
Toll-like Receptors in Viral Encephalitis

 , and
, and
Abstract
1. Viral Encephalitis
2. Toll-like Receptors and Toll-like Receptor Signaling
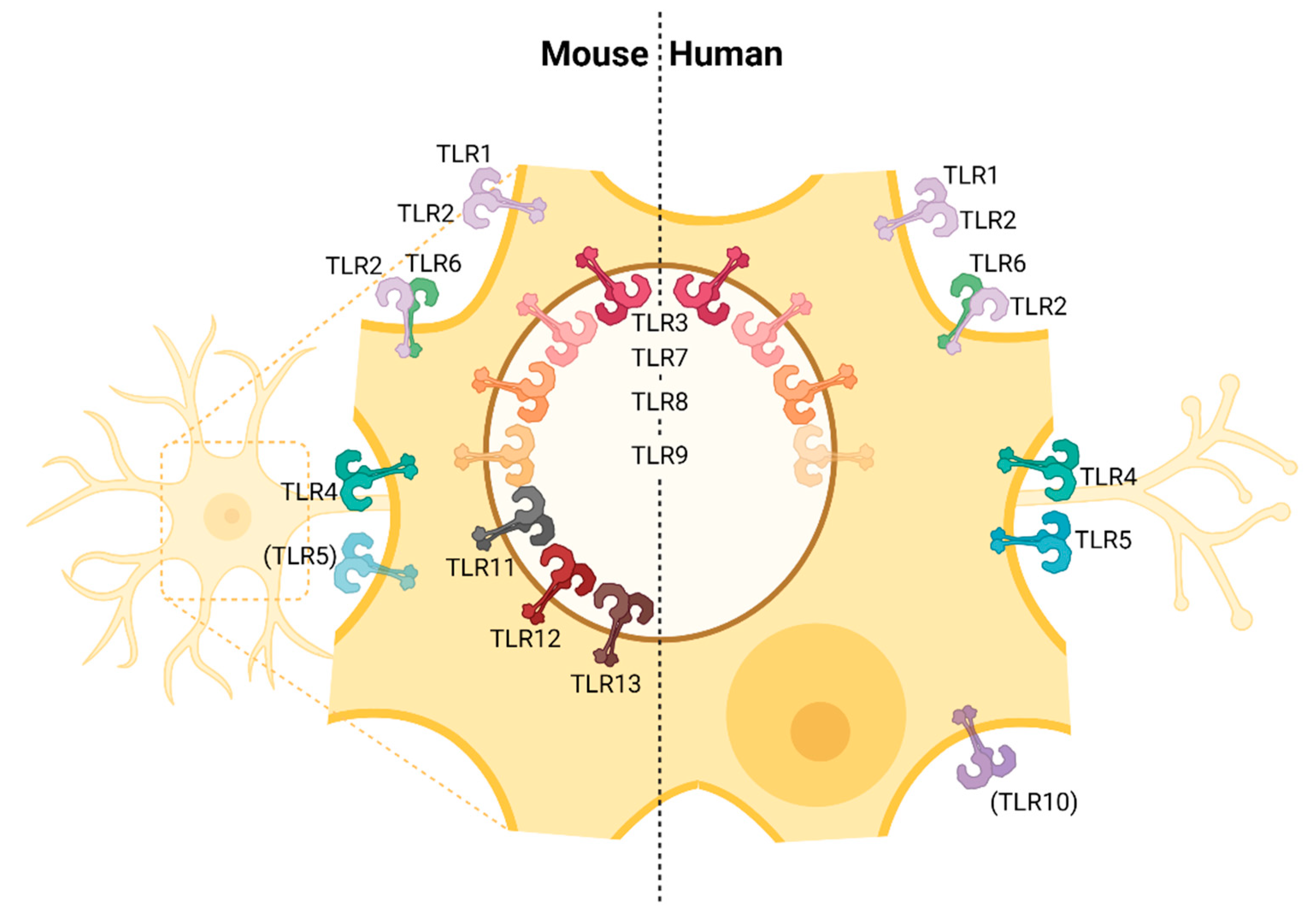
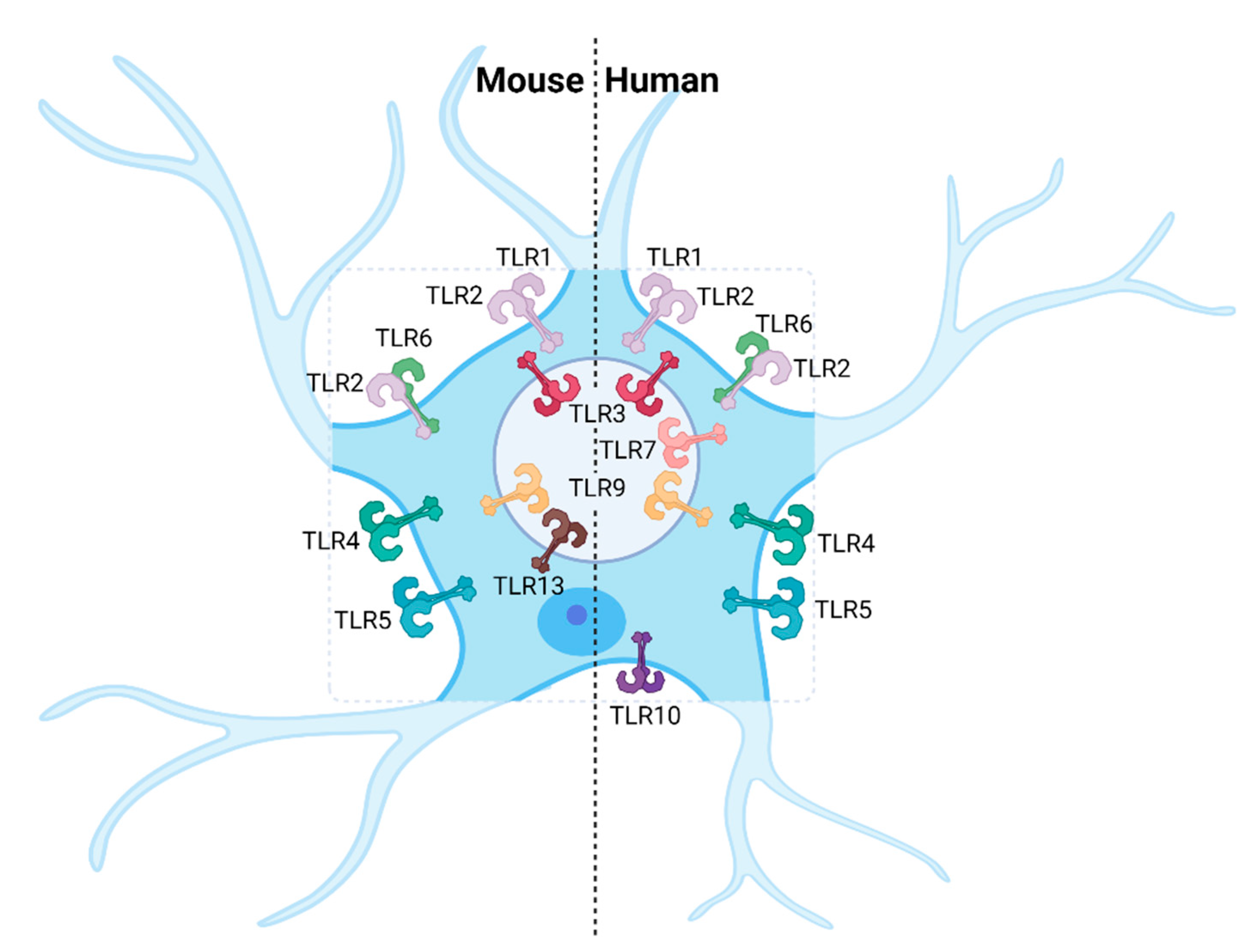
3. Expression of Toll-like Receptors in Cells of the Central Nervous System
3.1. Neurons
3.2. Astrocytes
3.3. Microglia
4. Immune Evasion of the TLR Pathway by Neurotropic Viruses
4.1. Herpes Simplex Virus 1
4.2. Flaviviruses
4.3. Rabies Virus
4.4. Influenza a Virus
5. Primary Defects in the TLR3-IFNAR Axis Can Affect the Clinical Outcome of Viral Encephalitis
6. Innovative TLR Agonist Mediated Intervention Strategies and Enhancement of Vaccine Responses against Neurotropic Virus Infections
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Autosomal dominant |
| AIM | 2-Absent in melanoma 2 |
| ALR | AIM-2-like receptor |
| AP1 | Activator protein 1 |
| AR | Autosomal recessive |
| BBB | Blood-brain barrier |
| CBP | CREB binding protein |
| CCL | Chemokine C-C motif ligand |
| cGAS | Cyclic guanosine monophosphate-adenosine monophosphate synthase |
| CLR | C-type lectin receptor |
| CNS | Central nervous system |
| COVID-19 | Coronavirus disease 2019 |
| CpG | 5′-Cytosine-phosphate-Guanine-3′ |
| CREB | Cyclic AMP-responsive element-binding protein |
| CSF | Cerebrospinal fluid |
| CXCL | Chemokine C-X-C motif ligand |
| DENV | Dengue virus |
| dsRNA | Double stranded RNA |
| EP | Envelope protein |
| ER | Endoplasmatic reticulum |
| FC | Flow cytometry |
| GP | Glycoprotein |
| HCMV | Human cytomegalovirus |
| HeV | Hendra virus |
| HIV-1 | Human immunodeficiency virus 1 |
| HSE | Herpes simplex encephalitis |
| HSV-1 | Herpes simplex virus 1 |
| IAV | Influenza A virus |
| ICC | Immunocytochemistry |
| IF | Immunofluorescence |
| IFN | Interferon |
| IFNAR | IFN-I receptor |
| IFN-I | Type-I interferons |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| iPSC | Induced pluripotent stem cell |
| IR | Immunoreactivity |
| IRAK | IL-1R-associated kinase |
| IRF | Interferon-regulatory factor |
| ISG | IFN stimulated gene |
| ISRE | IFN-stimulated response elements |
| JAK1 | Janus kinase 1 |
| JEV | Japanese encephalitis virus |
| LGTV | Langat virus |
| LPS | Lipopolysaccharide |
| LTA | Lipoteichoic acid |
| mAb | Monoclonal antibody |
| MAPK | Mitogen-activated protein kinase |
| MEF | Mouse embryonic fibroblast |
| mTOR | Mammalian target of rapamycin |
| mTORC | mTOR complex |
| MyD88 | Myeloid differentiation primary response 88 |
| NF | κB-Nuclear factor-κB |
| NiV | Nipah virus |
| NLR | NOD-like receptor |
| NOD | Nucleotide-binding oligomerization domain |
| NS | Nonstructural protein |
| ODN | Oligodeoxynucleotide |
| PAMP | Pathogen associated molecular pattern |
| PBMC | Peripheral blood mononuclear cells |
| PGN | Peptidoglycan |
| PKR | Protein kinase R |
| PRR | Pattern recognition receptor |
| RABV | Rabies virus |
| RIG | I-Retinoic acid inducible gene I |
| RIP1 | Receptor-interacting protein 1 |
| RLR | RIG-I like receptor |
| RNAi | RNA interference |
| RT | PCR-Reverse transcription polymerase chain reaction |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| sfRNA | Subgenomic flavivirus RNA |
| ssRNA | Single stranded RNA |
| STAT | Signal transducers and activators of transcription |
| STING | Stimulator of interferon genes |
| TBE | Tick-borne encephalitis |
| TBEV | Tick-borne encephalitis virus |
| TBK1 | TANK-binding kinase 1 |
| TCA | Tricarboxylic acid |
| TLR | Toll-like receptor |
| TMEV | Theiler’s murine encephalomyelitis virus |
| TNF-α | Tumor necrosis factor α |
| TRAF | TNF receptor-associated factor |
| TRIF | TIR-domain-containing adapter-inducing interferon-? |
| TYK2 | Tyrosine kinase 2 |
| VSV | Vesicular stomatitis virus |
| VZV | Varicella-zoster virus |
| WB | Western blot |
| WNV | West Nile virus |
| WNV-E | WNV envelope protein |
| ZIKV | Zika Virus |
References
- Kennedy, P.G.E.; Quan, P.-L.; Lipkin, W.I. Viral Encephalitis of Unknown Cause: Current Perspective and Recent Advances. Viruses 2017, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, A.; Michael, B.; Probasco, J.C.; Geocadin, R.G.; Solomon, T. Acute encephalitis in immunocompetent adults. Lancet 2019, 393, 702–716. [Google Scholar] [CrossRef]
- Hasbun, R. Meningitis and Encephalitis; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar] [CrossRef]
- Whitley, R.J. Herpes simplex encephalitis: Adolescents and adults. Antivir. Res. 2006, 71, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Baringer, J.R.; Swoveland, P. Recovery of Herpes-Simplex Virus from Human Trigeminal Ganglions. N. Engl. J. Med. 1973, 288, 648–650. [Google Scholar] [CrossRef] [PubMed]
- Shukla, N.D.; Tiwari, V.; Valyi-Nagy, T. Nectin-1-specific entry of herpes simplex virus 1 is sufficient for infection of the cornea and viral spread to the trigeminal ganglia. Mol. Vis. 2012, 18, 2711–2716. [Google Scholar] [PubMed]
- Mori, I.; Nishiyama, Y.; Yokochi, T.; Kimura, Y. Olfactory transmission of neurotropic viruses. J. Neurovirol. 2005, 11, 129–137. [Google Scholar] [CrossRef]
- Jennische, E.; Eriksson, C.; Lange, S.; Trybala, E.; Bergström, T. The anterior commissure is a pathway for contralateral spread of herpes simplex virus type 1 after olfactory tract infection. J. Neurovirol. 2015, 21, 129–147. [Google Scholar] [CrossRef]
- Granerod, J.; Ambrose, H.E.; Davies, N.W.S.; Clewley, J.P.; Walsh, A.L.; Morgan, D.; Cunningham, R.; Zuckerman, M.; Mutton, K.J.; Solomon, T.; et al. Causes of encephalitis and differences in their clinical presentations in England: A multicentre, population-based prospective study. Lancet Infect. Dis. 2010, 10, 835–844. [Google Scholar] [CrossRef]
- Harpaz, R.; Ortega-Sanchez, I.R.; Seward, J.F. Advisory Committee on Immunization Practices Centers for Disease. Prevention of herpes zoster: Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm. Rep. 2008, 57, 1–30. [Google Scholar]
- Chen, S.-Y.; Suaya, J.A.; Li, Q.; Galindo, C.M.; Misurski, D.; Burstin, S.; Levin, M.J. Incidence of herpes zoster in patients with altered immune function. Infection 2014, 42, 325–334. [Google Scholar] [CrossRef]
- Gerada, C.; Campbell, T.M.; Kennedy, J.; McSharry, B.; Steain, M.; Slobedman, B.; Abendroth, A. Manipulation of the Innate Immune Response by Varicella Zoster Virus. Front. Immunol. 2020, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Freer, G.; Pistello, M. Varicella-zoster virus infection: Natural history, clinical manifestations, immunity and current and future vaccination strategies. New Microbiol. 2018, 41, 95–105. [Google Scholar] [PubMed]
- Nagel, M.A.; Gilden, D. Complications of Varicella Zoster Virus Reactivation. Curr. Treat. Options Neurol. 2013, 15, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Zuhair, M.; Smit, G.S.A.; Wallis, G.; Jabbar, F.; Smith, C.; Devleesschauwer, B.; Griffiths, P. Estimation of the worldwide seroprevalence of cytomegalovirus: A systematic review and meta-analysis. Rev. Med. Virol. 2019, 29, e2034. [Google Scholar] [CrossRef] [PubMed]
- Teira, P.; Battiwalla, M.; Ramanathan, M.; Barrett, A.J.; Ahn, K.W.; Chen, M.; Green, J.S.; Saad, A.; Antin, J.H.; Savani, B.N.; et al. Early cytomegalovirus reactivation remains associated with increased transplant-related mortality in the current era: A CIBMTR analysis. Blood 2016, 127, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Arribas, J.; Storch, G.A.; Clifford, D.B.; Tselis, A.C. Cytomegalovirus Encephalitis. Ann. Intern. Med. 1996, 125, 577. [Google Scholar] [CrossRef] [PubMed]
- Fooks, A.R.; Banyard, A.C.; Horton, D.; Johnson, N.; McElhinney, L.M.; Jackson, A. Current status of rabies and prospects for elimination. Lancet 2014, 384, 1389–1399. [Google Scholar] [CrossRef]
- Hemachudha, T.; Ugolini, G.; Wacharapluesadee, S.; Sungkarat, W.; Shuangshoti, S.; Laothamatas, J. Human rabies: Neuropathogenesis, diagnosis, and management. Lancet Neurol. 2013, 12, 498–513. [Google Scholar] [CrossRef]
- Turtle, L.; Griffiths, M.J.; Solomon, T. Encephalitis caused by flaviviruses. QJM Int. J. Med. 2012, 105, 219–223. [Google Scholar] [CrossRef]
- Bhatt, S.; Gething, P.; Brady, O.; Messina, J.P.; Farlow, A.W.; Moyes, C.; Drake, J.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef]
- Chambers, T.J.; Diamond, M.S. Pathogenesis of flavivirus encephalitis. Adv. Virus Res. 2003, 60, 273–342. [Google Scholar] [CrossRef]
- Filgueira, L.; Lannes, N. Review of Emerging Japanese Encephalitis Virus: New Aspects and Concepts about Entry into the Brain and Inter-Cellular Spreading. Pathogens 2019, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Suen, W.W.; Prow, N.A.; Hall, R.A.; Bielefeldt-Ohmann, H. Mechanism of West Nile Virus Neuroinvasion: A Critical Appraisal. Viruses 2014, 6, 2796–2825. [Google Scholar] [CrossRef] [PubMed]
- Cain, M.D.; Salimi, H.; Diamond, M.S.; Klein, R.S. Mechanisms of Pathogen Invasion into the Central Nervous System. Neuron 2019, 103, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Hooper, P.T.; Williamson, M.M. Hendra and Nipah Virus Infections. Vet. Clin. N. Am. Equine Pract. 2000, 16, 597–603. [Google Scholar] [CrossRef]
- Middleton, D. Hendra Virus. Vet. Clin. N. Am. Equine Pract. 2014, 30, 579–589. [Google Scholar] [CrossRef] [PubMed]
- CDC Hendra Virus Disease (HeV). Available online: https://www.cdc.gov/vhf/hendra/index.html (accessed on 30 September 2021).
- Ang, B.S.P.; Lim, T.C.C.; Wang, L. Nipah Virus Infection. J. Clin. Microbiol. 2018, 56, e01875-17. [Google Scholar] [CrossRef]
- Field, H.; De Jong, C.; Melville, D.; Smith, C.; Smith, I.; Broos, A.; Kung, Y.H.; McLaughlin, A.; Zeddeman, A. Hendra Virus Infection Dynamics in Australian Fruit Bats. PLoS ONE 2011, 6, e28678. [Google Scholar] [CrossRef]
- Anderson, K.V.; Jürgens, G.; Nüsslein-Volhard, C. Establishment of dorsal-ventral polarity in the Drosophila embryo: Genetic studies on the role of the Toll gene product. Cell 1985, 42, 779–789. [Google Scholar] [CrossRef]
- Lemaitre, B.; Nicolas, E.; Michaut, L.; Reichhart, J.-M.; Hoffmann, J.A. The Dorsoventral Regulatory Gene Cassette spätzle/Toll/cactus Controls the Potent Antifungal Response in Drosophila Adults. Cell 1996, 86, 973–983. [Google Scholar] [CrossRef]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.-Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS Signaling in C3H/HeJ and C57BL/10ScCr Mice: Mutations in Tlr4 Gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 1. Nat. Cell Biol. 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Aliprantis, A.O.; Yang, R.-B.; Mark, M.R.; Suggett, S.; Devaux, B.; Radolf, J.D.; Klimpel, G.R.; Godowski, P.; Zychlinsky, A. Cell Activation and Apoptosis by Bacterial Lipoproteins Through Toll-like Receptor-1. Science 1999, 285, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; e Sousa, C.R. Innate Antiviral Responses by Means of TLR7-Mediated Recognition of Single-Stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, F.; Smith, K.D.; Ozinsky, A.; Hawn, T.R.; Yi, E.C.; Goodlett, D.R.; Eng, J.; Akira, S.; Underhill, D.; Aderem, A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor. Nat. Cell Biol. 2001, 410, 1099–1103. [Google Scholar] [CrossRef]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nat. Cell Biol. 2000, 408, 740–745. [Google Scholar] [CrossRef]
- Takeuchi, O.; Kawai, T.; Mühlradt, P.F.; Morr, M.; Radolf, J.D.; Zychlinsky, A.; Takeda, K.; Akira, S. Discrimination of bacterial lipoproteins by Toll-like receptor 1. Int. Immunol. 2001, 13, 933–940. [Google Scholar] [CrossRef]
- Takeuchi, O.; Sato, S.; Horiuchi, T.; Hoshino, K.; Takeda, K.; Dong, Z.; Modlin, R.; Akira, S. Cutting Edge: Role of Toll-Like Receptor 1 in Mediating Immune Response to Microbial Lipoproteins. J. Immunol. 2002, 169, 10–14. [Google Scholar] [CrossRef]
- Wyllie, D.H.; Kiss-Toth, E.; Visintin, A.; Smith, S.C.; Boussouf, S.; Segal, D.M.; Duff, G.W.; Dower, S.K. Evidence for an Accessory Protein Function for Toll-Like Receptor 1 in Anti-Bacterial Responses. J. Immunol. 2000, 165, 7125–7132. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, G.; Hayden, M.S.; Greenblatt, M.B.; Bussey, C.; Flavell, R.A.; Ghosh, S. A Toll-like Receptor That Prevents Infection by Uropathogenic Bacteria. Science 2004, 303, 1522–1526. [Google Scholar] [CrossRef] [PubMed]
- Hidmark, A.; Paul, A.V.S.; Dalpke, A.H. Cutting Edge: TLR13 Is a Receptor for Bacterial RNA. J. Immunol. 2012, 189, 2717–2721. [Google Scholar] [CrossRef]
- Gianni, T.; Campadelli-Fiume, G. The Epithelial αvβ3-Integrin Boosts the MYD88-Dependent TLR2 Signaling in Response to Viral and Bacterial Components. PLoS Pathog. 2014, 10, e1004477. [Google Scholar] [CrossRef]
- Leoni, V.; Gianni, T.; Salvioli, S.; Campadelli-Fiume, G. Herpes Simplex Virus Glycoproteins gH/gL and gB Bind Toll-Like Receptor 2, and Soluble gH/gL Is Sufficient to Activate NF-κB. J. Virol. 2012, 86, 6555–6562. [Google Scholar] [CrossRef]
- Li, Y.; Yin, S.; Chen, Y.; Zhang, Q.; Huang, R.; Jia, B.; Jie, W.; Yao, K.; Wang, J.; Tong, X.; et al. Hepatitis B virus-induced hyperactivation of B cells in chronic hepatitis B patients via TLR. J. Cell. Mol. Med. 2020, 24, 6096–6106. [Google Scholar] [CrossRef]
- Arribillaga, L.; Echeverria, I.; Belsue, V.; Gomez, T.; Lozano, T.; Casares, N.; Villanueva, L.; Domingos-Pereira, S.; Romero, P.J.; Nardelli-Haefliger, D.; et al. Bivalent therapeutic vaccine against HPV16/18 genotypes consisting of a fusion protein between the extra domain A from human fibronectin and HPV16/18 E7 viral antigens. J. Immunother. Cancer 2020, 8, e000704. [Google Scholar] [CrossRef]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, I.; Ostuni, R.; Marek, L.R.; Barresi, S.; Barbalat, R.; Barton, G.; Granucci, F.; Kagan, J.C. CD14 Controls the LPS-Induced Endocytosis of Toll-like Receptor 1. Cell 2011, 147, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-Y.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; et al. TLR3 Deficiency in Patients with Herpes Simplex Encephalitis. Science 2007, 317, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.-K.; Schlee, M.; et al. 5’-Triphosphate RNA Is the Ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Näslund, T.I.; Liljestrom, P.; Weber, F.; Sousa, C.R. RIG-I-Mediated Antiviral Responses to Single-Stranded RNA Bearing 5′-Phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid–inducible gene-I and melanoma differentiation–associated gene. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Gawanbacht, A.; Habjan, M.; Rang, A.; Borner, C.; Schmidt, A.M.; Veitinger, S.; Jacob, R.; Devignot, S.; Kochs, G.; et al. Incoming RNA Virus Nucleocapsids Containing a 5′-Triphosphorylated Genome Activate RIG-I and Antiviral Signaling. Cell Host Microbe 2013, 13, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Hato, S.V.; Langereis, M.; Zoll, J.; Virgen-Slane, R.; Peisley, A.; Hur, S.; Semler, B.L.; van Rij, R.P.; van Kuppeveld, F.J. MDA5 Detects the Double-Stranded RNA Replicative Form in Picornavirus-Infected Cells. Cell Rep. 2012, 2, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Goubau, D.; Schlee, M.; Deddouche, S.; Pruijssers, A.J.; Zillinger, T.; Goldeck, M.; Schuberth, C.; Van Der Veen, A.G.; Fujimura, T.; Rehwinkel, J.; et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nat. Cell Biol. 2014, 514, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Wu, Z.; Sun, L.; Hashioka, S.; Yu, S.; Schwab, C.; Okada, R.; Hayashi, Y.; McGeer, P.L.; Nakanishi, H. Differential pathways for interleukin-1β production activated by chromogranin A and amyloid β in microglia. Neurobiol. Aging 2013, 34, 2715–2725. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Shaw, M.H.; Kim, Y.-G.; Nuñez, G. NOD-Like Receptors: Role in Innate Immunity and Inflammatory Disease. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 365–398. [Google Scholar] [CrossRef] [PubMed]
- Lugrin, J.; Martinon, F. The AIM2 inflammasome: Sensor of pathogens and cellular perturbations. Immunol. Rev. 2017, 281, 99–114. [Google Scholar] [CrossRef]
- Roberts, T.L.; Idris, A.; Dunn, J.A.; Kelly, G.M.; Burnton, C.M.; Hodgson, S.; Hardy, L.L.; Garceau, V.; Sweet, M.J.; Ross, I.L.; et al. HIN-200 Proteins Regulate Caspase Activation in Response to Foreign Cytoplasmic DNA. Science 2009, 323, 1057–1060. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.H.; Gringhuis, S.I. Signalling through C-type lectin receptors: Shaping immune responses. Nat. Rev. Immunol. 2009, 9, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Reuven, E.M.; Fink, A.; Shai, Y. Regulation of innate immune responses by transmembrane interactions: Lessons from the TLR family. Biochim. Biophys. Acta Biomembr. 2014, 1838, 1586–1593. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Golenbock, D.T.; Bowie, A. The history of Toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C.; Su, T.; Horng, T.; Chow, A.; Akira, S.; Medzhitov, R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat. Immunol. 2008, 9, 361–368. [Google Scholar] [CrossRef]
- Hess, N.J.; Jiang, S.; Li, X.; Guan, Y.; Tapping, R.I. TLR10 Is a B Cell Intrinsic Suppressor of Adaptive Immune Responses. J. Immunol. 2017, 198, 699–707. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Jouanguy, E.; Béziat, V.; Mogensen, T.H.; Casanova, J.-L.; Tangye, S.G.; Zhang, S.-Y. Human inborn errors of immunity to herpes viruses. Curr. Opin. Immunol. 2020, 62, 106–122. [Google Scholar] [CrossRef]
- Sato, R.; Kato, A.; Chimura, T.; Saitoh, S.-I.; Shibata, T.; Murakami, Y.; Fukui, R.; Liu, K.; Zhang, Y.; Arii, J.; et al. Combating herpesvirus encephalitis by potentiating a TLR3–mTORC2 axis. Nat. Immunol. 2018, 19, 1071–1082. [Google Scholar] [CrossRef]
- Wang, T.; Town, T.; Alexopoulou, L.; Anderson, J.F.; Fikrig, E.; Flavell, R. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med. 2004, 10, 1366–1373. [Google Scholar] [CrossRef]
- Menasria, R.; Boivin, N.; Lebel, M.; Piret, J.; Gosselin, J.; Boivin, G. Both TRIF and IPS-1 Adaptor Proteins Contribute to the Cerebral Innate Immune Response against Herpes Simplex Virus 1 Infection. J. Virol. 2013, 87, 7301–7308. [Google Scholar] [CrossRef] [PubMed]
- Daffis, S.; Samuel, M.A.; Suthar, M.S.; Gale, M.; Diamond, M.S. Toll-Like Receptor 3 Has a Protective Role against West Nile Virus Infection. J. Virol. 2008, 82, 10349–10358. [Google Scholar] [CrossRef]
- Han, Y.W.; Choi, J.Y.; Uyangaa, E.; Kim, S.B.; Kim, J.H.; Kim, B.S.; Kim, K.; Eo, S.K. Distinct Dictation of Japanese Encephalitis Virus-Induced Neuroinflammation and Lethality via Triggering TLR3 and TLR4 Signal Pathways. PLoS Pathog. 2014, 10, e1004319. [Google Scholar] [CrossRef]
- Jin, Y.-H.; Kaneyama, T.; Kang, M.H.; Kang, H.S.; Koh, C.-S.; Kim, B.S. TLR3 signaling is either protective or pathogenic for the development of Theiler’s virus-induced demyelinating disease depending on the time of viral infection. J. Neuroinflamm. 2011, 8, 178. [Google Scholar] [CrossRef]
- Wang, X.; Majumdar, T.; Kessler, P.; Ozhegov, E.; Zhang, Y.; Chattopadhyay, S.; Barik, S.; Sen, G.C. STING Requires the Adaptor TRIF to Trigger Innate Immune Responses to Microbial Infection. Cell Host Microbe 2016, 20, 329–341. [Google Scholar] [CrossRef]
- Reinert, L.; Lopušná, K.; Winther, H.; Sun, C.; Thomsen, M.; Nandakumar, R.; Mogensen, T.H.; Meyer, M.; Vægter, C.; Nyengaard, J.R.; et al. Sensing of HSV-1 by the cGAS–STING pathway in microglia orchestrates antiviral defence in the CNS. Nat. Commun. 2016, 7, 13348. [Google Scholar] [CrossRef] [PubMed]
- Parker, Z.M.; Murphy, A.A.; Leib, D.A. Role of the DNA Sensor STING in Protection from Lethal Infection following Corneal and Intracerebral Challenge with Herpes Simplex Virus 1. J. Virol. 2015, 89, 11080–11091. [Google Scholar] [CrossRef]
- Reinert, L.; Harder, L.A.; Holm, C.; Iversen, M.B.; Horan, K.A.; Dagnæs-Hansen, F.; Ulhøi, B.P.; Holm, T.H.; Mogensen, T.H.; Owens, T.; et al. TLR3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates establishment of CNS infection in mice. J. Clin. Investig. 2012, 122, 1368–1376. [Google Scholar] [CrossRef]
- Ghita, L.; Spanier, J.; Chhatbar, C.; Mulenge, F.; Pavlou, A.; Larsen, P.-K.; Waltl, I.; Lueder, Y.; Kohls, M.; Jung, K.; et al. MyD88 signaling by neurons induces chemokines that recruit protective leukocytes to the virus-infected CNS. Sci. Immunol. 2021, 6, eabc9165. [Google Scholar] [CrossRef]
- Szretter, K.J.; Daffis, S.; Patel, J.; Suthar, M.S.; Klein, R.S.; Gale, M.; Diamond, M.S. The Innate Immune Adaptor Molecule MyD88 Restricts West Nile Virus Replication and Spread in Neurons of the Central Nervous System. J. Virol. 2010, 84, 12125–12138. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Kurt-Jones, E.A.; Fitzgerald, K.; Wang, J.; Cerny, A.M.; Chan, M.; Finberg, R.W. Role of MyD88 in Route-Dependent Susceptibility to Vesicular Stomatitis Virus Infection. J. Immunol. 2007, 178, 5173–5181. [Google Scholar] [CrossRef] [PubMed]
- Sabouri, A.H.; Marcondes, M.C.G.; Flynn, C.; Berger, M.; Xiao, N.; Fox, H.; Sarvetnick, N.E. TLR signaling controls lethal encephalitis in WNV-infected brain. Brain Res. 2014, 1574, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Dups, J.; Middleton, D.; Long, F.; Arkinstall, R.; Marsh, G.; Wang, L.-F. Subclinical infection without encephalitis in mice following intranasal exposure to Nipah virus-Malaysia and Nipah virus-Bangladesh. Virol. J. 2014, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Iampietro, M.; Aurine, N.; Dhondt, K.P.; Dumont, C.; Pelissier, R.; Spanier, J.; Vallve, A.; Raoul, H.; Kalinke, U.; Horvat, B. Control of Nipah Virus Infection in Mice by the Host Adaptors Mitochondrial Antiviral Signaling Protein (MAVS) and Myeloid Differentiation Primary Response 88 (MyD88). J. Infect. Dis. 2019, 221, S401–S406. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef] [PubMed]
- Awais, M.; Wang, K.; Lin, X.; Qian, W.; Zhang, N.; Wang, C.; Wang, K.; Zhao, L.; Fu, Z.F.; Cui, M. TLR7 Deficiency Leads to TLR8 Compensative Regulation of Immune Response against JEV in Mice. Front. Immunol. 2017, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, L.N.; Reinert, L.S.; Malmgaard, L.; Bartholdy, C.; Thomsen, A.R.; Paludan, S.R. TLR2 and TLR9 Synergistically Control Herpes Simplex Virus Infection in the Brain. J. Immunol. 2008, 181, 8604–8612. [Google Scholar] [CrossRef] [PubMed]
- Sarangi, P.P.; Kim, B.; Kurt-Jones, E.; Rouse, B.T. Innate Recognition Network Driving Herpes Simplex Virus-Induced Corneal Immunopathology: Role of the Toll Pathway in Early Inflammatory Events in Stromal Keratitis. J. Virol. 2007, 81, 11128–11138. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.P.; Bowen, G.N.; Zhou, S.; Cerny, A.; Zacharia, A.; Knipe, D.M.; Finberg, R.W.; Kurt-Jones, E.A. Role of Specific Innate Immune Responses in Herpes Simplex Virus Infection of the Central Nervous System. J. Virol. 2011, 86, 2273–2281. [Google Scholar] [CrossRef]
- Kurt-Jones, E.A.; Chan, M.; Zhou, S.; Wang, J.; Reed, G.; Bronson, R.; Arnold, M.; Knipe, D.M.; Finberg, R.W. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc. Natl. Acad. Sci. USA 2004, 101, 1315–1320. [Google Scholar] [CrossRef]
- Renaud, J.; Thérien, H.-M.; Plouffe, M.; Martinoli, M.-G. La neuro-inflammation. Méd. Sci. 2015, 31, 979–988. [Google Scholar] [CrossRef][Green Version]
- Moseman, E.A.; Blanchard, A.C.; Nayak, D.; McGAVERN, D.B. T cell engagement of cross-presenting microglia protects the brain from a nasal virus infection. Sci. Immunol. 2020, 5, eabb1817. [Google Scholar] [CrossRef]
- Solmaz, G.; Puttur, F.; Francozo, M.; Lindenberg, M.; Guderian, M.; Swallow, M.; Duhan, V.; Khairnar, V.; Kalinke, U.; Ludewig, B.; et al. TLR7 Controls VSV Replication in CD169+ SCS Macrophages and Associated Viral Neuroinvasion. Front. Immunol. 2019, 10, 466. [Google Scholar] [CrossRef] [PubMed]
- Iannacone, M.; Moseman, E.A.; Tonti, E.; Bosurgi, L.; Junt, T.; Henrickson, S.; Whelan, S.; Guidotti, L.G.; Von Andrian, U.H. Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nat. Cell Biol. 2010, 465, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Rua, R.; Lee, J.Y.; Silva, A.B.; Swafford, I.S.; Maric, D.; Johnson, K.R.; McGavern, D.B. Infection drives meningeal engraftment by inflammatory monocytes that impairs CNS immunity. Nat Immunol. 2019, 20, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Silvia, S.K. Lymphocytic choriomeningitis infection of the central nervous system. Front. Biosci. 2008, 13, 4529–4543. [Google Scholar] [CrossRef]
- Schwendemann, G.; Löhler, J.; Lehmann-Grube, F. Evidence for cytotoxic T-lymphocyte-target cell interaction in brains of mice infected intracerebrally with lymphocytic choriomeningitis virus. Acta Neuropathol. 1983, 61, 183–195. [Google Scholar] [CrossRef]
- Rowe, W.P.; Black, P.H.; Levey, R.H. Protective Effect of Neonatal Thymectomy on Mouse LCM Infection. Exp. Biol. Med. 1963, 114, 248–251. [Google Scholar] [CrossRef]
- Lynch, F.; Doherty, P.C.; Ceredig, R. Phenotypic and functional analysis of the cellular response in regional lymphoid tissue during an acute virus infection. J. Immunol. 1989, 142, 3592–3598. [Google Scholar]
- Zhou, S.; Halle, A.; Kurt-Jones, E.A.; Cerny, A.M.; Porpiglia, E.; Rogers, M.; Golenbock, D.T.; Finberg, R.W. Lymphocytic Choriomeningitis Virus (LCMV) infection of CNS glial cells results in TLR2-MyD88/Mal-dependent inflammatory responses. J. Neuroimmunol. 2008, 194, 70–82. [Google Scholar] [CrossRef]
- Zhou, S.; Kurt-Jones, E.A.; Mandell, L.; Cerny, A.; Chan, M.; Golenbock, D.T.; Finberg, R.W. MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur. J. Immunol. 2005, 35, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Kurt-Jones, E.A.; Cerny, A.M.; Chan, M.; Bronson, R.T.; Finberg, R.W. MyD88 Intrinsically Regulates CD4 T-Cell Responses. J. Virol. 2009, 83, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337. [Google Scholar] [CrossRef] [PubMed]
- De Lima, K.A.; Rustenhoven, J.; Kipnis, J. Meningeal Immunity and Its Function in Maintenance of the Central Nervous System in Health and Disease. Annu. Rev. Immunol. 2020, 38, 597–620. [Google Scholar] [CrossRef]
- Kim, J.V.; Kang, S.S.; Dustin, M.L.; McGavern, D. Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nat. Cell Biol. 2008, 457, 191–195. [Google Scholar] [CrossRef]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef]
- Raper, D.; Louveau, A.; Kipnis, J. How Do Meningeal Lymphatic Vessels Drain the CNS? Trends Neurosci. 2016, 39, 581–586. [Google Scholar] [CrossRef]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Kivisäkk, P.; Imitola, J.; Rasmussen, S.; Elyaman, W.; Zhu, B.; Ransohoff, R.M.; Khoury, S.J. Localizing central nervous system immune surveillance: Meningeal antigen-presenting cells activate T cells during experimental autoimmune encephalomyelitis. Ann. Neurol. 2009, 65, 457–469. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Schläger, C.; Körner, H.; Krueger, M.; Vidoli, S.; Haberl, M.; Mielke, D.; Brylla, E.; Issekutz, T.; Cabañas, C.; Nelson, P.J.; et al. Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nat. Cell Biol. 2016, 530, 349–353. [Google Scholar] [CrossRef]
- Peferoen, L.; Kipp, M.; Van Der Valk, P.; van Noort, J.; Amor, S. Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology 2014, 141, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Nutma, E.; Van Gent, D.; Amor, S.; Peferoen, L.A.N. Astrocyte and Oligodendrocyte Cross-Talk in the Central Nervous System. Cells 2020, 9, 600. [Google Scholar] [CrossRef] [PubMed]
- Bsibsi, M.; Ravid, R.; Gveric, D.; van Noort, J.M. Broad Expression of Toll-Like Receptors in the Human Central Nervous System. J. Neuropathol. Exp. Neurol. 2002, 61, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Bsibsi, M.; Nomden, A.; van Noort, J.; Baron, W. Toll-like receptors 2 and 3 agonists differentially affect oligodendrocyte survival, differentiation, and myelin membrane formation. J. Neurosci. Res. 2011, 90, 388–398. [Google Scholar] [CrossRef]
- Sloane, J.A.; Batt, C.; Ma, Y.; Harris, Z.M.; Trapp, B.; Vartanian, T. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc. Natl. Acad. Sci. USA 2010, 107, 11555–11560. [Google Scholar] [CrossRef]
- Wang, F.-X.; Liu, S.-Y.; Zheng, X.; Chen, X.; Lu, L.-X.; Chen, B.; Xiong, X.-Y.; Shu, H.-F.; Yang, Q.-W.; Yang, H. TLR1 expression in mouse brain was increased in a KA-induced seizure model. Inflamm. Res. 2015, 64, 487–495. [Google Scholar] [CrossRef]
- Carpentier, P.A.; Begolka, W.S.; Olson, J.K.; Elhofy, A.; Karpus, W.J.; Miller, S.D. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia 2005, 49, 360–374. [Google Scholar] [CrossRef]
- Lafon, M.; Megret, F.; Lafage, M.; Prehaud, C. The Innate Immune Facet of Brain: Human Neurons Express TLR-3 and Sense Viral dsRNA. J. Mol. Neurosci. 2006, 29, 185–194. [Google Scholar] [CrossRef]
- Zhou, Y.; Ye, L.; Wan, Q.; Zhou, L.; Wang, X.; Li, J.; Hu, S.; Zhou, D.; Ho, W. Activation of Toll-like receptors inhibits herpes simplex virus-1 infection of human neuronal cells. J. Neurosci. Res. 2009, 87, 2916–2925. [Google Scholar] [CrossRef]
- Jack, C.S.; Arbour, N.; Manusow, J.; Montgrain, V.; Blain, M.; McCrea, E.; Shapiro, A.; Antel, J. TLR Signaling Tailors Innate Immune Responses in Human Microglia and Astrocytes. J. Immunol. 2005, 175, 4320–4330. [Google Scholar] [CrossRef]
- Takeuchi, O.; Hoshino, K.; Kawai, T.; Sanjo, H.; Takada, H.; Ogawa, T.; Takeda, K.; Akira, S. Differential Roles of TLR2 and TLR4 in Recognition of Gram-Negative and Gram-Positive Bacterial Cell Wall Components. Immunity 1999, 11, 443–451. [Google Scholar] [CrossRef]
- Schwandner, R.; Dziarski, R.; Wesche, H.; Rothe, M.; Kirschning, C.J. Peptidoglycan- and Lipoteichoic Acid-induced Cell Activation Is Mediated by Toll-like Receptor. J. Biol. Chem. 1999, 274, 17406–17409. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.-C.; Arumugam, T.V.; Xu, X.; Cheng, A.; Mughal, M.R.; Jo, D.-G.; Lathia, J.D.; Siler, D.A.; Chigurupati, S.; Ouyang, X.; et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 13798–13803. [Google Scholar] [CrossRef]
- Kim, C.; Rockenstein, E.; Spencer, B.; Kim, H.-K.; Adame, A.; Trejo, M.; Stafa, K.; Lee, H.-J.; Lee, S.-J.; Masliah, E. Antagonizing Neuronal Toll-like Receptor 2 Prevents Synucleinopathy by Activating Autophagy. Cell Rep. 2015, 13, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Bowman, C.C.; Rasley, A.; Tranguch, S.L.; Marriott, I. Cultured astrocytes express toll-like receptors for bacterial products. Glia 2003, 43, 281–291. [Google Scholar] [CrossRef]
- Hu, F.; Dzaye, O.D.; Hahn, A.; Yu, Y.; Scavetta, R.J.; Dittmar, G.; Kaczmarek, A.K.; Dunning, K.; Ricciardelli, C.; Rinnenthal, J.L.; et al. Glioma-derived versican promotes tumor expansion via glioma-associated microglial/macrophages Toll-like receptor 2 signaling. Neuro-Oncology 2014, 17, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Lawrimore, C.J.; Crews, F.T. Ethanol, TLR3, and TLR4 Agonists Have Unique Innate Immune Responses in Neuron-Like SH-SY5Y and Microglia-Like BV1. Alcohol. Clin. Exp. Res. 2017, 41, 939–954. [Google Scholar] [CrossRef]
- Gao, D.; Ciancanelli, M.J.; Zhang, P.; Harschnitz, O.; Bondet, V.; Hasek, M.; Chen, J.; Mu, X.; Itan, Y.; Cobat, A.; et al. TLR3 controls constitutive IFN-β antiviral immunity in human fibroblasts and cortical neurons. J. Clin. Investig. 2021, 131, 134529. [Google Scholar] [CrossRef]
- Hoshino, K.; Takeuchi, O.; Kawai, T.; Sanjo, H.; Ogawa, T.; Takeda, Y.; Takeda, K.; Akira, S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J. Immunol. 1999, 162, 3749–3752. [Google Scholar] [PubMed]
- Leow-Dyke, S.; Allen, C.; Denes, A.; Nilsson, O.; Maysami, S.; Bowie, A.G.; Rothwell, N.J.; Pinteaux, E. Neuronal toll-like receptor 4 signaling induces brain endothelial activation and neutrophil transmigration in vitro. J. Neuroinflamm. 2012, 9, 230. [Google Scholar] [CrossRef]
- Ifuku, M.; Hinkelmann, L.; Kuhrt, L.D.; Efe, I.E.; Kumbol, V.; Buonfiglioli, A.; Krüger, C.; Jordan, P.; Fulde, M.; Noda, M.; et al. Activation of Toll-like receptor 5 in microglia modulates their function and triggers neuronal injury. Acta Neuropathol. Commun. 2020, 8, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-T.; Xu, X.-Y.; Lin, W.; Hu, D.-D.; Shi, W.; Jia, X.; Wang, H.; Song, N.-J.; Zhang, Y.-Q.; Zhang, L. Activation of Different Heterodimers of TLR2 Distinctly Mediates Pain and Itch. Neuroscience 2020, 429, 245–255. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Han, G.; Wu, S.; Du, S.; Zhang, Y.; Liu, W.; Jiang, B.; Zhang, L.; Xia, S.; Jia, S.; et al. Toll-like receptor 7 contributes to neuropathic pain by activating NF-κB in primary sensory neurons. Brain Behav. Immun. 2020, 87, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.-F.; Chen, C.-Y.; Shih, Y.-C.; Liu, H.-Y.; Huang, C.-M.; Hsueh, Y.-P. Endosomal TLR3, TLR7, and TLR8 control neuronal morphology through different transcriptional programs. J. Cell Biol. 2018, 217, 2727–2742. [Google Scholar] [CrossRef]
- Sun, Y.; Xiao, Q.; Luo, C.; Zhao, Y.; Pu, D.; Zhao, K.; Chen, J.; Wang, M.; Liao, Z. High-glucose induces tau hyperphosphorylation through activation of TLR9-P38MAPK pathway. Exp. Cell Res. 2017, 359, 312–318. [Google Scholar] [CrossRef]
- Maatouk, L.; Compagnion, A.-C.; Sauvage, M.-A.C.-D.; Bemelmans, A.-P.; Leclere-Turbant, S.; Cirotteau, V.; Tohme, M.; Beke, A.; Trichet, M.; Bazin, V.; et al. TLR9 activation via microglial glucocorticoid receptors contributes to degeneration of midbrain dopamine neurons. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Chuang, T.-H.; Ulevitch, R.J. Identification of hTLR10: A novel human Toll-like receptor preferentially expressed in immune cells. Biochim. Biophys. Acta Gene Struct. Expr. 2001, 1518, 157–161. [Google Scholar] [CrossRef]
- Van Le, H.; Kim, J.Y. Stable Toll-Like Receptor 10 Knockdown in THP-1 Cells Reduces TLR-Ligand-Induced Proinflammatory Cytokine Expression. Int. J. Mol. Sci. 2016, 17, 859. [Google Scholar] [CrossRef]
- Lee, S.M.Y.; Kok, K.-H.; Jaume, M.; Cheung, T.K.W.; Yip, T.-F.; Lai, J.C.C.; Guan, Y.; Webster, R.G.; Jin, D.-Y.; Peiris, J.S.M. Toll-like receptor 10 is involved in induction of innate immune responses to influenza virus infection. Proc. Natl. Acad. Sci. USA 2014, 111, 3793–3798. [Google Scholar] [CrossRef]
- Galatro, T.; Holtman, I.R.; Lerario, A.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Möller, T.; et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171. [Google Scholar] [CrossRef]
- Yarovinsky, F.; Zhang, D.; Andersen, J.F.; Bannenberg, G.L.; Serhan, C.N.; Hayden, M.S.; Hieny, S.; Sutterwala, F.S.; Flavell, R.A.; Ghosh, S.; et al. TLR11 Activation of Dendritic Cells by a Protozoan Profilin-Like Protein. Science 2005, 308, 1626–1629. [Google Scholar] [CrossRef]
- Mishra, B.B.; Gundra, U.M.; Teale, J.M. Expression and distribution of Toll-like receptors 11–13 in the brain during murine neurocysticercosis. J. Neuroinflamm. 2008, 5, 53. [Google Scholar] [CrossRef]
- Koblansky, A.A.; Jankovic, D.; Oh, H.; Hieny, S.; Sungnak, W.; Mathur, R.; Hayden, M.; Akira, S.; Sher, A.; Ghosh, S. Recognition of Profilin by Toll-like Receptor 12 Is Critical for Host Resistance to Toxoplasma gondii. Immunity 2013, 38, 119–130. [Google Scholar] [CrossRef]
- Lord, K.; Hoffman-Liebermann, B.; Liebermann, D. Nucleotide sequence and expression of a cDNA encoding MyD88, a novel myeloid differentiation primary response gene induced by IL1. Oncogene 1990, 5, 1095–1097. [Google Scholar]
- Fan, H.; Zhang, K.; Shan, L.; Kuang, F.; Chen, K.; Zhu, K.; Ma, H.; Ju, G.; Wang, Y.-Z. Reactive astrocytes undergo M1 microglia/macrohpages-induced necroptosis in spinal cord injury. Mol. Neurodegener. 2016, 11, 1–16. [Google Scholar] [CrossRef]
- Esen, N.; Kielian, T. Central Role for MyD88 in the Responses of Microglia to Pathogen-Associated Molecular Patterns. J. Immunol. 2006, 176, 6802–6811. [Google Scholar] [CrossRef]
- Lan, X.; Han, X.; Li, Q.; Gao, Y.; Cheng, T.; Wan, J.; Zhu, W.; Wang, J. Pinocembrin protects hemorrhagic brain primarily by inhibiting toll-like receptor 4 and reducing M1 phenotype microglia. Brain Behav. Immun. 2017, 61, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Sun, C.; Ma, Y.; Wang, S.; Wang, X.; Zhang, Y. Inhibition of TLR4 Induces M2 Microglial Polarization and Provides Neuroprotection via the NLRP3 Inflammasome in Alzheimer’s Disease. Front. Neurosci. 2020, 14, 444. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Kim, K.K.; Park, B.S.; Kim, D.H.; Jeong, B.; Kang, D.; Lee, T.H.; Park, J.W.; Kim, J.G.; Lee, B.J. Function of astrocyte MyD88 in high-fat-diet-induced hypothalamic inflammation. J. Neuroinflamm. 2020, 17, 195. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Sanjo, H.; Uematsu, S.; Kaisho, T.; Hoshino, K.; Takeuchi, O.; Kobayashi, M.; Fujita, T.; et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nat. Cell Biol. 2002, 420, 324–329. [Google Scholar] [CrossRef]
- Wang, F.-X.; Yang, X.-L.; Ma, Y.-S.; Wei, Y.-J.; Yang, M.-H.; Chen, X.; Chen, B.; He, Q.; Yang, Q.-W.; Yang, H.; et al. TRIF contributes to epileptogenesis in temporal lobe epilepsy during TLR4 activation. Brain Behav. Immun. 2018, 67, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Soung, A.; Klein, R.S. Viral Encephalitis and Neurologic Diseases: Focus on Astrocytes. Trends Mol. Med. 2018, 24, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, O.O.; Hogue, I.; Enquist, L.W. Virus Infections in the Nervous System. Cell Host Microbe 2013, 13, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-Y.; Chen, C.-Y.; Hsueh, Y.-P. Innate immune responses regulate morphogenesis and degeneration: Roles of Toll-like receptors and Sarm1 in neurons. Neurosci. Bull. 2014, 30, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.S.; Hunter, C.A. Protective and Pathological Immunity during Central Nervous System Infections. Immunity 2017, 46, 891–909. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, C.; Chen, O.; Ji, R.-R. How Do Sensory Neurons Sense Danger Signals? Trends Neurosci. 2020, 43, 822–838. [Google Scholar] [CrossRef]
- Lehnardt, S.; Massillon, L.; Follett, P.; Jensen, F.E.; Ratan, R.; Rosenberg, P.; Volpe, J.J.; Vartanian, T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 8514–8519. [Google Scholar] [CrossRef]
- Rietdijk, C.D.; Van Wezel, R.J.A.; Garssen, J.; Kraneveld, A.D. Neuronal toll-like receptors and neuro-immunity in Parkinson’s disease, Alzheimer’s disease and stroke. Neuroimmunol. Neuroinflamm. 2016, 3, 27. [Google Scholar] [CrossRef]
- Préhaud, C.; Mégret, F.; Lafage, M.; Lafon, M. Virus Infection Switches TLR-3-Positive Human Neurons to Become Strong Producers of Beta Interferon. J. Virol. 2005, 79, 12893–12904. [Google Scholar] [CrossRef]
- Delhaye, S.; Paul, S.; Blakqori, G.; Minet, M.; Weber, F.; Staeheli, P.; Michiels, T. Neurons produce type I interferon during viral encephalitis. Proc. Natl. Acad. Sci. USA 2006, 103, 7835–7840. [Google Scholar] [CrossRef]
- Liu, H.-Y.; Hong, Y.-F.; Huang, C.-M.; Chen, C.-Y.; Huang, T.-N.; Hsueh, Y.-P. TLR7 Negatively Regulates Dendrite Outgrowth through the Myd88-c-Fos-IL-6 Pathway. J. Neurosci. 2013, 33, 11479–11493. [Google Scholar] [CrossRef] [PubMed]
- Di Liberto, G.; Pantelyushin, S.; Kreutzfeldt, M.; Page, N.; Musardo, S.; Coras, R.; Steinbach, K.; Vincenti, I.; Klimek, B.; Lingner, T.; et al. Neurons under T Cell Attack Coordinate Phagocyte-Mediated Synaptic Stripping. Cell 2018, 175, 458–471.e19. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.S.; Garber, C.; Funk, K.; Salimi, H.; Soung, A.; Kanmogne, M.; Manivasagam, S.; Agner, S.; Cain, M. Neuroinflammation During RNA Viral Infections. Annu. Rev. Immunol. 2019, 37, 73–95. [Google Scholar] [CrossRef] [PubMed]
- Priya, R.; Dhanwani, R.; Patro, I.; Rao, P.; Parida, M. Differential regulation of TLR mediated innate immune response of mouse neuronal cells following infection with novel ECSA genotype of Chikungunya virus with and without E1:A226V mutation. Infect. Genet. Evol. 2013, 20, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, B.; Ewaleifoh, O.; Harschnitz, O.; Lee, Y.-S.; Peneau, C.; McAlpine, J.L.; Liu, B.; Tchieu, J.; Steinbeck, J.A.; Lafaille, F.; et al. Human iPSC-derived trigeminal neurons lack constitutive TLR3-dependent immunity that protects cortical neurons from HSV-1 infection. Proc. Natl. Acad. Sci. USA 2018, 115, E8775–E8782. [Google Scholar] [CrossRef]
- Matias, I.; Morgado, J.; Gomes, F.C.A. Astrocyte Heterogeneity: Impact to Brain Aging and Disease. Front. Aging Neurosci. 2019, 11, 59. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2015, 7, a020420. [Google Scholar] [CrossRef]
- Detje, C.N.; Lienenklaus, S.; Chhatbar, C.; Spanier, J.; Prajeeth, C.K.; Soldner, C.; Tovey, M.; Schlüter, D.; Weiss, S.; Stangel, M.; et al. Upon Intranasal Vesicular Stomatitis Virus Infection, Astrocytes in the Olfactory Bulb Are Important Interferon Beta Producers That Protect from Lethal Encephalitis. J. Virol. 2014, 89, 2731–2738. [Google Scholar] [CrossRef]
- Pfefferkorn, C.; Kallfass, C.; Lienenklaus, S.; Spanier, J.; Kalinke, U.; Rieder, M.; Conzelmann, K.-K.; Michiels, T.; Staeheli, P. Abortively Infected Astrocytes Appear to Represent the Main Source of Interferon Beta in the Virus-Infected Brain. J. Virol. 2016, 90, 2031–2038. [Google Scholar] [CrossRef]
- Lindqvist, R.; Mundt, F.; Gilthorpe, J.D.; Wölfel, S.; Gekara, N.O.; Kröger, A.; Överby, A.K. Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J. Neuroinflamm. 2016, 13, 277. [Google Scholar] [CrossRef]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Bsibsi, M.; Persoon-Deen, C.; Verwer, R.W.; Meeuwsen, S.; Ravid, R.; Van Noort, J.M. Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia 2006, 53, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Trudler, D.; Farfara, D.; Frenkel, D. Toll-Like Receptors Expression and Signaling in Glia Cells in Neuro-Amyloidogenic Diseases: Towards Future Therapeutic Application. Mediat. Inflamm. 2010, 2010, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-Stranded RNA Is Produced by Positive-Strand RNA Viruses and DNA Viruses but Not in Detectable Amounts by Negative-Strand RNA Viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef]
- Liu, Z.; Guan, Y.; Sun, X.; Shi, L.; Liang, R.; Lv, X.; Xin, W. HSV-1 activates NF-kappaB in mouse astrocytes and increases TNF-alpha and IL-6 expressionviaToll-like receptor 1. Neurol. Res. 2013, 35, 755–762. [Google Scholar] [CrossRef]
- Ojha, C.R.; Rodriguez, M.; Karuppan, M.K.M.; Lapierre, J.; Kashanchi, F.; El-Hage, N. Toll-like receptor 3 regulates Zika virus infection and associated host inflammatory response in primary human astrocytes. PLoS ONE 2019, 14, e0208543. [Google Scholar] [CrossRef]
- Ghita, L.; Breitkopf, V.; Mulenge, F.; Pavlou, A.; Gern, O.L.; Durán, V.; Prajeeth, C.K.; Kohls, M.; Jung, K.; Stangel, M.; et al. Sequential MAVS and MyD88/TRIF signaling triggers anti-viral responses of tick-borne encephalitis virus-infected murine astrocytes. J. Neurosci. Res. 2021. [Google Scholar] [CrossRef]
- Carpentier, P.A.; Williams, B.R.; Miller, S.D. Distinct roles of protein kinase R and toll-like receptor 3 in the activation of astrocytes by viral stimuli. Glia 2007, 55, 239–252. [Google Scholar] [CrossRef]
- So, E.Y.; Kang, M.H.; Kim, B.S. Induction of chemokine and cytokine genes in astrocytes following infection with Theiler’s murine encephalomyelitis virus is mediated by the Toll-like receptor. Glia 2006, 53, 858–867. [Google Scholar] [CrossRef]
- Zhu, J.; Martinez, J.; Huang, X.; Yang, Y. Innate immunity against vaccinia virus is mediated by TLR2 and requires TLR-independent production of IFN-β. Blood 2006, 109, 619–625. [Google Scholar] [CrossRef] [PubMed]
- So, E.Y.; Kim, B.S. Theiler’s virus infection induces TLR3-dependent upregulation of TLR2 critical for proinflammatory cytokine production. Glia 2009, 57, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Korn, T.; Kuchroo, V.K. Th17: The third member of the effector T cell trilogy. Curr. Opin. Immunol. 2007, 19, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, N.; Rivest, S. Toll-like receptor 4: The missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J. 2001, 15, 155–163. [Google Scholar] [CrossRef]
- Gorina, R.; Font-Nieves, M.; Márquez-Kisinousky, L.; Santalucia, T.; Planas, A.M. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia 2011, 59, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Villalba, M.; Hott, M.; Martin, C.; Aguila, B.; Valdivia, S.; Quezada, C.; Zambrano, Á.; Concha, M.I.; Otth, C. Herpes simplex virus type 1 induces simultaneous activation of Toll-like receptors 2 and 4 and expression of the endogenous ligand serum amyloid A in astrocytes. Med. Microbiol. Immunol. 2012, 201, 371–379. [Google Scholar] [CrossRef]
- Hosoi, T.; Suzuki, S.; Nomura, J.; Ono, A.; Okuma, Y.; Akira, S.; Nomura, Y. Bacterial DNA induced iNOS expression through MyD88-p38 MAP kinase in mouse primary cultured glial cells. Mol. Brain Res. 2004, 124, 159–164. [Google Scholar] [CrossRef]
- Li, L.; Ni, L.; Heary, R.F.; Elkabes, S. Astroglial TLR9 antagonism promotes chemotaxis and alternative activation of macrophages via modulation of astrocyte-derived signals: Implications for spinal cord injury. J. Neuroinflamm. 2020, 17, 1–18. [Google Scholar] [CrossRef]
- Hamel, R.; Ferraris, P.; Wichit, S.; Diop, F.; Talignani, L.; Pompon, J.; Garcia, D.; Liegeois, F.; Sall, A.A.; Yssel, H.; et al. African and Asian Zika virus strains differentially induce early antiviral responses in primary human astrocytes. Infect. Genet. Evol. 2017, 49, 134–137. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Prinz, M.; Masuda, T.; Wheeler, M.A.; Quintana, F.J. Microglia and Central Nervous System–Associated Macrophages—From Origin to Disease Modulation. Annu. Rev. Immunol. 2021, 39, 251–277. [Google Scholar] [CrossRef] [PubMed]
- Fekete, R.; Cserép, C.; Lénárt, N.; Tóth, K.; Orsolits, B.; Martinecz, B.; Méhes, E.; Szabó, B.; Németh, V.; Gönci, B.; et al. Microglia control the spread of neurotropic virus infection via P2Y12 signalling and recruit monocytes through P2Y12-independent mechanisms. Acta Neuropathol. 2018, 136, 461–482. [Google Scholar] [CrossRef] [PubMed]
- Reinert, L.S.; Rashidi, A.S.; Tran, D.N.; Katzilieris-Petras, G.; Hvidt, A.K.; Gohr, M.; Fruhwürth, S.; Bodda, C.; Thomsen, M.K.; Vendelbo, M.H.; et al. Brain immune cells undergo cGAS/STING-dependent apoptosis during herpes simplex virus type 1 infection to limit type I IFN production. J. Clin. Investig. 2021, 131, 136824. [Google Scholar] [CrossRef] [PubMed]
- Chhatbar, C.; Detje, C.N.; Grabski, E.; Borst, K.; Spanier, J.; Ghita, L.; Elliott, D.A.; Jordão, M.J.C.; Mueller, N.; Sutton, J.; et al. Type I Interferon Receptor Signaling of Neurons and Astrocytes Regulates Microglia Activation during Viral Encephalitis. Cell Rep. 2018, 25, 118–129.e4. [Google Scholar] [CrossRef]
- Seitz, S.; Clarke, P.; Tyler, K.L. Pharmacologic Depletion of Microglia Increases Viral Load in the Brain and Enhances Mortality in Murine Models of Flavivirus-Induced Encephalitis. J. Virol. 2018, 92, 525. [Google Scholar] [CrossRef]
- Waltl, I.; Käufer, C.; Gerhauser, I.; Chhatbar, C.; Ghita, L.; Kalinke, U.; Löscher, W. Microglia have a protective role in viral encephalitis-induced seizure development and hippocampal damage. Brain Behav. Immun. 2018, 74, 186–204. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Sariol, A.; Meyerholz, D.K.; Perlman, S. Microglia are required for protection against lethal coronavirus encephalitis in mice. J. Clin. Investig. 2018, 128, 931–943. [Google Scholar] [CrossRef]
- Wang, Y.; Szretter, K.; Vermi, W.; Gilfillan, S.; Rossini, C.; Cella, M.; Barrow, A.; Diamond, M.S.; Colonna, M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 2012, 13, 753–760. [Google Scholar] [CrossRef]
- Funk, K.; Klein, R.S. CSF1R antagonism limits local restimulation of antiviral CD8+ T cells during viral encephalitis. J. Neuroinflamm. 2019, 16, 1–19. [Google Scholar] [CrossRef]
- Town, T.; Jeng, D.; Alexopoulou, L.; Tan, J.; Flavell, R.A. Microglia Recognize Double-Stranded RNA via TLR3. J. Immunol. 2006, 176, 3804–3812. [Google Scholar] [CrossRef]
- Olson, J.K.; Miller, S.D. Microglia Initiate Central Nervous System Innate and Adaptive Immune Responses through Multiple TLRs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Welte, T.; Wang, J.; Whiteman, M.C.; Wicker, J.A.; Saxena, V.; Cong, Y.; Barrett, A.D.; Wang, T. A West Nile virus NS4BP38G mutant strain induces adaptive immunity via TLR7-MyD88-dependent and independent signaling pathways. Vaccine 2013, 31, 4143–4151. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Ye, J.; Zhu, B.; Song, Y.; Chen, H.; Cao, S. Roles of TLR3 and RIG-I in Mediating the Inflammatory Response in Mouse Microglia following Japanese Encephalitis Virus Infection. J. Immunol. Res. 2014, 2014, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jhan, M.-K.; Tsai, T.-T.; Chen, C.-L.; Tsai, C.-C.; Cheng, Y.-L.; Lee, Y.-C.; Ko, C.-Y.; Lin, Y.-S.; Chang, C.-P.; Lin, L.-T.; et al. Dengue virus infection increases microglial cell migration. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Davey, G.M.; Wojtasiak, M.; Proietto, A.I.; Carbone, F.R.; Heath, W.R.; Bedoui, S. Cutting Edge: Priming of CD8 T Cell Immunity to Herpes Simplex Virus Type 1 Requires Cognate TLR3 Expression InVivo. J. Immunol. 2010, 184, 2243–2246. [Google Scholar] [CrossRef] [PubMed]
- Town, T.; Bai, F.; Wang, T.; Kaplan, A.; Qian, F.; Montgomery, R.; Anderson, J.F.; Flavell, R.A.; Fikrig, E. Toll-like Receptor 7 Mitigates Lethal West Nile Encephalitis via Interleukin 23-Dependent Immune Cell Infiltration and Homing. Immunity 2009, 30, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Aravalli, R.N.; Hu, S.; Lokensgard, J.R. Toll-like receptor 2 signaling is a mediator of apoptosis in herpes simplex virus-infected microglia. J. Neuroinflamm. 2007, 4, 11. [Google Scholar] [CrossRef]
- Fernández-Arjona, M.D.M.; Grondona, J.M.; Fernández-Llebrez, P.; López-Ávalos, M.D. Microglial activation by microbial neuraminidase through TLR2 and TLR4 receptors. J. Neuroinflamm. 2019, 16, 1–14. [Google Scholar] [CrossRef]
- Lv, X.; Wang, H.; Su, A.; Xu, S.; Chu, Y. Herpes simplex virus type 2 infection triggers AP-1 transcription activity through TLR4 signaling in genital epithelial cells. Virol. J. 2018, 15, 173. [Google Scholar] [CrossRef]
- Xiao, Y.; Jin, J.; Chang, M.; Chang, J.-H.; Hu, H.; Zhou, X.; Brittain, G.C.; Stansberg, C.; Torkildsen, Ø.; Wang, X.; et al. Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation. Nat. Med. 2013, 19, 595–602. [Google Scholar] [CrossRef]
- Luo, H.; Winkelmann, E.R.; Zhu, S.; Ru, W.; Mays, E.; Silvas, J.; Vollmer, L.L.; Gao, J.; Peng, B.-H.; Bopp, N.E.; et al. Peli1 facilitates virus replication and promotes neuroinflammation during West Nile virus infection. J. Clin. Investig. 2018, 128, 4980–4991. [Google Scholar] [CrossRef]
- Li, H.; Zhang, J.; Kumar, A.; Zheng, M.; Atherton, S.S.; Yu, F.S. Herpes simplex virus 1 infection induces the expression of proinflammatory cytokines, interferons and TLR7 in human corneal epithelial cells. Immunology 2006, 117, 167–176. [Google Scholar] [CrossRef]
- Jacquemont, B.; Roizman, B. RNA synthesis in cells infected with herpes simplex virus. X. Properties of viral symmetric transcripts and of double-stranded RNA prepared from them. J. Virol. 1975, 15, 707–713. [Google Scholar] [CrossRef]
- Whitley, R.J.; Kimberlin, D.W.; Roizman, B. Herpes Simplex Viruses. Clin. Infect. Dis. 1998, 26, 541–553. [Google Scholar] [CrossRef]
- Zhu, H.; Zheng, C. The Race between Host Antiviral Innate Immunity and the Immune Evasion Strategies of Herpes Simplex Virus. Microbiol. Mol. Biol. Rev. 2020, 84, 99. [Google Scholar] [CrossRef]
- van Lint, A.L.; Murawski, M.R.; Goodbody, R.E.; Severa, M.; Fitzgerald, K.A.; Finberg, R.W.; Knipe, D.M.; Kurt-Jones, E.A. Herpes Simplex Virus Immediate-Early ICP0 Protein Inhibits Toll-Like Receptor 2-Dependent Inflammatory Responses and NF-κB Signaling. J. Virol. 2010, 84, 10802–10811. [Google Scholar] [CrossRef]
- Sen, J.; Liu, X.; Roller, R.; Knipe, D.M. Herpes simplex virus US3 tegument protein inhibits Toll-like receptor 2 signaling at or before TRAF6 ubiquitination. Virology 2013, 439, 65–73. [Google Scholar] [CrossRef]
- Peri, P.; Mattila, R.K.; Kantola, H.; Broberg, E.; Karttunen, H.S.; Waris, M.; Vuorinen, T.; Hukkanen, V. Herpes Simplex Virus Type 1 Us3 Gene Deletion Influences Toll-like Receptor Responses in Cultured Monocytic Cells. Virol. J. 2008, 5, 140. [Google Scholar] [CrossRef]
- Xing, J.; Ni, L.; Wang, S.; Wang, K.; Lin, R.; Zheng, C. Herpes Simplex Virus 1-Encoded Tegument Protein VP16 Abrogates the Production of Beta Interferon (IFN) by Inhibiting NF- B Activation and Blocking IFN Regulatory Factor 3 To Recruit Its Coactivator CBP. J. Virol. 2013, 87, 9788–9801. [Google Scholar] [CrossRef]
- Iwamoto, M.; Jernigan, D.B.; Guasch, A.; Trepka, M.J.; Blackmore, C.G.; Hellinger, W.C.; Pham, S.M.; Zaki, S.; Lanciotti, R.S.; Lance-Parker, S.E.; et al. Transmission of West Nile Virus from an Organ Donor to Four Transplant Recipients. N. Engl. J. Med. 2003, 348, 2196–2203. [Google Scholar] [CrossRef]
- Green, A.M.; Beatty, P.R.; Hadjilaou, A.; Harris, E. Innate Immunity to Dengue Virus Infection and Subversion of Antiviral Responses. J. Mol. Biol. 2014, 426, 1148–1160. [Google Scholar] [CrossRef]
- Tsai, Y.-T.; Chang, S.-Y.; Lee, C.-N.; Kao, C.-L. Human TLR3 recognizes dengue virus and modulates viral replicationin vitro. Cell. Microbiol. 2009, 11, 604–615. [Google Scholar] [CrossRef]
- Aguirre, S.; Maestre, A.M.; Pagni, S.; Patel, J.; Savage, T.; Gutman, D.; Maringer, K.; Bernal-Rubio, D.; Shabman, R.; Simon, V.; et al. DENV Inhibits Type I IFN Production in Infected Cells by Cleaving Human STING. PLoS Pathog. 2012, 8, e1002934. [Google Scholar] [CrossRef]
- Muñoz-Jordán, J.L.; Laurent-Rolle, M.; Ashour, J.; Martínez-Sobrido, L.; Ashok, M.; Lipkin, W.I.; García-Sastre, A. Inhibition of Alpha/Beta Interferon Signaling by the NS4B Protein of Flaviviruses. J. Virol. 2005, 79, 8004–8013. [Google Scholar] [CrossRef]
- Schuessler, A.; Funk, A.; Lazear, H.; Cooper, D.; Torres, S.; Daffis, S.; Jha, B.K.; Kumagai, Y.; Takeuchi, O.; Hertzog, P.; et al. West Nile Virus Noncoding Subgenomic RNA Contributes to Viral Evasion of the Type I Interferon-Mediated Antiviral Response. J. Virol. 2012, 86, 5708–5718. [Google Scholar] [CrossRef]
- Arjona, A.; Ledizet, M.; Anthony, K.; Bonafé, N.; Modis, Y.; Town, T.; Fikrig, E. West Nile virus envelope protein inhibits dsRNA-induced innate immune responses. J. Immunol. 2007, 179, 8403–8409. [Google Scholar] [CrossRef]
- Best, S.M.; Morris, K.L.; Shannon, J.G.; Robertson, S.J.; Mitzel, D.N.; Park, G.S.; Boer, E.; Wolfinbarger, J.B.; Bloom, M.E. Inhibition of Interferon-Stimulated JAK-STAT Signaling by a Tick-Borne Flavivirus and Identification of NS5 as an Interferon Antagonist. J. Virol. 2005, 79, 12828–12839. [Google Scholar] [CrossRef]
- Laurent-Rolle, M.; Boer, E.; Lubick, K.J.; Wolfinbarger, J.B.; Carmody, A.B.; Rockx, B.; Liu, W.; Ashour, J.; Shupert, W.L.; Holbrook, M.R.; et al. The NS5 Protein of the Virulent West Nile Virus NY99 Strain Is a Potent Antagonist of Type I Interferon-Mediated JAK-STAT Signaling. J. Virol. 2010, 84, 3503–3515. [Google Scholar] [CrossRef]
- Ito, N.; Moseley, G.W.; Sugiyama, M. The importance of immune evasion in the pathogenesis of rabies virus. J. Vet. Med. Sci. 2016, 78, 1089–1098. [Google Scholar] [CrossRef]
- Brzózka, K.; Finke, S.; Conzelmann, K.-K. Identification of the Rabies Virus Alpha/Beta Interferon Antagonist: Phosphoprotein P Interferes with Phosphorylation of Interferon Regulatory Factor. J. Virol. 2005, 79, 7673–7681. [Google Scholar] [CrossRef]
- Wiltzer, L.; Okada, K.; Yamaoka, S.; Larrous, F.; Kuusisto, H.V.; Sugiyama, M.; Blondel, D.; Bourhy, H.; Jans, D.; Ito, N.; et al. Interaction of Rabies Virus P-Protein with STAT Proteins is Critical to Lethal Rabies Disease. J. Infect. Dis. 2014, 209, 1744–1753. [Google Scholar] [CrossRef]
- Ben Khalifa, Y.; Luco, S.; Besson, B.; Sonthonnax, F.; Archambaud, M.; Grimes, J.M.; Larrous, F.; Bourhy, H. The matrix protein of rabies virus binds to RelAp43 to modulate NF-κB-dependent gene expression related to innate immunity. Sci. Rep. 2016, 6, 39420. [Google Scholar] [CrossRef]
- Kouznetzoff, A.; Buckle, M.; Tordo, N. Identification of a region of the rabies virus N protein involved in direct binding to the viral RNA. J. Gen. Virol. 1998, 79, 1005–1013. [Google Scholar] [CrossRef]
- Okabe, N.; Yamashita, K.; Inouye, K.T.A.S. Influenza surveillance system of Japan and acute encephalitis and encephalopathy in the influenza season. Pediatr. Int. 2000, 42, 187–191. [Google Scholar] [CrossRef]
- Studahl, M. Influenza virus and CNS manifestations. J. Clin. Virol. 2003, 28, 225–232. [Google Scholar] [CrossRef]
- Hatada, E.; Fukuda, R. Binding of influenza A virus NS1 protein to dsRNA in vitro. J. Gen. Virol. 1992, 73, 3325–3329. [Google Scholar] [CrossRef]
- Abel, L.; Plancoulaine, S.; Jouanguy, E.; Zhang, S.-Y.; Mahfoufi, N.; Nicolas, N.; Sancho-Shimizu, V.; Alcaïs, A.; Guo, Y.; Cardon, A.; et al. Age-Dependent Mendelian Predisposition to Herpes Simplex Virus Type 1 Encephalitis in Childhood. J. Pediatr. 2010, 157, 623–629.e1. [Google Scholar] [CrossRef]
- Boisson-Dupuis, S.; Jouanguy, E.; Al-Hajjar, S.; Fieschi, C.; Al-Mohsen, I.Z.; Al-Jumaah, S.; Yang, K.; Chapgier, A.; Eidenschenk, C.; Eid, P.; et al. Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency. Nat. Genet. 2003, 33, 388–391. [Google Scholar] [CrossRef]
- Bastard, P.; Manry, J.; Chen, J.; Rosain, J.; Seeleuthner, Y.; AbuZaitun, O.; Lorenzo, L.; Khan, T.; Hasek, M.; Hernandez, N.; et al. Herpes simplex encephalitis in a patient with a distinctive form of inherited IFNAR1 deficiency. J. Clin. Investig. 2021, 131, 139980. [Google Scholar] [CrossRef]
- Tabeta, K.; Hoebe, K.; Janssen, E.M.; Du, X.; Georgel, P.; Crozat, K.; Mudd, S.; Mann, N.; Sovath, S.; Goode, J.R.; et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat. Immunol. 2006, 7, 156–164. [Google Scholar] [CrossRef]
- Casrouge, A.; Zhang, S.-Y.; Eidenschenk, C.; Jouanguy, E.; Puel, A.; Yang, K.; Alcais, A.; Picard, C.; Mahfoufi, N.; Nicolas, N.; et al. Herpes Simplex Virus Encephalitis in Human UNC-93B Deficiency. Science 2006, 314, 308–312. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Brinkmann, M.M.; Paquet, M.-E.; Ploegh, H.L. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nat. Cell Biol. 2008, 452, 234–238. [Google Scholar] [CrossRef]
- Lee, B.L.; Moon, J.; Shu, J.H.; Yuan, L.; Newman, Z.R.; Schekman, R.; Barton, G.M. UNC93B1 mediates differential trafficking of endosomal TLRs. eLife 2013, 2, e00291. [Google Scholar] [CrossRef]
- Pelka, K.; Bertheloot, D.; Reimer, E.; Phulphagar, K.; Schmidt, S.V.; Christ, A.; Stahl, R.; Watson, N.; Miyake, K.; Hacohen, N.; et al. The Chaperone UNC93B1 Regulates Toll-like Receptor Stability Independently of Endosomal TLR Transport. Immunity 2018, 48, 911–922.e7. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Audry, M.; Ciancanelli, M.; Alsina, L.; Azevedo, J.; Herman, M.; Anguiano, E.; Sancho-Shimizu, V.; Lorenzo, L.; Pauwels, E.; et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J. Exp. Med. 2011, 208, 2083–2098. [Google Scholar] [CrossRef]
- Lafaille, F.G.; Pessach, I.M.; Zhang, S.-Y.; Ciancanelli, M.; Herman, M.; Abhyankar, A.; Ying, S.-W.; Keros, S.; Goldstein, P.; Mostoslavsky, G.; et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nat. Cell Biol. 2012, 491, 769–773. [Google Scholar] [CrossRef]
- Sancho-Shimizu, V.; de Diego, R.P.; Lorenzo, L.; Halwani, R.; Alangari, A.; Israelsson, E.; Fabrega, S.; Cardon, A.; Maluenda, J.; Tatematsu, M.; et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J. Clin. Investig. 2011, 121, 4889–4902. [Google Scholar] [CrossRef]
- Herman, M.; Ciancanelli, M.; Ou, Y.-H.; Lorenzo, L.; Klaudel-Dreszler, M.; Pauwels, E.; Sancho-Shimizu, V.; de Diego, R.P.; Abhyankar, A.; Israelsson, E.; et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J. Exp. Med. 2012, 209, 1567–1582. [Google Scholar] [CrossRef] [PubMed]
- de Diego, R.P.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; Plancoulaine, S.; Picard, C.; Herman, M.; Cardon, A.; Durandy, A.; Bustamante, J.; et al. Human TRAF3 Adaptor Molecule Deficiency Leads to Impaired Toll-like Receptor 3 Response and Susceptibility to Herpes Simplex Encephalitis. Immunity 2010, 33, 400–411. [Google Scholar] [CrossRef]
- Andersen, L.L.; Mørk, N.; Reinert, L.; Kofod-Olsen, E.; Narita, R.; Jørgensen, S.E.; Skipper, K.; Höning, K.; Gad, H.H.; Østergaard, L.; et al. Functional IRF3 deficiency in a patient with herpes simplex encephalitis. J. Exp. Med. 2015, 212, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Sironi, M.; Peri, A.M.; Cagliani, R.; Forni, D.; Riva, S.; Biasin, M.; Clerici, M.; Gori, A. TLR3 Mutations in Adult Patients with Herpes Simplex Virus and Varicella-Zoster Virus Encephalitis. J. Infect. Dis. 2017, 215, 1430–1434. [Google Scholar] [CrossRef]
- Kindberg, E.; Vene, S.; Mickiene, A.; Lundkvist, Å.; Lindquist, L.; Svensson, L. A Functional Toll-Like Receptor 3 Gene (TLR3) May Be a Risk Factor for Tick-borne Encephalitis Virus (TBEV) Infection. J. Infect. Dis. 2011, 203, 523–528. [Google Scholar] [CrossRef]
- Gorbea, C.; Makar, K.A.; Pauschinger, M.; Pratt, G.; Bersola, J.L.F.; Varela, J.; David, R.M.; Banks, L.; Huang, C.-H.; Li, H.; et al. A Role for Toll-like Receptor 3 Variants in Host Susceptibility to Enteroviral Myocarditis and Dilated Cardiomyopathy. J. Biol. Chem. 2010, 285, 23208–23223. [Google Scholar] [CrossRef]
- Ranjith-Kumar, C.T.; Miller, W.; Sun, J.; Xiong, J.; Santos, J.; Yarbrough, I.; Lamb, R.J.; Mills, J.; Duffy, K.E.; Hoose, S.; et al. Effects of Single Nucleotide Polymorphisms on Toll-like Receptor 3 Activity and Expression in Cultured Cells. J. Biol. Chem. 2007, 282, 17696–17705. [Google Scholar] [CrossRef]
- Ishizaki, Y.; Takemoto, M.; Kira, R.; Kusuhara, K.; Torisu, H.; Sakai, Y.; Sanefuji, M.; Yukaya, N.; Hara, T. Association of toll-like receptor 3 gene polymorphism with subacute sclerosing panencephalitis. J. NeuroVirol. 2008, 14, 486–491. [Google Scholar] [CrossRef]
- Hidaka, F.; Matsuo, S.; Muta, T.; Takeshige, K.; Mizukami, T.; Nunoi, H. A missense mutation of the Toll-like receptor 3 gene in a patient with influenza-associated encephalopathy. Clin. Immunol. 2006, 119, 188–194. [Google Scholar] [CrossRef]
- Sato-Kaneko, F.; Yao, S.; Ahmadi, A.; Zhang, S.S.; Hosoya, T.; Kaneda, M.M.; Varner, J.A.; Pu, M.; Messer, K.S.; Guiducci, C.; et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight 2017, 2, 93397. [Google Scholar] [CrossRef]
- Kuai, R.; Sun, X.; Yuan, W.; Ochyl, L.J.; Xu, Y.; Najafabadi, A.H.; Scheetz, L.; Yu, M.; Balwani, I.; Schwendeman, A.; et al. Dual TLR agonist nanodiscs as a strong adjuvant system for vaccines and immunotherapy. J. Control. Release 2018, 282, 131–139. [Google Scholar] [CrossRef]
- Ziegler, A.; Soldner, C.; Lienenklaus, S.; Spanier, J.; Trittel, S.; Riese, P.; Kramps, T.; Weiss, S.; Heidenreich, R.; Jasny, E.; et al. A New RNA-Based Adjuvant Enhances Virus-Specific Vaccine Responses by Locally Triggering TLR- and RLH-Dependent Effects. J. Immunol. 2017, 198, 1595–1605. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Li, H.; Wang, H.; Zhang, T.; Hutchinson, M.; Yin, H.; Wang, X. Small-Molecule Modulators of Toll-like Receptors. Acc. Chem. Res. 2020, 53, 1046–1055. [Google Scholar] [CrossRef]
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M.; Rosensweig, C.; Faden, L.B.; Dewitz, M.C. Monoclonal antibody successes in the clinic. Nat. Biotechnol. 2005, 23, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- FDA. 2018. Available online: https://www.fda.gov/media/117240/download (accessed on 30 September 2021).
- Chutivongse, S.; Supich, C.; Wilde, H. Acceptability and Efficacy of Purified Vero-Cell Rabies Vaccine in Thai Children Exposed to Rabies. Asia-Pac. J. Public Health 1988, 2, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-E.; Pan, M.-J.; Tseng, H.-F.; Liau, M.-Y. The efficacy of mouse-brain inactivated Nakayama strain Japanese encephalitis vaccine—Results from 30 years experience in Taiwan. Vaccine 2006, 24, 2669–2673. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, S.; Strom, B.; Bilker, W.; Zhengle, L.; Chao-Min, W.; Hui-Lian, L.; Tai-Xiang, W.; Hong-Ji, Y.; Qi-Mau, L.; Tsai, T.; et al. Effectiveness of live-attenuated Japanese encephalitis vaccine (SA14-14-2): A case-control study. Lancet 1996, 347, 1583–1586. [Google Scholar] [CrossRef]
- Hu, J.; Wang, S.; Zhou, R.; Liu, H.; Gan, X.; Wei, M.; Zhu, F.; Meng, F.; Hou, W. Long-term immunity and the effect of one or two booster doses with a lyophilized human rabies vaccine (human diploid cells) at 10 years post primary vaccination in China. Hum. Vaccines Immunother. 2021, 17, 3162–3168. [Google Scholar] [CrossRef]
- CDC Rabies Vaccine: What You Need to Know. Available online: https://www.cdc.gov/vaccines/hcp/vis/vis-statements/rabies.pdf (accessed on 30 September 2021).
- Kubinski, M.; Beicht, J.; Gerlach, T.; Volz, A.; Sutter, G.; Rimmelzwaan, G.F. Tick-Borne Encephalitis Virus: A Quest for Better Vaccines against a Virus on the Rise. Vaccines 2020, 8, 451. [Google Scholar] [CrossRef] [PubMed]
- Hegde, N.R.; Gore, M.M. Japanese encephalitis vaccines: Immunogenicity, protective efficacy, effectiveness, and impact on the burden of disease. Hum. Vaccines Immunother. 2017, 13, 1320–1337. [Google Scholar] [CrossRef]
- Woods, C.W.; Sanchez, A.M.; Swamy, G.K.; McClain, M.T.; Harrington, L.; Freeman, D.; Poore, E.A.; Slifka, D.K.; DeRaad, D.; Amanna, I.J.; et al. An observer blinded, randomized, placebo-controlled, phase I dose escalation trial to evaluate the safety and immunogenicity of an inactivated West Nile virus Vaccine, HydroVax-001, in healthy adults. Vaccine 2019, 37, 4222–4230. [Google Scholar] [CrossRef]
- Van Hoeven, N.; Joshi, S.W.; Nana, G.I.; Bosco-Lauth, A.; Fox, C.; Bowen, R.A.; Clements, D.E.; Martyak, T.; Parks, D.E.; Baldwin, S.; et al. A Novel Synthetic TLR-4 Agonist Adjuvant Increases the Protective Response to a Clinical-Stage West Nile Virus Vaccine Antigen in Multiple Formulations. PLoS ONE 2016, 11, e0149610. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Matschke, J.; Lütgehetmann, M.; Hagel, C.; Sperhake, J.P.; Schröder, A.S.; Edler, C.; Mushumba, H.; Fitzek, A.; Allweiss, L.; Dandri, M.; et al. Neuropathology of patients with COVID-19 in Germany: A post-mortem case series. Lancet Neurol. 2020, 19, 919–929. [Google Scholar] [CrossRef]
- Moriguchi, T.; Harii, N.; Goto, J.; Harada, D.; Sugawara, H.; Takamino, J.; Ueno, M.; Sakata, H.; Kondo, K.; Myose, N.; et al. A first case of meningitis/encephalitis associated with SARS-Coronavirus-19. Int. J. Infect. Dis. 2020, 94, 55–58. [Google Scholar] [CrossRef]
- Ellul, M.A.; Benjamin, L.; Singh, B.; Lant, S.; Michael, B.D.; Easton, A.; Kneen, R.; Defres, S.; Sejvar, J.; Solomon, T. Neurological associations of COVID-19. Lancet Neurol. 2020, 19, 767–783. [Google Scholar] [CrossRef]
- Song, E.; Zhang, C.; Israelow, B.; Lu-Culligan, A.; Prado, A.V.; Skriabine, S.; Lu, P.; Weizman, O.-E.; Liu, F.; Dai, Y.; et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med. 2021, 218, 218. [Google Scholar] [CrossRef]
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Neely, G.; Yaghubian-Malhami, R.; Perkmann, T.; Van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.C.; Wang, H.; et al. Identification of Oxidative Stress and Toll-like Receptor 4 Signaling as a Key Pathway of Acute Lung Injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef]
- Shirey, K.A.; Lai, W.; Scott, A.; Lipsky, M.; Mistry, P.; Pletneva, L.M.; Karp, C.; McAlees, J.; Gioannini, T.L.; Weiss, J.; et al. The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nat. Cell Biol. 2013, 497, 498–502. [Google Scholar] [CrossRef]
- Aravalli, R.N.; Hu, S.; Rowen, T.N.; Palmquist, J.M.; Lokensgard, J.R. Cutting Edge: TLR2-Mediated Proinflammatory Cytokine and Chemokine Production by Microglial Cells in Response to Herpes Simplex Virus. J. Immunol. 2005, 175, 4189–4193. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Receptor | Ligand | Expression on CNS-Resident Cells | |||||
| Mouse | Human | ||||||
| Neurons | Astrocytes | Microglia | Neurons | Astrocytes | Microglia | ||
| TLR1 [41] | Bacterial lipo-proteins [41] | + IF (in vivo) [111] | + RT-PCR (in vitro, primary astrocytes), IF (in vivo) [111,112] | + IF (in vivo) [111] | + RT-PCR (in vitro, cell line) [113,114] | + (low) RT-PCR (in vitro, primary astrocytes) [115] | + RT-PCR (in vitro, primary microglia) [108,115] |
| TLR2 [35] | PGN, LTA [116,117], viral EPs, GPs, core proteins | + IF (in vivo), IF (in vitro, primary cortical neurons) [118,119] | + RT-PCR, FC, WB (in vitro, primary astrocytes), FC (ex vivo) [112,120,121] | + RT-PCR (in vitro, cell line), FC (ex vivo) [121,122] | + RT-PCR (in vitro, cell lines) [113,114,122] | + (low) RT-PCR (in vitro, primary astrocytes) [108,115] | + RT-PCR, IF (in vitro, primary microglia) [108,115] |
| TLR3 [34] | dsRNA, poly(I:C) [34] | + FC (in vitro, primary neurons) [70] | + RT-PCR, FC (in vitro, primary astrocytes) [70,112] | + RT-PCR (in vitro, cell line), FC (in vitro, primary microglia) [70,122] | + RT-PCR (in vitro, primary neurons and cell lines), WB (in vitro, hiPSC neurons) [113,114,122,123] | + RT-PCR, FC (in vitro, primary astrocytes) [108,115] | + (low) RT-PCR, FC (in vitro, primary microglia) [108,115] |
| TLR4 [33,124] | LPS [33,124], GPs, EPs, fusion proteins [47,48] | + RT-PCR (in vitro, primary neurons) [125] | + RT-PCR, FC (in vitro, primary astrocytes) [112,120,125] | + RT-PCR, WB (in vitro, cell line), RT-PCR (in vitro, primary microglia) [122,125] | + RT-PCR, WB (in vitro, cell lines and primary microglia) [113,114,122] | + (low) RT-PCR (in vitro, primary astrocytes) [115] | + RT-PCR, IF (in vitro, primary microglia) [108,115] |
| TLR5 [37] | Flagellin, profilin [37] | − IF (in vitro, primary neurons) [126] + microarray (in vitro, primary cortical neurons) [118] | + RT-PCR, IF (in vitro, primary astrocytes) [112,120,126] | + IF (in vitro, primary microglia) [126] | + RT-PCR (in vitro, primary neurons) [114] | + (low) RT-PCR (in vitro, primary astrocytes) [115] | + (low) RT-PCR (in vitro, primary microglia) [108,115] |
| TLR6 [40] | Diacyl lipo-peptides [40] | + IF (in vitro, primary DRG neurons) [127] | + RT-PCR (in vitro, primary astrocytes), FC (ex vivo) [112,121] | − FC (ex vivo) [121] | + RT-PCR (in vitro, primary neurons and cell lines ) [114] | + (low) RT-PCR (in vitro, primary astrocytes) [115] | + (low) RT-PCR (in vitro, primary microglia) [108,115] |
| TLR7 [36,38] | ssRNA [36,38] | + WB, RT-PCR (in vitro, primary DRG and CNS neurons) [128,129] | − RT-PCR (in vitro, primary astrocytes) [112] | + RT-PCR (in vitro, cell line) [122] | + (low) RT-PCR (in vitro, cell lines and primary neurons) [114,122] | + (low) RT-PCR (in vitro, primary astrocytes) [115] | + RT-PCR (in vitro, primary microglia) [108,115] |
| TLR8 [38] | ssRNA [38] | + WB, RT-PCR (in vitro, primary DRG and CNS neurons) [128,129] | − RT-PCR (in vitro, primary astrocytes) [112] | + RT-PCR (in vitro, cell line) [122] | + (low) RT-PCR (in vitro, cell lines and primary neurons) [114,122] | − RT-PCR (in vitro, primary astrocytes) [115] | + (low) RT-PCR (in vitro, primary microglia) [108,115] |
| TLR9 [39] | Unmethylated CpG DNA [39] | + IF, WB, RT-PCR (in vitro, primary neurons) [130] | + (low) RT-PCR (in vitro, primary astrocytes) Very low on basal level [120], present upon inflammatory stimulation [112,120] | + WB (ex vivo) [131] | + RT-PCR (in vitro, primary cells, cell line) [114] − RT-PCR (in vitro, cell line) [113] | + (low) RT-PCR (in vitro, primary astrocytes) [115] | + (low) RT-PCR (in vitro, primary microglia) [115] |
| TLR10 [132] | Triacyl lipopeptides, flagellin [133], EPs, RNA [134] | - | - | - | + RT-PCR (in vitro, primary neurons) [114] − RT-PCR (in vitro, cell line) [113] | + (low) RT-PCR (in vitro, primary astrocytes) [115] | − RT-PCR (in vitro, primary microglia) [115] + RNAseq (ex vivo) [135] |
| TLR11 [43] | Profilin [136] | + In situ IF (in vivo) [137] | − In situ IF (in vivo) [137] | − In situ IF (in vivo) [137] | - | - | - |
| TLR12 [138] | Profilin [138] | + In situ IF (in vivo) [137] | − In situ IF (in vivo) [137] | − In situ IF (in vivo) [137] | - | - | - |
| TLR13 [44] | Unmethylated bacterial RNA [44] | + In situ IF (in vivo) [137] | + In situ IF (in vivo) [137] | + (low) In situ IF (in vivo) [137] | - | - | - |
| Adaptor | Connected TLR | Expression on CNS-Resident Cells | |||||
| Mouse | Human | ||||||
| Neurons | Astrocytes | Microglia | Neurons | Astrocytes | Microglia | ||
| MyD88 [139] | TLR1, TLR2, TLR4-9, TLR11-13 | + IF (in vitro, primary neurons) [118] | + WB (in vitro) IF (in vivo, spinal cord astrocytes) [140] | + RT-PCR (in vitro, cell line), WB (in vitro, primary microglia and cell line) [122,141,142] | + RT-PCR, WB (in vitro, cell line) [122,143] | + RT-PCR (in vivo) [144] | + RNAseq (ex vivo) [135] |
| TRIF [145] | TLR3, TLR4 | + IF, WB (in vivo, in vitro, primary hippocampal neurons) [146] | + (low) IF (in vivo) [146] | + (low) IF(in vivo), WB (in vitro, cell line) [142,146] | + IR (in vivo) [146] | + IR (in vivo) [146] | + IR (in vivo) [146] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gern, O.L.; Mulenge, F.; Pavlou, A.; Ghita, L.; Steffen, I.; Stangel, M.; Kalinke, U. Toll-like Receptors in Viral Encephalitis. Viruses 2021, 13, 2065. https://doi.org/10.3390/v13102065
Gern OL, Mulenge F, Pavlou A, Ghita L, Steffen I, Stangel M, Kalinke U. Toll-like Receptors in Viral Encephalitis. Viruses. 2021; 13(10):2065. https://doi.org/10.3390/v13102065
Chicago/Turabian StyleGern, Olivia Luise, Felix Mulenge, Andreas Pavlou, Luca Ghita, Imke Steffen, Martin Stangel, and Ulrich Kalinke. 2021. "Toll-like Receptors in Viral Encephalitis" Viruses 13, no. 10: 2065. https://doi.org/10.3390/v13102065
APA StyleGern, O. L., Mulenge, F., Pavlou, A., Ghita, L., Steffen, I., Stangel, M., & Kalinke, U. (2021). Toll-like Receptors in Viral Encephalitis. Viruses, 13(10), 2065. https://doi.org/10.3390/v13102065