
CRISPR Tackles Emerging Viral Pathogens


Abstract
1. Introduction
2. Origins of CRISPR Genome Editing Technology

{kind=link}
{kind=link}
{kind=link}
| Nuclease Domains | PAM | Substrate | Cleavage | Collateral Cleavage? | Use in Virus Research | ||
|---|---|---|---|---|---|---|---|
| Cas9 |  | RuvC, HNH | 5′-NGG-3′ | dsDNA | Blunt ends | No | Utilised in numerous genome-wide and target-specific CRISPR screens to identify and characterise the relationship between cellular host factors and viruses (refer to Table 2) Used in studies aiming to inactivate integrated viral DNA that results in chronic infection (e.g., HBV) [53] |
| Cas12 |  | RuvC, Nuc | 5′-TTTN-3′ | dsDNA | 5′ staggered overhang of 5 bp | Yes—ssDNA Not in mammalian or plant culture | Forms the basis of the DETECTR diagnostic method used for detection of viral nucleic acids (e.g., SARS-CoV-2) [40] Potential alternative for genome-wide screens due to alternative PAM sequence requirements, allowing potential to target T’-rich gene sequences [35] |
| Cas13 |  | 2× HEPN | Subspecies and culture model dependent. Preference for 5′ “Protospacer Flanking Sequence” | ssRNA | Cleavage at uracil | Yes—ssRNA Not in mammalian or plant culture | Forms the basis of the SHERLOCK diagnostic method for detection of viral nucleic acids (e.g., SARS-CoV-2) [51] Shown in cell culture models to cleave viral RNA and inhibit replication [50] Development of dCas13 as a method to track viral RNA movement in the cell throughout viral replication [52] |
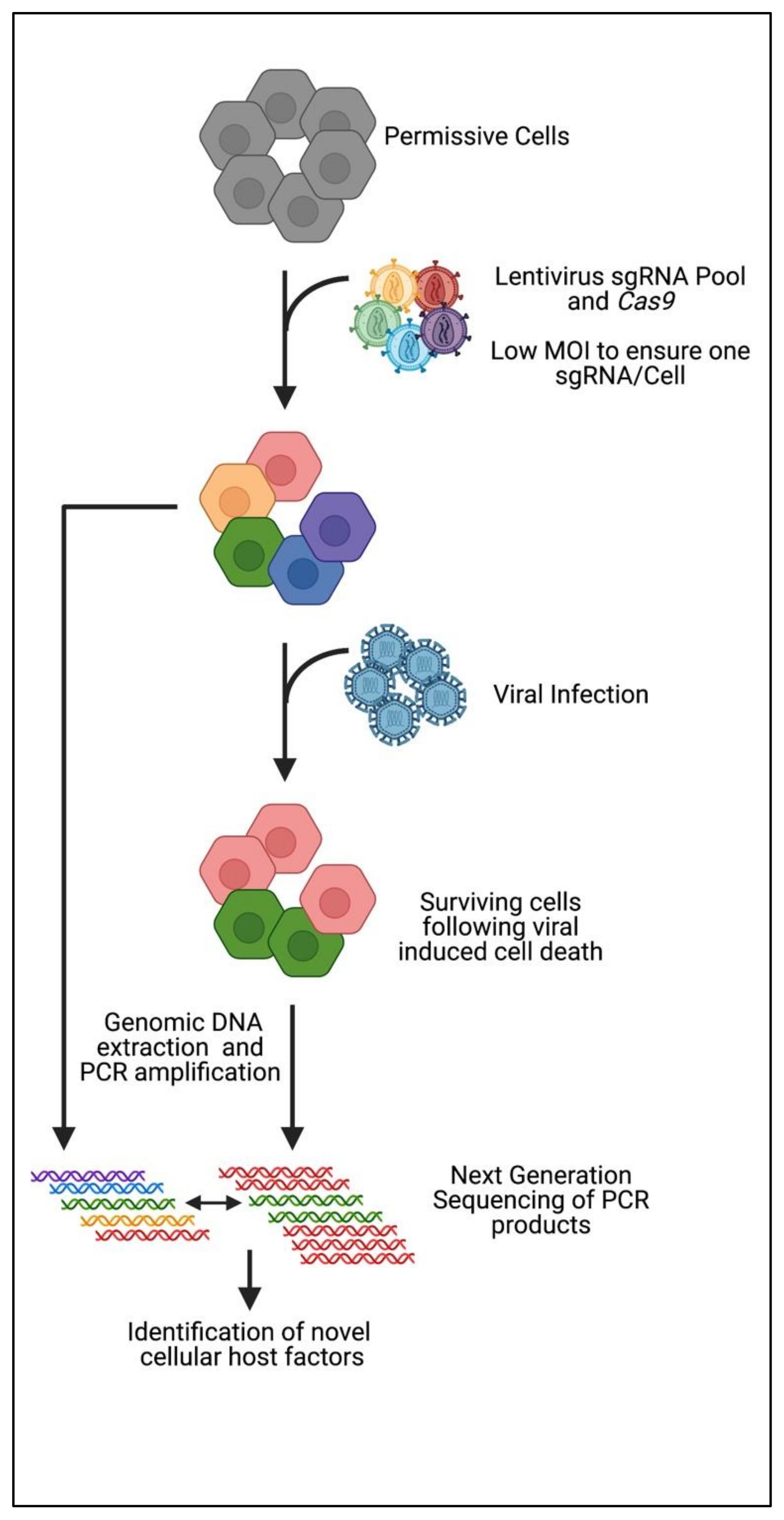
3. CRISPR Knockout Screening
4. CRISPR Activation Screening
4.1. CRISPR-VPR
4.2. CRISPR-Synergistic Activation Mediator (SAM)
4.3. CRISPR-SunTag
4.4. Zika Virus
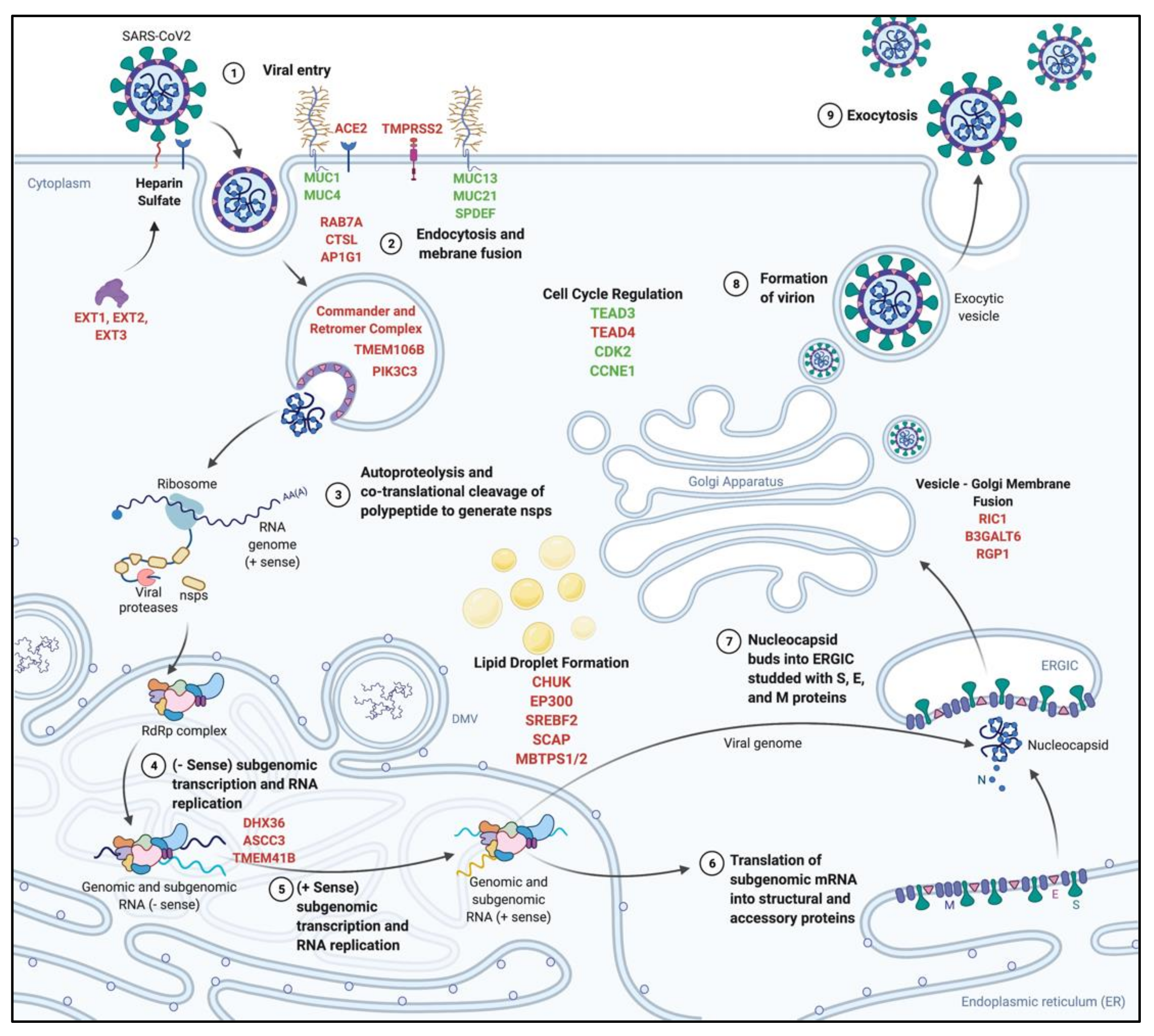
4.5. SARS-CoV-2
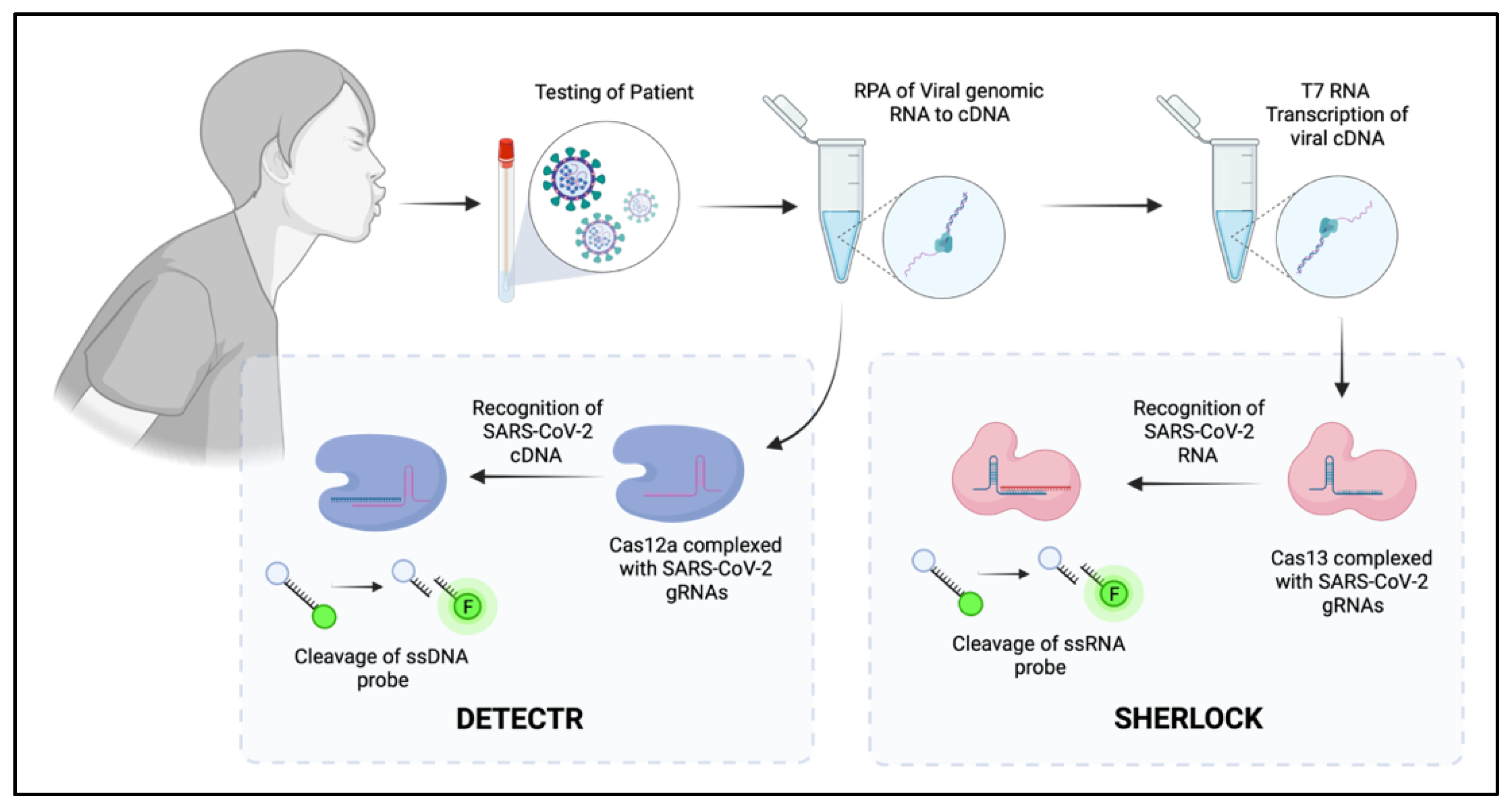
5. CRISPR as the Future of Diagnostic Screening
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 19 June 2021).
- Zhu, Z.; Lian, X.; Su, X.; Wu, W.; Marraro, G.A.; Zeng, Y. From SARS and MERS to COVID-19: A brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir. Res. 2020, 21, 224. [Google Scholar] [CrossRef]
- Yun, S.I.; Lee, Y.M. Zika virus: An emerging flavivirus. J. Microbiol. 2017, 55, 204–219. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.G.; Gubler, D.J.; Izquierdo, A.; Martinez, E.; Halstead, S.B. Dengue infection. Nat. Rev. Dis. Primers 2016, 2, 16055. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Morens, D.M. 1918 Influenza: The Mother of All Pandemics. Emerg. Infect. Dis. 2006, 12, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV infection. Nat. Rev. Dis. Primers 2015, 1, 15035. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Dang, Y.; Wu, Y.; Jia, G.; Anaya, E.; Zhang, J.; Abraham, S.; Choi, J.G.; Shi, G.; Qi, L.; et al. A CRISPR-Based Screen Identifies Genes Essential for West-Nile-Virus-Induced Cell Death. Cell Rep. 2015, 12, 673–683. [Google Scholar] [CrossRef]
- Marceau, C.D.; Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Brewer, S.M.; Fuchs, G.; Swaminathan, K.; Mata, M.A.; Elias, J.E.; Sarnow, P.; et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 2016, 535, 159–163. [Google Scholar] [CrossRef]
- Zhang, R.; Miner, J.J.; Gorman, M.J.; Rausch, K.; Ramage, H.; White, J.P.; Zuiani, A.; Zhang, P.; Fernandez, E.; Zhang, Q.; et al. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 2016, 535, 164–168. [Google Scholar] [CrossRef]
- Pichlmair, A.; Reis e Sousa, C. Innate recognition of viruses. Immunity 2007, 27, 370–383. [Google Scholar] [CrossRef]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef]
- Lee, A.S.; Burdeinick-Kerr, R.; Whelan, S.P. A genome-wide small interfering RNA screen identifies host factors required for vesicular stomatitis virus infection. J. Virol. 2014, 88, 8355–8360. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martin, S.; Chiramel, A.I.; Schmidt, M.L.; Chen, Y.C.; Whitt, N.; Watt, A.; Dunham, E.C.; Shifflett, K.; Traeger, S.; Leske, A.; et al. A genome-wide siRNA screen identifies a druggable host pathway essential for the Ebola virus life cycle. Genome Med. 2018, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Radoshitzky, S.R.; Pegoraro, G.; Chi, X.O.; Dong, L.; Chiang, C.Y.; Jozwick, L.; Clester, J.C.; Cooper, C.L.; Courier, D.; Langan, D.P.; et al. siRNA Screen Identifies Trafficking Host Factors that Modulate Alphavirus Infection. PLoS Pathog. 2016, 12, e1005466. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Brass, A.L.; Ng, A.; Hu, Z.; Xavier, R.J.; Liang, T.J.; Elledge, S.J. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl. Acad. Sci. USA 2009, 106, 16410–16415. [Google Scholar] [CrossRef]
- Ramanan, V.; Shlomai, A.; Cox, D.B.; Schwartz, R.E.; Michailidis, E.; Bhatta, A.; Scott, D.A.; Zhang, F.; Rice, C.M.; Bhatia, S.N. CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Sci. Rep. 2015, 5, 10833. [Google Scholar] [CrossRef]
- Seeger, C.; Sohn, J.A. Targeting Hepatitis B Virus with CRISPR/Cas9. Mol. Ther. Nucleic Acids 2014, 3, e216. [Google Scholar] [CrossRef]
- Dash, P.K.; Kaminski, R.; Bella, R.; Su, H.; Mathews, S.; Ahooyi, T.M.; Chen, C.; Mancuso, P.; Sariyer, R.; Ferrante, P.; et al. Sequential LASER ART and CRISPR Treatments Eliminate HIV-1 in a Subset of Infected Humanized Mice. Nat. Commun. 2019, 10, 2753. [Google Scholar] [CrossRef]
- Xiao, Q.; Guo, D.; Chen, S. Application of CRISPR/Cas9-Based Gene Editing in HIV-1/AIDS Therapy. Front. Cell Infect. Microbiol. 2019, 9, 69. [Google Scholar] [CrossRef]
- Li, H.; Sheng, C.; Wang, S.; Yang, L.; Liang, Y.; Huang, Y.; Liu, H.; Li, P.; Yang, C.; Yang, X.; et al. Removal of Integrated Hepatitis B Virus DNA Using CRISPR-Cas9. Front. Cell Infect. Microbiol. 2017, 7, 91. [Google Scholar] [CrossRef]
- Stone, D.; Long, K.R.; Loprieno, M.A.; De Silva Feelixge, H.S.; Kenkel, E.J.; Liley, R.M.; Rapp, S.; Roychoudhury, P.; Nguyen, T.; Stensland, L.; et al. CRISPR-Cas9 gene editing of hepatitis B virus in chronically infected humanized mice. Mol. Ther. Methods Clin. Dev. 2021, 20, 258–275. [Google Scholar] [CrossRef]
- Das, A.T.; Binda, C.S.; Berkhout, B. Elimination of infectious HIV DNA by CRISPR-Cas9. Curr. Opin. Virol. 2019, 38, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, R.; Moineau, S.; Romero, D.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Sci. Rep. 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.; Wolf, Y.I.; Koonin, E. Classification and Nomenclature of CRISPR-Cas Systems: Where from Here? CRISPR J. 2018, 1, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Shmakov, S.; Smargon, A.; Scott, D.; Cox, D.; Pyzocha, N.; Yan, W.; Abudayyeh, O.O.; Gootenberg, J.S.; Makarova, K.S.; Wolf, Y.I.; et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol. 2017, 15, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR-Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentie, E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Lim, Y.; Bak, S.Y.; Sung, K.; Jeong, E.; Lee, S.H.; Kim, J.S.; Bae, S.; Kim, S.K. Structural roles of guide RNAs in the nuclease activity of Cas9 endonuclease. Nat. Commun. 2016, 7, 13350. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, 2579–2586. [Google Scholar] [CrossRef]
- Chen, H.; Choi, J.; Bailey, S. Cut Site Selection by the Two Nuclease Domains of the Cas9 RNA-guided Endonuclease. J. Biol. Chem. 2014, 289, 13284–13294. [Google Scholar] [CrossRef]
- Andersson, A.; Banfield, J. Virus Population Dynamics and Acquired Virus Resistance in Natural Microbial Communities. Science 2008, 320, 1047–1049. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Proudfoot, C.; Whitelaw, B.; Lillico, S. Comparison of CRISPR/Cas9 and TALENs on editing an integrated EGFP gene in the genome of HEK293FT cells. SpringerPlus 2016, 5, 814. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Guerrero, A.; Sanchez-Hernandez, S.; Galvani, G.; Pinedo-Gomez, J.; Martin-Guerra, R.; Sanchez-Gilabert, A.; Aguilar-González, A.; Cobo, M.; Gregory, P.; Holmes, M.; et al. Comparison of Zinc Finger Nucleases Versus CRISPR-Specific Nucleases for Genome Editing of the Wiskott-Aldrich Syndrome Locus. Hum. Gene Ther. 2018, 29, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Stella, S.; Alcon, P.; Montoya, G. Structure of the Cpf1 endonuclease R-loop complex after target DNA cleavage. Nature 2017, 546, 559–563. [Google Scholar] [CrossRef]
- Fonfara, I.; Richter, H.; Bratovic, M.; Le Rhun, A.; Charpentier, E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 2016, 532, 517–521. [Google Scholar] [CrossRef]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Cosa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Kellner, M.J.; Joung, J.; Collins, J.J.; Zhang, F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a and Csm6. Science 2018, 360, 439–444. [Google Scholar] [CrossRef]
- Broughton, J.P.; Deng, X.; Yu, G.; Fasching, C.L.; Servellita, V.; Singh, J.; Miao, X.; Streithorst, J.A.; Granados, A.; Sotomayor-Gonzalez, A.; et al. CRISPR-Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020, 38, 870–874. [Google Scholar] [CrossRef]
- Zhang, B.; Ye, Y.; Ye, W.; Perculija, V.; Jiang, H.; Chen, Y.; Li, Y.; Chen, J.; Lin, J.; Wang, S.; et al. Two HEPN domains dictate CRISPR RNA maturation and target cleavage in Cas13d. Nat. Commun. 2019, 10, 2544. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef] [PubMed]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 019–1027. [Google Scholar] [CrossRef]
- East-Seletsky, A.; O’Connell, M.R.; Knight, S.C.; Burstein, D.; Cate, J.H.; Tjian, R.; Doudna, J.A. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 2016, 538, 270–273. [Google Scholar] [CrossRef]
- Konermann, S.; Lotfy, P.; Brideau, N.J.; Oki, J.; Shokhirev, M.N.; Hsu, P.D. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 2018, 173, 665–676.e614. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Zhou, Y.; Xiao, Q.; He, B.; Geng, G.; Wang, Z.; Cao, B.; Dong, X.; Bai, W.; Wang, Y.; et al. Programmable RNA editing with compact CRISPR-Cas13 systems from uncultivated microbes. Nat. Methods 2021, 18, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, C.M.; Myhrvold, C.; Thakku, S.G.; Freije, C.A.; Metsky, H.C.; Yang, D.K.; Ye, S.H.; Boehm, C.K.; Kosoko-Thoroddsen, T.F.; Kehe, J.; et al. Massively multiplexed nucleic acid detection with Cas13. Nature 2020, 582, 277–282. [Google Scholar] [CrossRef]
- Myhrvold, C.; Freije, C.A.; Gootenberg, J.S.; Abudayyeh, O.O.; Metsky, H.C.; Durbin, A.F.; Kellner, M.J.; Tan, A.L.; Paul, L.M.; Parham, L.A.; et al. field-deployable viral diagnostics using CRISPR-Cas13. Science 2018, 360, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Freije, C.A.; Myhrvold, C.; Boehm, C.K.; Lin, A.E.; Welch, N.L.; Carter, A.; Metsky, H.C.; Luo, C.Y.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. Programmable Inhibition and Detection of RNA Viruses Using Cas13. Mol. Cell 2019, 76, 826–837.e811. [Google Scholar] [CrossRef]
- Patchsung, M.; Jantarug, K.; Pattama, A.; Aphicho, K.; Suraritdechachai, S.; Meesawat, P.; Sappakhaw, K.; Leelahakorn, N.; Ruenkam, T.; Wongsatit, T.; et al. Clinical validation of a Cas13-based assay for the detection of SARS-CoV-2 RNA. Nat. Biomed. Eng. 2020, 4, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.Z.; Wang, Y.; Li, S.Q.; Yao, R.W.; Luan, P.F.; Wu, H.; Carmichael, G.G.; Chen, L.L. Dynamic Imaging of RNA in Living Cells by CRISPR-Cas13 Systems. Mol. Cell 2019, 76, 981–997.e987. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Chen, Y.H.; Kao, J.H.; Ching, C.; Liu, I.J.; Wang, C.C.; Tsai, C.H.; Wu, F.Y.; Liu, C.J.; Chen, P.J.; et al. Permanent Inactivation of HBV Genomes by CRISPR/Cas9-Mediated Non-cleavage Base Editing. Mol. Ther. Nucleic Acids 2020, 20, 480–490. [Google Scholar] [CrossRef]
- Pillay, S.; Carette, J.E. Hunting Viral Receptors Using Haploid Cells. Annu. Rev. Virol. 2015, 2, 219–239. [Google Scholar] [CrossRef]
- Luteijn, R.D.; van Diemen, F.; Blomen, V.A.; Boer, I.G.J.; Manikam Sadasivam, S.; van Kuppevelt, T.H.; Drexler, I.; Brummelkamp, T.R.; Lebbink, R.J.; Wiertz, E.J. A Genome-Wide Haploid Genetic Screen Identifies Heparan Sulfate-Associated Genes and the Macropinocytosis Modulator TMED10 as Factors Supporting Vaccinia Virus Infection. J. Virol. 2019, 93, e02160-18. [Google Scholar] [CrossRef] [PubMed]
- Timms, R.T.; Duncan, L.M.; Tchasovnikarova, I.A.; Antrobus, R.; Smith, D.L.; Dougan, G.; Weekes, M.P.; Lehner, P.J. Haploid genetic screens identify an essential role for PLP2 in the downregulation of novel plasma membrane targets by viral E3 ubiquitin ligases. PLoS Pathog. 2013, 9, e1003772. [Google Scholar] [CrossRef] [PubMed]
- Ramage, H.; Cherry, S. Virus-Host Interactions: From Unbiased Genetic Screens to Function. Annu. Rev. Virol. 2015, 2, 497–524. [Google Scholar] [CrossRef] [PubMed]
- Kurreck, J. siRNA efficiency: Structure or sequence-that is the question. J. Biomed. Biotechnol. 2006, 2006, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, M.; McManus, M.T. Choosing the Right Tool for the Job: RNAi, TALEN, or CRISPR. Mol. Cell 2015, 58, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Tuschl, T. Mechanisms of gene silencing by double-stranded RNA. Nature 2004, 431, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, A.J. The use of RNAi-based screens to identify host proteins involved in viral replication. Future Microbiol. 2010, 5, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Karlas, A.; Machuy, N.; Shin, Y.; Pleissner, K.P.; Artarini, A.; Heuer, D.; Becker, D.; Khalil, H.; Ogilvie, L.A.; Hess, S.; et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 2010, 463, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Xu, M.; Huang, Q.; Gates, A.T.; Zhang, X.D.; Castle, J.C.; Stec, E.; Ferrer, M.; Strulovici, B.; Hazuda, D.J.; et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008, 4, 495–504. [Google Scholar] [CrossRef] [PubMed]
- McCormick, D.; Lin, Y.; Grey, F. Idenification of Host Factors Involved in Human Cytomegalovirus Replication, Assembly and Egress Using a Two-Step Small Interfering RNA Screen. mBio 2018, 9, e00716-18. [Google Scholar] [CrossRef] [PubMed]
- Judge, A.D.; Sood, V.; Shaw, J.R.; Fang, D.; McClintock, K.; MacLachlan, I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol. 2005, 23, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Guenthner-Biller, M.; Bourquin, C.; Ablasser, A.; Schlee, M.; Uematsu, S.; Noronha, A.; Manoharan, M.; Akira, S.; de Fougerolles, A.; et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 2005, 11, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Clohisey, S.M.; Chia, B.S.; Wang, B.; Cui, A.; Eisenhaure, T.; Schweitzer, L.D.; Hoover, P.; Parkinson, N.J.; Nachshon, A.; et al. Genome-wide CRISPR screen identifies host dependency factors for influenza A virus infection. Nat. Commun. 2020, 11, 164. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Perez, J.T.; Chen, C.; Li, Y.; Benitez, A.; Kandasamy, M.; Lee, Y.; Andrade, J.; tenOever, B.; Manicassamy, B. Genome-wide CRISPR/Cas9 Screen Identifies Host Factors Essential for Influenza Virus Replication. Cell Rep. 2018, 23, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Heaton, B.E.; Kennedy, E.M.; Dumm, R.E.; Harding, A.T.; Sacco, M.T.; Sachs, D.; Heaton, N.S. A CRISPR Activation Screen Identifies a Pan-avian Influenza Virus Inhibitory Host Factor. Cell Rep. 2017, 20, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Muffat, J.; Omer Javed, A.; Keys, H.R.; Lungjangwa, T.; Bosch, I.; Khan, M.; Virgilio, M.C.; Gehrke, L.; Sabatini, D.M.; et al. Genome-wide CRISPR screen for Zika virus resistance in human neural cells. Proc. Natl. Acad. Sci. USA 2019, 116, 9527–9532. [Google Scholar] [CrossRef]
- Savidis, G.; McDougall, W.M.; Meraner, P.; Perreira, J.M.; Portmann, J.M.; Trincucci, G.; John, S.P.; Aker, A.M.; Renzette, N.; Robbins, D.R.; et al. Identification of Zika Virus and Dengue Virus Dependency Factors using Functional Genomics. Cell Rep. 2016, 16, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, Q.; Tiwari, S.K.; Lichinchi, G.; Yau, E.H.; Hui, H.; Li, W.; Furnari, F.; Rana, T.M. Integrin alphavbeta5 Internalizes Zika Virus during Neural Stem Cells Infection and Provides a Promising Target for Antiviral Therapy. Cell Rep. 2020, 30, 969–983. [Google Scholar] [CrossRef] [PubMed]
- Dukhovny, A.; Lamkiewicz, K.; Chen, Q.; Fricke, M.; Jabrane-Ferrat, N.; Marz, M.; Jung, J.U.; Sklan, E.H. A CRISPR Activation Screen Identifies Genes That Protect against Zika Virus Infection. J. Virol. 2019, 93, e00211-19. [Google Scholar] [CrossRef] [PubMed]
- Labeau, A.; Simon-Loriere, E.; Hafirassou, M.L.; Bonnet-Madin, L.; Tessier, S.; Zamborlini, A.; Dupre, T.; Seta, N.; Schwartz, O.; Chaix, M.L.; et al. A Genome-Wide CRISPR-Cas9 Screen Identifies the Dolichol-Phosphate Mannose Synthase Complex as a Host Dependency Factor for Dengue Virus Infection. J. Virol. 2020, 94, e01751-19. [Google Scholar] [CrossRef] [PubMed]
- OhAinle, M.; Helms, L.; Vermeire, J.; Roesch, F.; Humes, D.; Basom, R.; Delrow, J.J.; Overbaugh, J.; Emerman, M. A virus-packageable CRISPR screen identifies host factors mediating interferon inhibition of HIV. Elife 2018, 7, e39823. [Google Scholar] [CrossRef] [PubMed]
- Krasnopolsky, S.; Kuzmina, A.; Taube, R. Genome-wide CRISPR knockout screen identifies ZNF304 as a silencer of HIV transcription that promotes viral latency. PLoS Pathog. 2020, 16, e1008834. [Google Scholar] [CrossRef]
- Park, R.J.; Wang, T.; Koundakjian, D.; Hultquist, J.F.; Lamothe-Molina, P.; Monel, B.; Schumann, K.; Yu, H.; Krupzcak, K.M.; Garcia-Beltran, W.; et al. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 2017, 49, 193–203. [Google Scholar] [CrossRef]
- Flint, M.; Chatterjee, P.; Lin, D.L.; McMullan, L.K.; Shrivastava-Ranjan, P.; Bergeron, E.; Lo, M.K.; Welch, S.R.; Nichol, S.T.; Tai, A.W.; et al. A genome-wide CRISPR screen identifies N-acetylglucosamine-1-phosphate transferase as a potential antiviral target for Ebola virus. Nat. Commun. 2019, 10, 285. [Google Scholar] [CrossRef]
- Ma, Y.; Walsh, M.J.; Bernhardt, K.; Ashbaugh, C.W.; Trudeau, S.J.; Ashbaugh, I.Y.; Jiang, S.; Jiang, C.; Zhao, B.; Root, D.E.; et al. CRISPR/Cas9 Screens Reveal Epstein-Barr Virus-Transformed B Cell Host Dependency Factors. Cell Host Microbe 2017, 21, 580–591.e587. [Google Scholar] [CrossRef]
- Richardson, R.B.; Ohlson, M.B.; Eitson, J.L.; Kumar, A.; McDougal, M.B.; Boys, I.N.; Mar, K.B.; De La Cruz-Rivera, P.C.; Douglas, C.; Konopka, G.; et al. A CRISPR screen identifies IFI6 as an ER-resident interferon effector that blocks flavivirus replication. Nat. Microbiol. 2018, 3, 1214–1223. [Google Scholar] [CrossRef]
- Orchard, R.C.; Sullender, M.E.; Dunlap, B.F.; Balce, D.R.; Doench, J.G.; Virgin, H.W. Identification of Antinorovirus Genes in Human Cells Using Genome-Wide CRISPR Activation Screening. J. Virol. 2019, 93, e01324-18. [Google Scholar] [CrossRef]
- Orchard, R.C.; Wilen, C.B.; Doench, J.G.; Baldridge, M.T.; McCune, B.T.; Lee, Y.C.; Lee, S.; Pruett-Miller, S.M.; Nelson, C.A.; Fremont, D.H.; et al. Discovery of a proteinaceous cellular receptor for a norovirus. Science 2016, 353, 933–936. [Google Scholar] [CrossRef]
- Kulsuptrakul, J.; Wang, R.; Meyers, N.L.; Ott, M.; Puschnik, A.S. A CRISPR screen identifies UFMylation and TRAMP-like complexes required for hepatitis A virus infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Baggen, J.; Persoons, L.; Vanstreels, E.; Jansen, S.; Van Looveren, D.; Boeckx, B.; Geudens, V.; De Man, J.; Jochmans, D.; Wauters, J.; et al. Genome-wide CRISPR screening identifies TMEM106B as a proviral host factor for SARS-CoV-2. Nat. Genet. 2021, 53, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Biering, S.B.; Sarnik, S.A.; Wang, E.; Zengel, J.R.; Sathyan, V.; Nguyenla, X.; Van Dis, E.; Catamura, C.; Yamashiro, L.H.; Begeman, A.; et al. Genome-wide, bidirectional CRISPR screens identify mucins as critical host factors, modulating SARS-CoV-2 infection. bioRxiv 2021. [Google Scholar] [CrossRef]
- Daniloski, Z.; Jordan, T.X.; Wessels, H.H.; Hoagland, D.A.; Kasela, S.; Legut, M.; Maniatis, S.; Mimitou, E.P.; Lu, L.; Geller, E.; et al. Identification of Required Host Factors for SARS-CoV-2 Infection in Human Cells. Cell 2021, 184, 92–105.e116. [Google Scholar] [CrossRef]
- Wang, R.; Simoneau, C.R.; Kulsuptrakul, J.; Bouhaddou, M.; Travisano, K.A.; Hayashi, J.M.; Carlson-Stevermer, J.; Zengel, J.R.; Richards, C.M.; Fozouni, P.; et al. Genetic Screens Identify Host Factors for SARS-CoV-2 and Common Cold Coronaviruses. Cell 2021, 184, 106–119.e114. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Luna, J.M.; Hoffmann, H.H.; Sanchez-Rivera, F.J.; Leal, A.A.; Ashbrook, A.W.; Le Pen, J.; Ricardo-Lax, I.; Michailidis, E.; Peace, A.; et al. Genome-Scale Identification of SARS-CoV-2 and Pan-coronavirus Host Factor Networks. Cell 2021, 184, 120–132.e114. [Google Scholar] [CrossRef]
- Wei, J.; Alfajaro, M.M.; DeWeirdt, P.C.; Hanna, R.E.; Lu-Culligan, W.J.; Cai, W.L.; Strine, M.S.; Zhang, S.M.; Graziano, V.R.; Schmitz, C.O.; et al. Genome-wide CRISPR Screens Reveal Host Factors Critical for SARS-CoV-2 Infection. Cell 2020, 184, 76–91.e13. [Google Scholar] [CrossRef]
- Zhu, Y.; Feng, F.; Hu, G.; Wang, Y.; Yu, Y.; Zhu, Y.; Xu, W.; Cai, X.; Sun, Z.; Han, W.; et al. A genome-wide CRISPR screen identifies host factors that regulate SARS-CoV-2 entry. Nat. Commun. 2021, 12, 961. [Google Scholar] [CrossRef]
- Rebendenne, A.; Roy, P.; Bonaventure, B.; Chaves Valadao, A.L.; Desmarets, L.; Rouille, Y.; Tauziet, M.; Arnaud-Arnould, M.; Giovannini, D.; Lee, Y.; et al. Bidirectional genome-wide CRISPR screens reveal host factors regulating SARS-CoV-2, MERS-CoV and seasonal coronaviruses. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kratzel, A.; Kelly, J.N.; Brueggemann, Y.; Portmann, J.; V’kovski, P.; Todt, D.; Ebert, N.; Steinmann, E.; Dijkman, R.; Zimmer, G.; et al. A genome-wide CRISPR screen identifies interactors of the autophagy pathway as conserved coronavirus targets. bioRxiv 2021. [Google Scholar] [CrossRef]
- Trimarco, J.D.; Heaton, B.E.; Chaparian, R.R.; Burke, K.N.; Binder, R.A.; Gray, G.C.; Smith, C.M.; Menachery, V.D.; Heaton, N.S. TMEM41B is a host factor required for the replication of diverse coronaviruses including SARS-CoV-2. PLoS Pathog. 2021, 17, e1009599. [Google Scholar] [CrossRef] [PubMed]
- Edridge, A.W.D.; Kaczorowska, J.; Hoste, A.C.R.; Bakker, M.; Klein, M.; Loens, K.; Jebbink, M.F.; Matser, A.; Kinsella, C.M.; Rueda, P.; et al. Seasonal coronavirus protective immunity is short-lasting. Nat. Med. 2020, 26, 1691–1693. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.X.; Liang, J.Q.; Fung, T.S. Human Coronavirus-229E, -OC43, -NL63, and -HKU1 (Coronaviridae). In Encyclopedia of Virology, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2021; Volume 2, pp. 428–440. [Google Scholar]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef]
- Weiss, S.R. Forty years with coronaviruses. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Mecozzi, V.J.; Berman, D.E.; Simoes, S.; Vetanovetz, C.; Awal, M.R.; Patel, V.M.; Schneider, R.T.; Petsko, G.A.; Ringe, D.; Small, S.A. Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol. 2014, 10, 443–449. [Google Scholar] [CrossRef]
- Seaman, M.N.J.; Marcusson, E.G.; Cereghino, J.L.; Emr, S.D. Endosome to Golgi Retrieval of the Vaculaor Protein Sorting Receptor, Vps10p, Requires the Function of the VPS29, VPS30 and VPS35 Gene Products. J. Cell Biol. 1997, 137, 79–92. [Google Scholar] [CrossRef]
- Schwenk, B.M.; Lang, C.M.; Hogl, S.; Tahirovic, S.; Orozco, D.; Rentzsch, K.; Lichtenthaler, S.F.; Hoogenraad, C.C.; Capell, A.; Haass, C.; et al. The FTLD risk factor TMEM106B and MAP6 control dendritic trafficking of lysosomes. EMBO J. 2014, 33, 450–467. [Google Scholar] [CrossRef]
- Luningschror, P.; Werner, G.; Stroobants, S.; Kakuta, S.; Dombert, B.; Sinske, D.; Wanner, R.; Lullmann-Rauch, R.; Wefers, B.; Wurst, W.; et al. The FTLD Risk Factor TMEM106B Regulates the Transport of Lysosomes at the Axon Initial Segment of Motoneurons. Cell Rep. 2020, 30, 3506–3519.e3506. [Google Scholar] [CrossRef]
- Senay, C.; Lind, T.; Muguruma, K.; Tone, Y.; Kitagawa, H.; Sugahra, K.; Lidholt, K.; Lindahl, U.; Kusche-Gullberg, M. The EXT1/EXT2 tumor suppressors: Catalytic activities and role in heparan sulfate biosynthesis. EMBO Rep. 2000, 1, 282–286. [Google Scholar] [CrossRef]
- Busse-Wicher, M.; Wicher, K.B.; Kusche-Gullberg, M. The exostosin family: Proteins with many functions. Matrix Biol. 2014, 35, 25–33. [Google Scholar] [CrossRef]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057.e1015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, C.Z.; Swaroop, M.; Xu, M.; Wang, L.; Lee, J.; Wang, A.Q.; Pradhan, M.; Hagen, N.; Chen, L.; et al. Heparan sulfate assists SARS-CoV-2 in cell entry and can be targeted by approved drugs in vitro. Cell Discov. 2020, 6, 80. [Google Scholar] [CrossRef]
- Germi, R.; Crance, J.M.; Garin, D.; Guimet, J.; Lortat-Jacob, H.; Ruigrok, R.W.; Zarski, J.P.; Drouet, E. Heparan sulfate-mediated binding of infectious dengue virus type 2 and yellow fever virus. Virology 2002, 292, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, M.; Rusnati, M.; Presta, M.; Giacca, M. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J. Biol. Chem. 2001, 276, 3254–3261. [Google Scholar] [CrossRef]
- O’Donnell, C.D.; Kovacs, M.; Akhtar, J.; Valyi-Nagy, T.; Shukla, D. Expanding the role of 3-O sulfated heparan sulfate in herpes simplex virus type-1 entry. Virology 2010, 397, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Milewska, A.; Zarebski, M.; Nowak, P.; Stozek, K.; Potempa, J.; Pyrc, K. Human coronavirus NL63 utilizes heparan sulfate proteoglycans for attachment to target cells. J. Virol. 2014, 88, 13221–13230. [Google Scholar] [CrossRef]
- Hofmann, H.; Pyrc, K.; van der Hoek, L.; Geier, M.; Berkhout, B.; Pohlmann, S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. USA 2005, 102, 7988–7993. [Google Scholar] [CrossRef]
- Morita, K.; Hama, Y.; Izume, T.; Tamura, N.; Ueno, T.; Yamashita, Y.; Sakamaki, Y.; Mimura, K.; Morishita, H.; Shihoya, W.; et al. Genome-wide CRISPR screen identifies TMEM41B as a gene required for autophagosome formation. J. Cell Biol. 2018, 217, 3817–3828. [Google Scholar] [CrossRef]
- Moretti, F.; Bergman, P.; Dodgson, S.; Marcellin, D.; Claerr, I.; Goodwin, J.M.; DeJesus, R.; Kang, Z.; Antczak, C.; Begue, D.; et al. TMEM41B is a novel regulator of autophagy and lipid mobilization. EMBO Rep. 2018, 19, e45889. [Google Scholar] [CrossRef]
- Ghanbarpour, A.; Valverde, D.P.; Melia, T.J.; Reinisch, K.M. A model for a partnership of lipid transfer proteins and scramblases in membrane expansion and organelle biogenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2101562118. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, H.H.; Schneider, W.M.; Rozen-Gagnon, K.; Miles, L.A.; Schuster, F.; Razooky, B.; Jacobson, E.; Wu, X.; Yi, S.; Rudin, C.M.; et al. TMEM41B Is a Pan-flavivirus Host Factor. Cell 2021, 184, 133–148.e120. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Muller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef]
- Qiao, Y.; Wang, X.M.; Mannan, R.; Pitchiaya, S.; Zhang, Y.; Wotring, J.W.; Xiao, L.; Robinson, D.R.; Wu, Y.M.; Tien, J.C.; et al. Targeting transcriptional regulation of SARS-CoV-2 entry factors ACE2 and TMPRSS2. Proc. Natl. Acad. Sci. USA 2020, 118, e2021450118. [Google Scholar] [CrossRef] [PubMed]
- Limburg, H.; Harbig, A.; Bestle, D.; Stein, D.A.; Moulton, H.M.; Jaeger, J.; Janga, H.; Hardes, K.; Koepke, J.; Schulte, L.; et al. TMPRSS2 Is the Major Activating Protease of Influenza A Virus in Primary Human Airway Cells and Influenza B Virus in Human Type II Pneumocytes. J. Virol. 2019, 93, e00649-19. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.J.; Hsu, T.W.; Wu, S.R.; Lan, S.W.; Hsiao, T.F.; Lin, H.Y.; Lin, H.H.; Tu, H.F.; Lee, C.F.; Huang, C.C.; et al. Inhibition of TMPRSS2 by HAI-2 reduces prostate cancer cell invasion and metastasis. Oncogene 2020, 39, 5950–5963. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Laxman, B.; Varambally, S.; Cao, X.; Yu, J.; Helgeson, B.E.; Cao, Q.; Prensner, J.R.; Rubin, M.A.; Shah, R.B.; et al. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia 2008, 10, 177–188. [Google Scholar] [CrossRef]
- Reid, C.R.; Airo, A.M.; Hobman, T.C. The Virus-Host Interplay: Biogenesis of +RNA Replication Complexes. Viruses 2015, 7, 4385–4413. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.P.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.; Larson, M.; Morsut, L.; Liu, Z.; Brar, G.; Torres, S.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Horlbeck, M.; Gilbert, L.; Villalta, J.; Adamson, B.; Pak, R.; Chen, Y.; Fields, A.; Park, C.; Corn, J.; Kampmann, J.; et al. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLifesciences 2016, 5, e19760. [Google Scholar]
- Musso, D.; Gubler, D.J. Zika Virus. Clin. Microbiol. Rev. 2016, 29, 487–524. [Google Scholar] [CrossRef]
- de Araújo, T.V.B.; Ximenes, R.A.d.A.; Miranda-Filho, D.d.B.; Souza, W.V.; Montarroyos, U.R.; de Melo, A.P.L.; Valongueiro, S.; de Albuquerque, M.d.F.P.M.; Braga, C.; Filho, S.P.B.; et al. Association between microcephaly, Zika virus infection, and other risk factors in Brazil: Final report of a case-control study. Lancet Infect. Dis. 2018, 18, 328–336. [Google Scholar] [CrossRef]
- Miner, J.J.; Cao, B.; Govero, J.; Smith, A.M.; Fernandez, E.; Cabrera, O.H.; Garber, C.; Noll, M.; Klein, R.S.; Noguchi, K.K.; et al. Zika Virus Infection during Pregnancy in Mice Causes Placental Damage and Fetal Demise. Cell 2016, 165, 1081–1091. [Google Scholar] [CrossRef]
- Li, C.; Xu, D.; Ye, Q.; Hong, S.; Jiang, Y.; Liu, X.; Zhang, N.; Shi, L.; Qin, C.F.; Xu, Z. Zika Virus Disrupts Neural Progenitor Development and Leads to Microcephaly in Mice. Cell Stem Cell 2016, 19, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Cheriyath, V.; Kaur, J.; Davenport, A.; Khalel, A.; Chowdhury, N.; Gaddipati, L. G1P3 (IFI6), a mitochondrial localised antiapoptotic protein, promotes metastatic potential of breast cancer cells through mtROS. Br. J. Cancer 2018, 119, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Forloni, M.; Bisserier, M.; Dogra, S.K.; Yang, Q.; Wajapeyee, N. Interferon alpha-inducible protein 6 regulates NRASQ61K-induced melanomagenesis and growth. Elife 2016, 5, e16432. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Li, Y.; Zhang, Y.; Zhang, L.; Wang, Z.; Zhang, X.; Gui, L.; Huang, J. IFI6 Inhibits Apoptosis via Mitochondrial-Dependent Pathway in Dengue Virus 2 Infected Vascular Endothelial Cells. PLoS ONE 2015, 10, e0132743. [Google Scholar] [CrossRef]
- Cheriyath, V.; Kuhns, M.A.; Jacobs, B.S.; Evangelista, P.; Elson, P.; Downs-Kelly, E.; Tubbs, R.; Borden, E.C. G1P3, an interferon- and estrogen-induced survival protein contributes to hyperplasia, tamoxifen resistance and poor outcomes in breast cancer. Oncogene 2012, 31, 2222–2236. [Google Scholar] [CrossRef]
- Cheriyath, V.; Glaser, K.B.; Waring, J.F.; Baz, R.; Hussein, M.A.; Borden, E.C. G1P3, an IFN-induced survival factor, antagonizes TRAIL-induced apoptosis in human myeloma cells. J. Clin. Investig. 2007, 117, 3107–3117. [Google Scholar] [CrossRef] [PubMed]
- Tahara, E., Jr.; Tahara, H.; Kanno, M.; Naka, K.; Takeda, Y.; Matsuzaki, T.; Yamazaki, R.; Ishihara, H.; Yasui, W.; Barrett, J.C.; et al. G1P3, an interferon inducible gene 6-16, is expressed in gastric cancers and inhibits mitochondrial-mediated apoptosis in gastric cancer cell line TMK-1 cell. Cancer Immunol. Immunother. 2005, 54, 729–740. [Google Scholar] [CrossRef]
- Plouffe, S.W.; Meng, Z.; Lin, K.C.; Lin, B.; Hong, A.W.; Chun, J.V.; Guan, K.L. Characterization of Hippo Pathway Components by Gene Inactivation. Mol. Cell 2016, 64, 993–1008. [Google Scholar] [CrossRef]
- Han, Z.; Yu, Y.; Cai, B.; Xu, Z.; Bao, Z.; Zhang, Y.; Bamba, D.; Ma, W.; Gao, X.; Yuan, Y.; et al. YAP/TEAD3 signal mediates cardiac lineage commitment of human-induced pluripotent stem cells. J. Cell Physiol. 2020, 235, 2753–2760. [Google Scholar] [CrossRef] [PubMed]
- Li, F.Q.; Tam, J.P.; Liu, D.X. Cell cycle arrest and apoptosis induced by the coronavirus infectious bronchitis virus in the absence of p53. Virology 2007, 365, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Suryawanshi, R.K.; Koganti, R.; Agelidis, A.; Patil, C.D.; Shukla, D. Dysregulation of Cell Signaling by SARS-CoV-2. Trends Microbiol. 2021, 29, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Lei, R.; Wang, X.; Zhang, D.; Liu, Y.; Chen, Q.; Jiang, N. Rapid isothermal duplex real-time recombinase polymerase amplification (RPA) assay for the diagnosis of equine piroplasmosis. Sci. Rep. 2020, 10, 4096. [Google Scholar] [CrossRef]
- Rosser, A.; Rollinson, D.; Forrest, M.; Webster, B.L. Isothermal Recombinase Polymerase amplification (RPA) of Schistosoma haematobium DNA and oligochromatographic lateral flow detection. Parasit Vectors 2015, 8, 446. [Google Scholar] [CrossRef]
- Lobato, I.M.; O’Sullivan, C.K. Recombinase polymerase amplification: Basics, applications and recent advances. Trends Analyt. Chem. 2018, 98, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Arizti-Sanz, J.; Freije, C.A.; Stanton, A.C.; Boehm, C.K.; Petros, B.A.; Siddiqui, S.; Shaw, B.M.; Adams, G.; Kosoko-Thoroddsen, T.F.; Kemball, M.E.; et al. Integrated sample inactivation, amplification, and Cas13-based detection of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Fozouni, P.; Son, S.; Diaz de Leon Derby, M.; Knott, G.J.; Gray, C.N.; D’Ambrosio, M.V.; Zhao, C.; Switz, N.A.; Kumar, G.R.; Stephens, S.I.; et al. Amplification-free detection of SARS-CoV-2 with CRISPR-Cas13a and mobile phone microscopy. Cell 2020, 184, 323–333.e9. [Google Scholar] [CrossRef]
- Barnes, K.G.; Lachenauer, A.E.; Nitido, A.; Siddiqui, S.; Gross, R.; Beitzel, B.; Siddle, K.J.; Freije, C.A.; Dighero-Kemp, B.; Mehta, S.B.; et al. Deployable CRISPR-Cas13a diagnostic tools to detect and report Ebola and Lassa virus cases in real-time. Nat. Commun. 2020, 11, 4131. [Google Scholar] [CrossRef]



| Virus | CRISPR Screen | Cell Type | Host Factor Class | Top Candidates | Reference |
|---|---|---|---|---|---|
| Influenza A PR8 | KO | A549 | Pro-viral | WDR7, CCDC115, TMEM199, SLC35A1 | [67] |
| Influenza A H5N1 | KO | A549 | Pro-Viral | SLC35A1, GDF11, IRX3, C2CD4C, TRIM23, PIGN, ACADSB, GRAMD2 | [68] |
| Influenza A PR8 | Activator | A549 | Anti-Viral | B4GALNT2 | [69] |
| Zika Virus PRVABC59 and MR766 | KO | iPSC differentiated into NPC | Pro-Viral | WDR7, EMC1, EMC2, EMC4, ATP6V1A, MMGT1, TM9SF2, EXT2 EMC | [70] |
| Zika Virus MR766 | KO | HeLa | Pro-Viral | AXL, EMC, MMGT1, SSR3, STT3A, WDR7, RABGEF1 | [71] |
| Zika Virus MR766 | KO | GSC | Pro-Viral | SSR3, STT3A, MMGT1, SSR2, TMEM41B, OXGR1, EMC, OST4 | [72] |
| Zika Virus MR766 | Activator | Huh7.5 | Anti-Viral | IFI6, ISG20, ZCCHC6, IFN-λ2, IRF1, MAVS, TRIM25 | [73] |
| Dengue Virus 16681 | KO | Huh7.5 | Pro-Viral | SSR1-3, ERAD Pathway (EMC), MMGT1, AGT1, STT3B, RPN2, STT3A, OST4 | [8] |
| Dengue Virus Jamaican | KO | HAP1 | Pro-Viral | SLC35B2, PAPSS1, B4GALT7, EXT2, STT3A, B3GAT3, DPM1, DPM3 | [74] |
| Hepatitis C Virus JFH-1 | KO | Huh7.5 | Pro-Viral | CLDN1, OCLN, CD81, PPIA, RFK, FLAD1, ELAVL1, SRRD, ANKRD49, ZFB1 | [8] |
| Human Immunodeficiency Virus | KO | THP-1 | Pro-Viral | IFNAR1, IRF9, STAT1, STAT2, ZC3HAV1, TRIM25, N4BP1 | [75,76] |
| Human Immunodeficiency Virus | KO | GXR | Pro-Viral | CD4, CCR5, ALCAM, SLC35B2, TPST2 | [77] |
| Human Immunodeficiency Virus | KO | Jurkat T Cells | Pro-Viral (latency) | ZNF304, ARL16, ATF1, CGREF1, USMG5 | [76] |
| West Nile Virus B596 | KO | 293T | Pro-Viral | SPCS1, SPCS3, EMC, OST complex (STT3A), TRAP complex, SEL1L, HRD1 | [7] |
| West Nile Virus NY2000 | KO | 293T | Pro-Viral | STT3A, SEC63, SEC61B, OSTC, SPCS1, SPCS3, SERP1, EMC6, SEL1L, HSPA13, OST4, EMC4 | [9] |
| Ebola Virus Mayinga | KO | Huh7.5 | Pro-Viral | GNPTAB, NPC1, SPNS1, SLC30A1, VPS16, VPS33A and VPS18 | [78] |
| Epstein–Barr Virus B95.8 | KO | B Cell Lymphocytes | Pro-Viral | CD19, CD81, cFLIP, BATF, IRF4 and IRF2 | [79] |
| Yellow Fever Virus YFV-17D | KO | Huh7.5 | Pro-Viral | IFI6, BiP, IFN pathway (IFNAR, STAT2, JAK1), HSPA5 | [80] |
| Murine Norovirus MNoVCW3 MNoV CR6 | Activator | HeLa | Anti-Viral | TRIM7, HOXC11, MX1, DDX60, PITX1 | [81] |
| Murine Norovirus MNoVCW3 MNoV CR6 | KO | HeLa | Pro-Viral | CD300LF, G3BP1, KMT2D, CD300LH | [82] |
| Hepatitis A Virus HM175/18f | KO | Huh7.5 | Pro-Viral | SLC35A1, ZCCHC14, EIF4B, PTBP1, PDAP1, SCAP, A1CF, FXR1, UFM1, PAPD7, PAPD5, UGCG, ST3GAL5 | [83] |
| SARS-CoV-2 HCoV-229E | KO | Huh7 | Pro-Viral | SARS-CoV-2: TMEM41B, TMEM106B, KRT19, AHCYL1, PTDSS1, OSBPL9, GLUD1, DTD1, EXT1, ACE2 HCoV-229E: ANPEP, TMEM41B, PIK3C3, NUFIP2 | [84] |
| SARS-CoV-2 | KO | Calu-3 | Pro-Viral | AP1G1, ACE2, CHUK, TMPRSS2, AP1B1, RIPK4, ROCK1, AP1M2 | [85] |
| SARS-CoV-2 | Activator | Calu-3 | Anti-Viral | TEAD3, MUC21, MUC4, MUC1, CPNE3, SPDEF, LY6E, JDP2, CCNE1, ZNF275 | [85] |
| SARS-CoV-2 | KO | A549 | Pro-Viral | ACE2, ACTR2, ARPC3, ARPC4, RAB7A, CTSL, Retromer Complex, Commander Complex, PIK3C3, SPEN, SLTM, DPM3, ERMP1, PPID, CHST14 | [86] |
| SARS-CoV-2 HCoV-229E HCoV-OC43 | KO | Huh7.5 | Pro-Viral | SARS-CoV-2: TMEM106B, VAC14, SCAP, ACE2, EXT1, PCDH19, MBTPS2 HCoV-229E: ANPEP, TMEM41B, VPS11, -16, -18, RAB7A, PIK3C3, GPR89A, GPR89B HCoV-OC43: B3GALT6, SLC35B2, EXT1, EXT2, B3GAT3, B4GALT7, FAM20B | [87] |
| SARS-COV-2 HCoV-229E HCoV-NL63 HCoV-OC43 | KO | Huh7.5 | Pro-Viral | SARS-CoV-2: TMEM41B, DHX36, EXTL3, EXT1, EXT2, ACE2, MBTPS2, SCAP, TMEM106B, VAC14, SLC35B2 HCoV-229E: ANPEP, TMEM41B, PIK3C3, VPS11, RAB7A HCoV-NL63: CDX2, ACE2, NRIP1, SMAD4, BMPR1A, EP300, KMT2B, SETDB1, AVCR1, KDM6A HCoV-OC43: B3GAT3, EXT1, EXT2, SLC35B2, B4GALT7, RAB7A, TM9SF3, XYLT2, SCAP, MBTPS1, NDST1, TMEM41B | [88] |
| SARS-CoV-2 MERS-CoV HKU5-SARS-CoV-1-S | KO | Vero | Pro-Viral | SARS-CoV-2: ACE2, CTSL, ARID1A, KDM6A, SMARCC1, HMGB1, SMARCA4 MERS-CoV: DPP4, AXIN1, TMEM41B, MTF1, CTSL, ARID1A, KDM6A HKU5-SARS-CoV-1-S: CTSL, ACE2, SMARCA4, JMJD6, PHIP, KDM6A | [89] |
| SARS-CoV-2 | KO | A549 | Pro-Viral | Commander complex, Retromer Complex, ACE2, WDR81, ARPC4, NPC1, CTSL | [90] |
| SARS-CoV-2 | KO | Calu-3 | Pro-Viral | AP1G1, ACE2, TMPRSS2, KMT2C, ARID2, KDM6A | [91] |
| SARS-CoV-2 | Activator | Calu-3 | Anti-Viral | LY6E, MUC21, TEAD3, PLAGL1, MUC4, MUC1, JADE3 | [91] |
| MERS-CoV | KO | Huh7 | Pro-Viral | DPP4, HNF1A, PTBP1, CLCN5, PCTP, OR9K2 | [92] |
| HCoV-229E | KO | Huh7 | Pro-Viral | VMP1, ANPEP, PHGDH, TMEM41B, LAMB3, BCL21 | [92] |
| HCoV-229E | KO | Huh7 | Pro-Viral | ANPEP, TMEM41B | [93] |
| System | Components | |
|---|---|---|
| CRISPR VPR |  | dCas9 is fused with the transcriptional activators VP64 (HSV), p65 (cellular) and Rta (EBV) [124] |
| CRISPRaSAM |  | The sgRNA incorporates MS2 RNA aptamers in the stem–loop Helper complex, composed of MS2 coat protein, p65 (cellular) and Heat Shock Factor 1 (HSF1-cellular) dCas9 is fused with VP64 (HSV). Helper complex binds to the MS2 RNA aptamers [125] |
| CRISPRaSunTag |  | dCas9 is fused with a GCN4 repeating polypeptide, “SunTag” ScFv, fused with VP64 (HSV) and GFP, are raised against GCN4, allow recruitment of additional transcriptional activators to the promoter [126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirby, E.N.; Shue, B.; Thomas, P.Q.; Beard, M.R. CRISPR Tackles Emerging Viral Pathogens. Viruses 2021, 13, 2157. https://doi.org/10.3390/v13112157
Kirby EN, Shue B, Thomas PQ, Beard MR. CRISPR Tackles Emerging Viral Pathogens. Viruses. 2021; 13(11):2157. https://doi.org/10.3390/v13112157
Chicago/Turabian StyleKirby, Emily N., Byron Shue, Paul Q. Thomas, and Michael R. Beard. 2021. "CRISPR Tackles Emerging Viral Pathogens" Viruses 13, no. 11: 2157. https://doi.org/10.3390/v13112157
APA StyleKirby, E. N., Shue, B., Thomas, P. Q., & Beard, M. R. (2021). CRISPR Tackles Emerging Viral Pathogens. Viruses, 13(11), 2157. https://doi.org/10.3390/v13112157