C.; Ogden, S.C.; Madden, J.C.; Medina, A.; Xu, C.; Phillips, E.; Son, Y.; Cone, A.; Giovinazzi, S.; Didier, R.A.; et al. Zika Virus Infection Induces DNA Damage Response in Human Neural Progenitors That Enhances Viral Replication. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Lee, E.M.; Wen, Z.; Cheng, Y.; Huang, W.-K.; Qian, X.; Tcw, J.; Kouznetsova, J.; Ogden, S.C.; Hammack, C.; et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 2016, 22, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Broeke, C.V.D.; Jacob, T.; Favoreel, H.W. Rho’ing in and out of cells. Small GTPases 2014, 5, e28318. [Google Scholar] [CrossRef] [PubMed]
- Chenette, E.; Abo, A.; Der, C. Critical and Distinct Roles of Amino- and Carboxyl-terminal Sequences in Regulation of the Biological Activity of the Chp Atypical Rho GTPase. J. Biol. Chem. 2005, 280, 13784–13792. [Google Scholar] [CrossRef] [Green Version]
- Banyard, J.; Anand-Apte, B.; Symons, M.; Zetter, B.R. Motility and invasion are differentially modulated by Rho family GTPases. Oncogene 2000, 19, 580–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanni, C.; Ottaviano, C.; Guo, F.; Puppo, M.; Varesio, L.; Zheng, Y.; Eva, A. Constitutively Active Cdc42 Mutant Confers Growth Disadvantage in Cell Transformation. Cell Cycle 2005, 4, 1675–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuartas-López, A.M.; Hernández-Cuellar, C.E.; Gallego-Gómez, J.C. Disentangling the role of PI3K/Akt, Rho GTPase and the actin cytoskeleton on dengue virus infection. Virus Res. 2018, 256, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-L.; Zhang, J.-L.; Chen, W.; Xu, X.-F.; Gao, N.; Fan, D.-Y.; An, J. Roles of Small GTPase Rac1 in the Regulation of Actin Cytoskeleton during Dengue Virus Infection. PLoS Neglected Trop. Dis. 2010, 4, e809. [Google Scholar] [CrossRef] [Green Version]
- Zamudio-Meza, H.; Castillo, A.; Bonilla, C.G.; Meza, I. Cross-talk between Rac1 and Cdc42 GTPases regulates formation of filopodia required for dengue virus type-2 entry into HMEC-1 cells. J. Gen. Virol. 2009, 90, 2902–2911. [Google Scholar] [CrossRef]
- Kalia, M.; Khasa, R.; Sharma, M.; Nain, M.; Vrati, S. Japanese Encephalitis Virus Infects Neuronal Cells through a Clathrin-Independent Endocytic Mechanism. J. Virol. 2012, 87, 148–162. [Google Scholar] [CrossRef] [Green Version]
- Korobko, I.V.; Shepelev, M.V. Mutations in the Effector Domain of RhoV GTPase Impair Its Binding to Pak1 Protein Kinase. Mol. Biol. 2018, 52, 598–603. [Google Scholar] [CrossRef]
- Harms, F.L.; Kloth, K.; Bley, A.; Denecke, J.; Santer, R.; Lessel, D.; Hempel, M.; Kutsche, K. Activating Mutations in PAK1, Encoding p21-Activated Kinase 1, Cause a Neurodevelopmental Disorder. Am. J. Hum. Genet. 2018, 103, 579–591. [Google Scholar] [CrossRef] [Green Version]
- Moureau, G.; Cook, S.; Lemey, P.; Nougairede, A.; Forrester, N.L.; Khasnatinov, M.; Charrel, R.; Firth, A.; Gould, E.A.; De Lamballerie, X. New Insights into Flavivirus Evolution, Taxonomy and Biogeographic History, Extended by Analysis of Canonical and Alternative Coding Sequences. PLoS ONE 2015, 10, e0117849. [Google Scholar] [CrossRef] [Green Version]
- Agrelli, A.; de Moura, R.R.; Crovella, S.; Brandão, L.A.C. ZIKA virus entry mechanisms in human cells. Infect. Genet. Evol. 2019, 69, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Vancini, R.; Hernandez, R.; Brown, D. Alphavirus Entry into Host Cells. Prog. Mol. Biol. Transl. Sci. 2015, 129, 33–62. [Google Scholar] [CrossRef] [PubMed]
- Smit, J.M.; Moesker, B.; Rodenhuis-Zybert, I.; Wilschut, J. Flavivirus Cell Entry and Membrane Fusion. Viruses 2011, 3, 160–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radoshitzky, S.; Pegoraro, G.; Chī, X.; Dǒng, L.; Chiang, C.-Y.; Jozwick, L.; Clester, J.C.; Cooper, C.; Currier, D.; Langan, D.P.; et al. siRNA Screen Identifies Trafficking Host Factors that Modulate Alphavirus Infection. PLoS Pathog. 2016, 12, e1005466. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.I. Cdc42: An Essential Rho-Type GTPase Controlling Eukaryotic Cell Polarity. Microbiol. Mol. Biol. Rev. 1999, 63, 54–105. [Google Scholar] [CrossRef] [Green Version]
- Goulidaki, N.; Alarifi, S.; Alkahtani, S.H.; Al-Qahtani, A.; Spandidos, D.; Stournaras, C.; Sourvinos, G. RhoB is a component of the human cytomegalovirus assembly complex and is required for efficient viral production. Cell Cycle 2015, 14, 2748–2763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseliou, M.; Al-Qahtani, A.; Alarifi, S.; Alkahtani, S.H.; Stournaras, C.; Sourvinos, G. The Role of RhoA, RhoB and RhoC GTPases in Cell Morphology, Proliferation and Migration in Human Cytomegalovirus (HCMV) Infected Glioblastoma Cells. Cell. Physiol. Biochem. 2016, 38, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Quinn, K.; Brindley, M.A.; Weller, M.L.; Kaludov, N.; Kondratowicz, A.; Hunt, C.L.; Sinn, P.; McCray, P.; Stein, C.S.; Davidson, B.L.; et al. Rho GTPases Modulate Entry of Ebola Virus and Vesicular Stomatitis Virus Pseudotyped Vectors. J. Virol. 2009, 83, 10176–10186. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward (F) and Reverse (R) Primer Sequences |
|---|---|
| RhoV | F: 5′-CCTCATCGTCAGCTACACCTG-3′ R: 5′-GAACGAAGTCGGTCAAAATCCT-3′ |
| RhoU | F: 5′-GCTACCCCACCGAGTACATC-3′ R: 5′-GGCTCACGACACTGAAGCA-3′ |
| Cdc42 | F: 5′-CCATCGGAATATGTACCGACTG-3′ R: 5′-CTCAGCGGTCGTAATCTGTCA-3′ |
| RhoQ | F: 5′-CCACCGTCTTCGACCACTAC-3′ R: 5′-AGGCTGGATTTACCACCGAGA-3′ |
| RhoJ | F: 5′-AGGGGCAACGACGAGAAGA-3′ R: 5′-TTGGCGTAGCTCATCAGCAG-3′ |
| Rac1 | F: 5′-ATGTCCGTGCAAAGTGGTATC-3′ R: 5′-CTCGGATCGCTTCGTCAAACA-3′ |
| RhoA | F: 5′-GGAAAGCAGGTAGAGTTGGCT-3′ R: 5′-GGCTGTCGATGGAAAAACACAT-3′ |
| RhoB | F: 5′-CTGCTGATCGTGTTCAGTAAGG-3′ R: 5′-TCAATGTCGGCCACATAGTTC-3′ |
| RhoC | F: 5′-GGAGGTCTACGTCCCTACTGT-3′ R: 5′-CGCAGTCGATCATAGTCTTCC-3′ |
| Pak1 | F: 5′-CAGCCCCTCCGATGAGAAATA-3′ R: 5′-CAAAACCGACATGAATTGTGTGT-3′ |
| ZIKV | F: 5′-TTGTGGAAGGTATGTCAGGTG-3′ R: 5′-ATCTTACCTCCGCCATGTTG-3′ |
| RPS11 | F: 5′-GCCGAGACTATCTGCACTAC-3′ R: 5′-ATGTCCAGCCTCAGAACTTC-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luu, A.P.; Yao, Z.; Ramachandran, S.; Azzopardi, S.A.; Miles, L.A.; Schneider, W.M.; Hoffmann, H.-H.; Bozzacco, L.; Garcia, G., Jr.; Gong, D.; et al. A CRISPR Activation Screen Identifies an Atypical Rho GTPase That Enhances Zika Viral Entry. Viruses 2021, 13, 2113. https://doi.org/10.3390/v13112113
Luu AP, Yao Z, Ramachandran S, Azzopardi SA, Miles LA, Schneider WM, Hoffmann H-H, Bozzacco L, Garcia G Jr., Gong D, et al. A CRISPR Activation Screen Identifies an Atypical Rho GTPase That Enhances Zika Viral Entry. Viruses. 2021; 13(11):2113. https://doi.org/10.3390/v13112113
Chicago/Turabian StyleLuu, Anh Phuong, Zhenlan Yao, Sangeetha Ramachandran, Stephanie A. Azzopardi, Linde A. Miles, William M. Schneider, H.-Heinrich Hoffmann, Leonia Bozzacco, Gustavo Garcia, Jr., Danyang Gong, and et al. 2021. "A CRISPR Activation Screen Identifies an Atypical Rho GTPase That Enhances Zika Viral Entry" Viruses 13, no. 11: 2113. https://doi.org/10.3390/v13112113
APA StyleLuu, A. P., Yao, Z., Ramachandran, S., Azzopardi, S. A., Miles, L. A., Schneider, W. M., Hoffmann, H.-H., Bozzacco, L., Garcia, G., Jr., Gong, D., Damoiseaux, R., Tang, H., Morizono, K., Rudin, C. M., Sun, R., Arumugaswami, V., Poirier, J. T., MacDonald, M. R., Rice, C. M., & Li, M. M. H. (2021). A CRISPR Activation Screen Identifies an Atypical Rho GTPase That Enhances Zika Viral Entry. Viruses, 13(11), 2113. https://doi.org/10.3390/v13112113