
HCMV-Mediated Interference of Bortezomib-Induced Apoptosis in Colon Carcinoma Cell Line Caco-2


Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Proteasome Inhibitors
2.3. Immunofluorescence
2.4. Quantitative Analysis of Infected Cells by Flow Cytometry
2.5. Cell Proliferation Assay
2.6. Plaque Reduction Assay
2.7. Yield Assay
2.8. Real-Time Quantitative PCR
2.9. PAGE and Western Blot Analysis
2.10. Co-Culturing of Caco-2 and HELF Fi301
2.11. Apoptosis Assay
2.12. Mitochondrial Membrane Potential Assay
2.13. Cell-Death Detection
2.14. Necrosis Assay with PicoGreen
2.15. Cell Viability
2.16. Statistical Analysis
3. Results
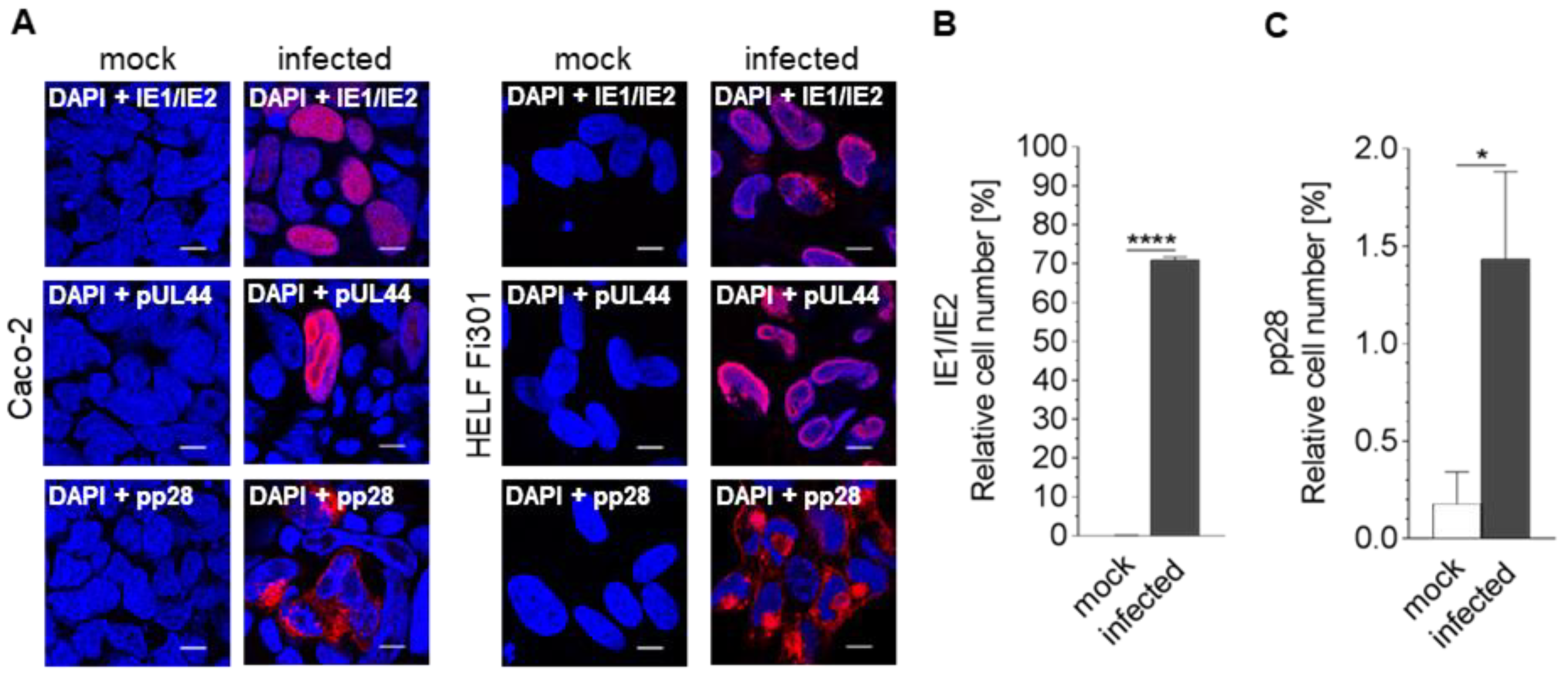
3.1. Susceptibility of Caco-2 Cells to HCMV
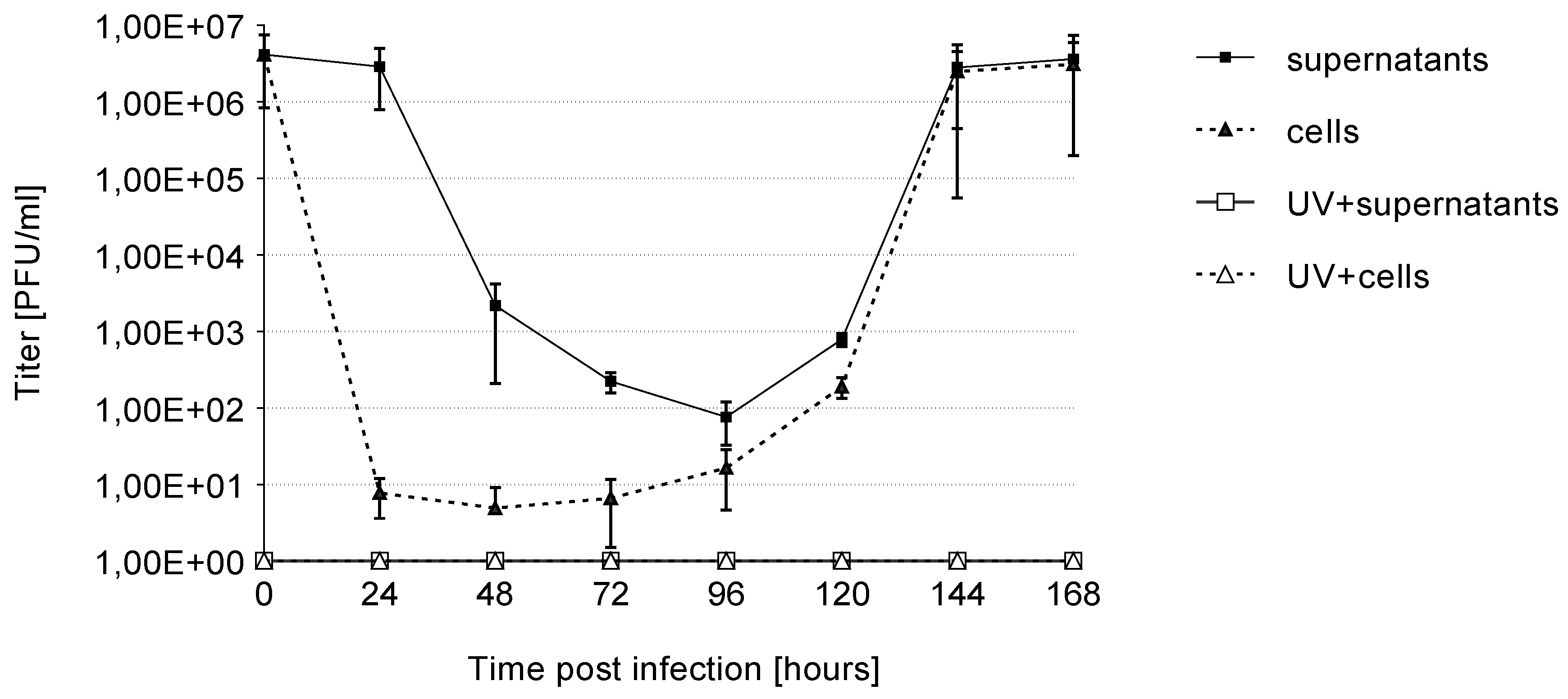
3.2. Growth Kinetics
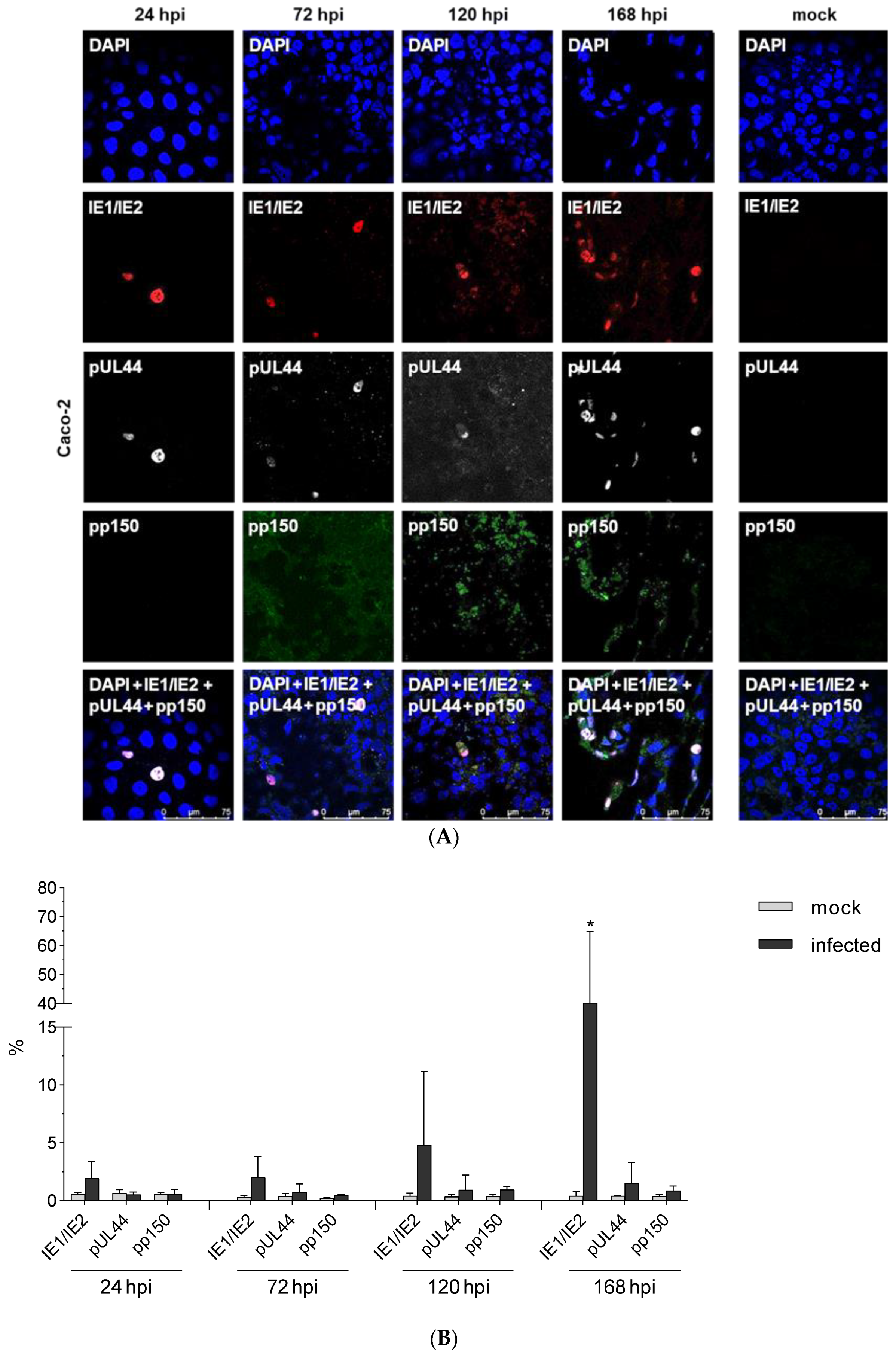
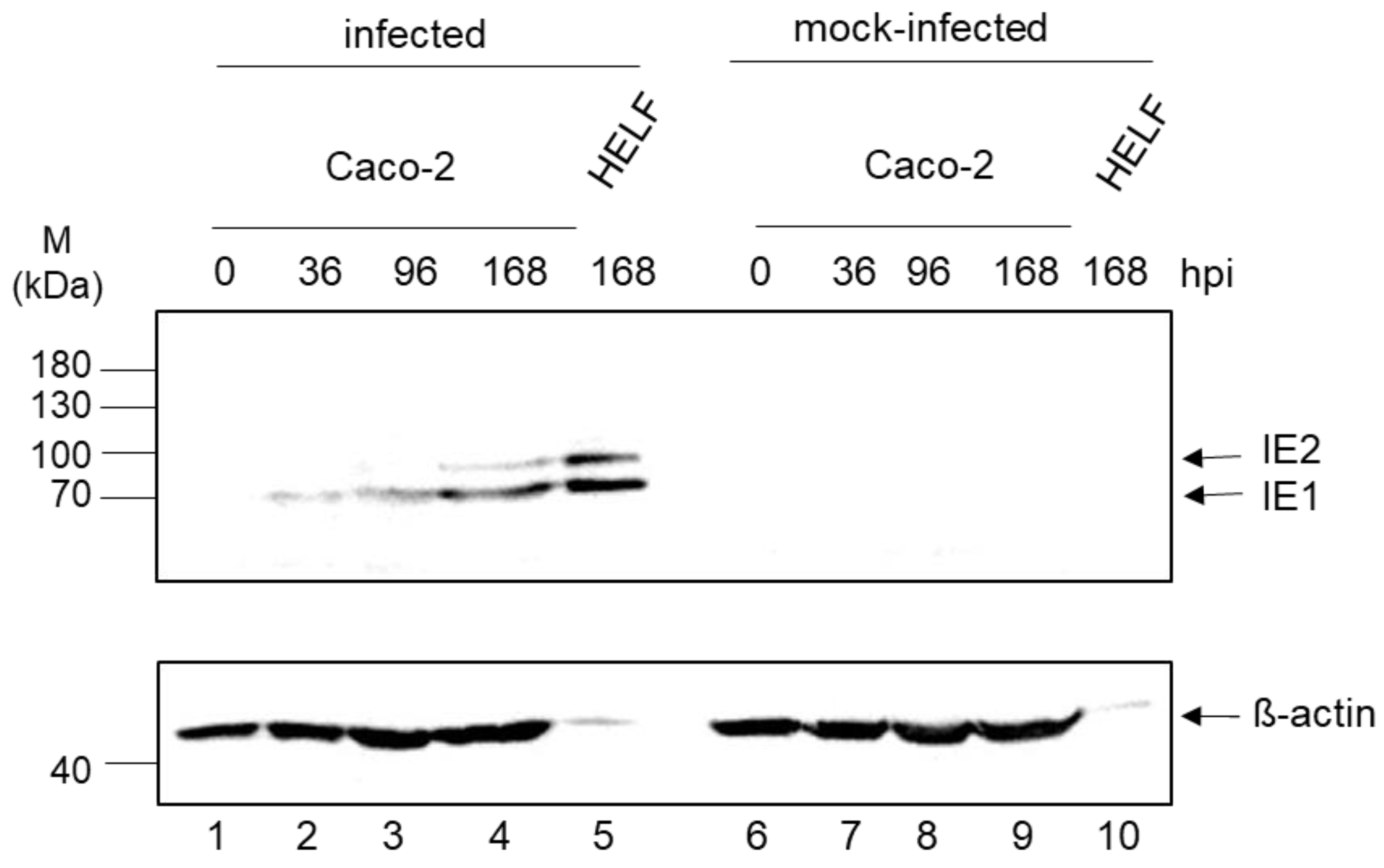
3.3. Expression Kinetic of IE1/2
3.4. Genome Quantification
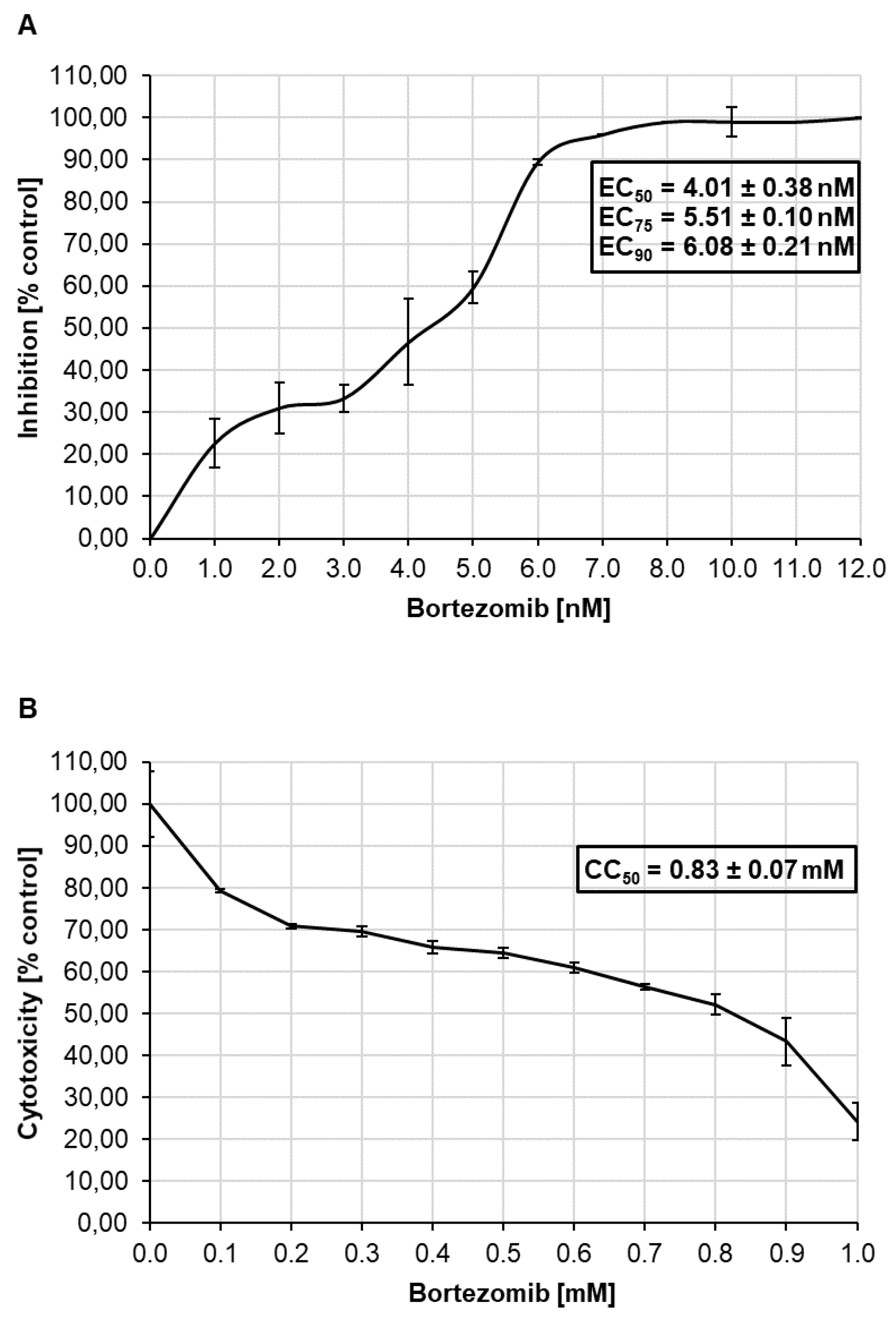
3.5. Antiviral Effect of Bortezomib on HCMV Infected Caco-2 Cells and Cytotoxicity
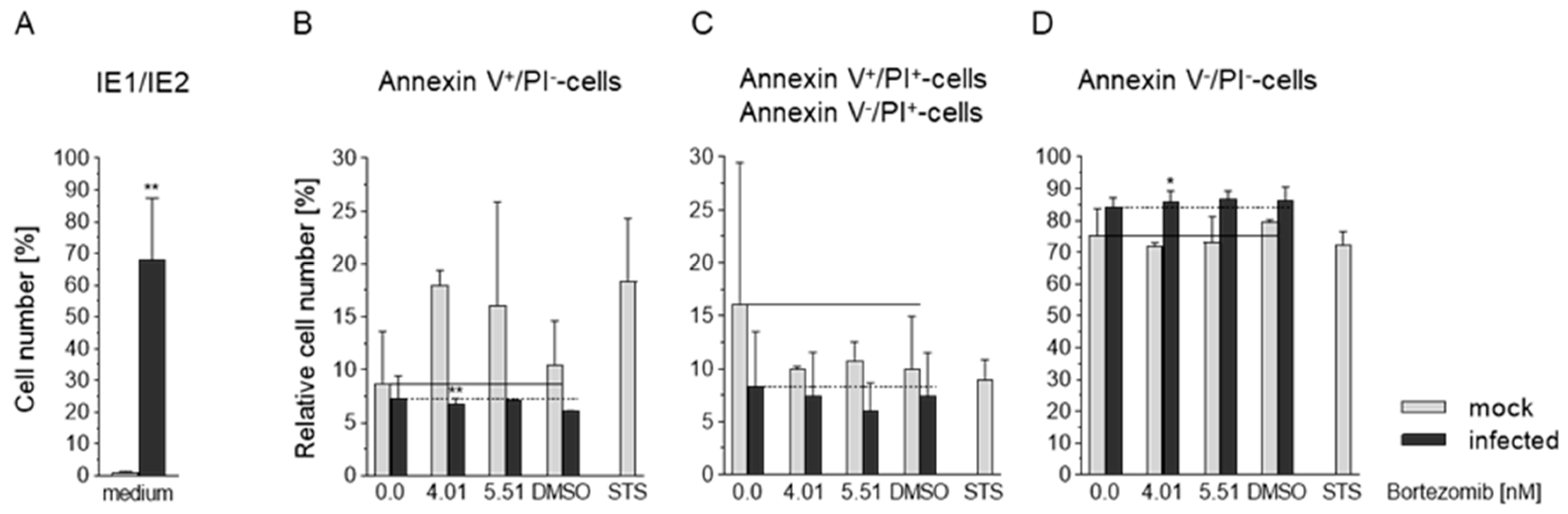
3.6. Effects of HCMV and Bortezomib on Apoptosis in Caco-2 Cells
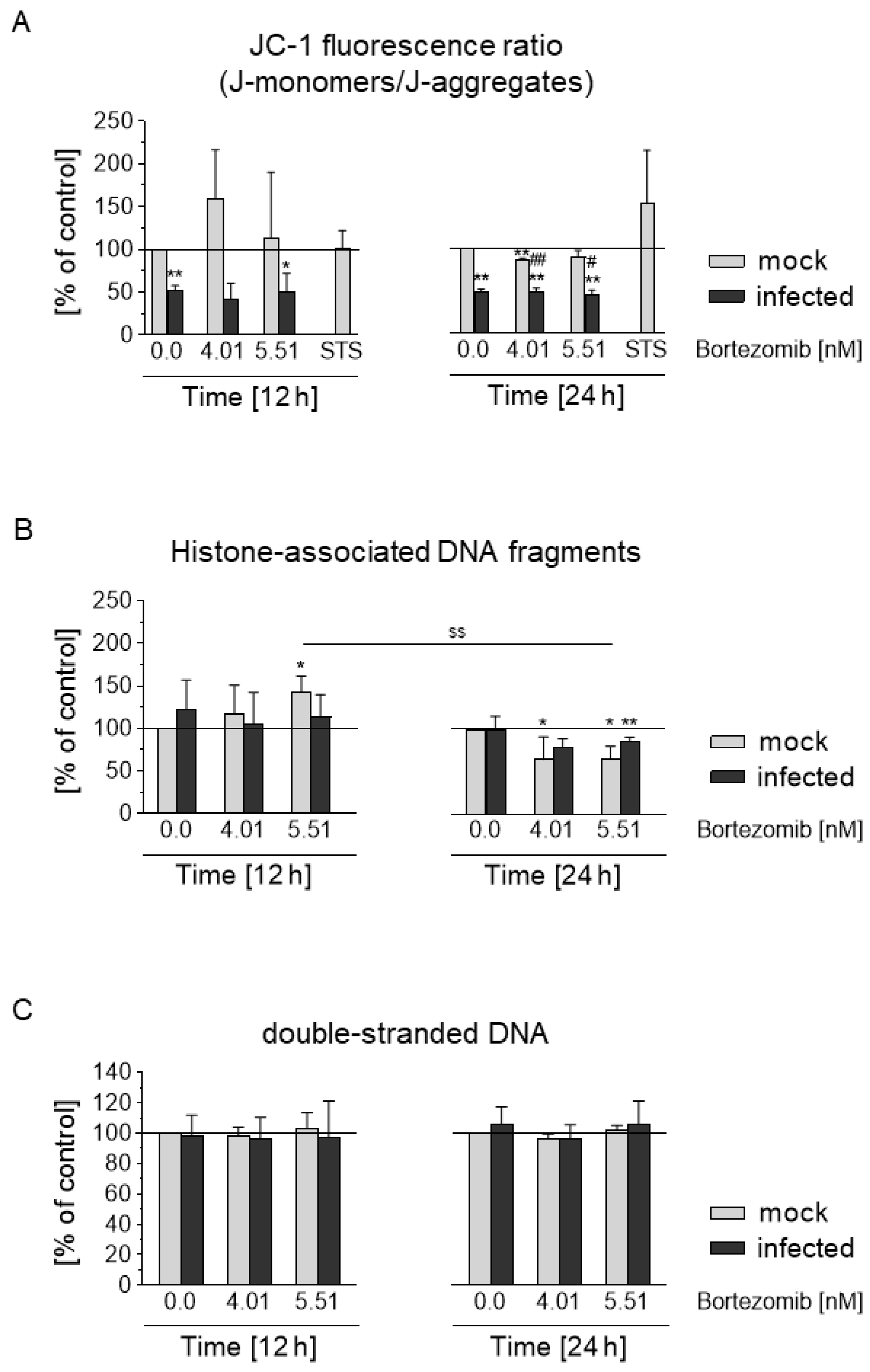
3.7. HCMV Prevents Depolarization of the Mitochondrial Membrane and Blocks the Apoptotic Effect of Bortezomib
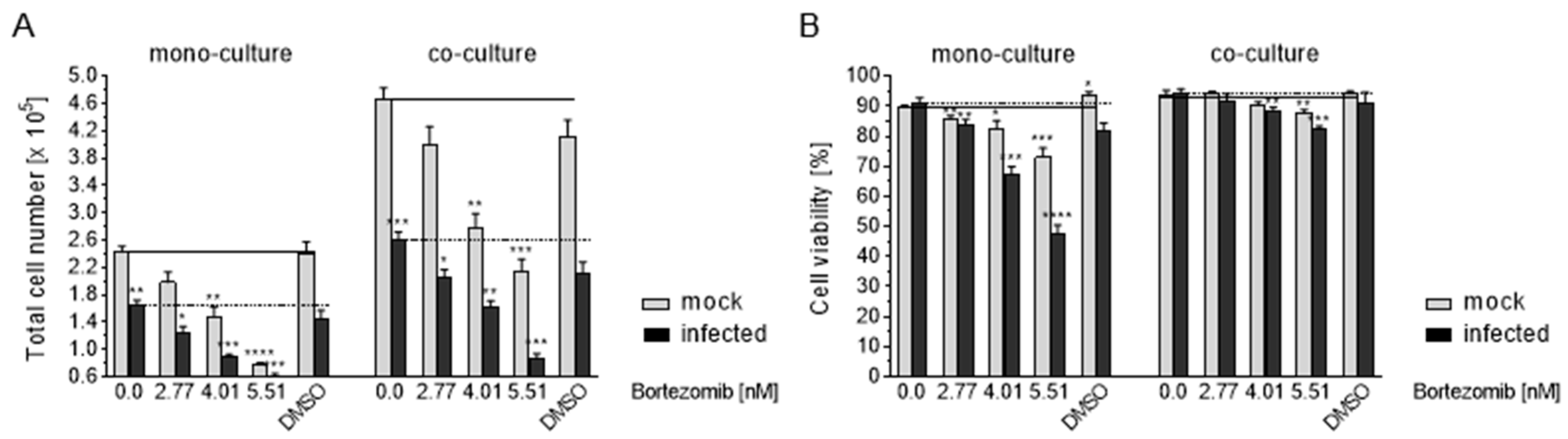
3.8. HCMV Infection of Caco-2 by Co-Culture with Infected HELF Fi301
3.9. Effects of Co-Culture on Apoptosis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Britt, W.J.; Alford, C.A. Cytomegalovirus. In Fields Virology, 3rd ed.; Fields, B.N., Knipe, D.M., Howley, P.M., Eds.; Raven Press: New York, NY, USA, 1996; pp. 2493–2523. [Google Scholar]
- Mocarski, E.S.; Shenk, T.; Griffiths, P.D.; Pass, R.F. Cytomegalovirus. In Fields Virology, 6th ed.; Knipe, D.M., Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 1960–2014. [Google Scholar]
- Adams, M.J.; Carstens, E.B. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses. Arch. Virol. 2012, 157, 1411–1422. [Google Scholar] [CrossRef] [PubMed]
- Cobbs, C.S.; Harkins, L.; Samanta, M.; Gillespie, G.Y.; Bharara, S.; King, P.H.; Nabors, L.B.; Cobbs, C.G.; Britt, W.J. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002, 62, 3347–3350. [Google Scholar] [PubMed]
- Harkins, L.; Volk, A.L.; Samanta, I.; Mikolaenko, I.; Britt, W.J.; Bland, I.; Cobbs, S. Specific localisation of human cytomegalovirus nucleic acids and proteins in human colorectal cancer. Lancet 2002, 360, 1557–1563. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Xie, W.; Schmittling, R.; Learn, C.; Friedman, A.; McLendon, R.E.; Sampson, J.H. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro. Oncol. 2008, 10, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Samanta, M.; Harkins, L.; Klem, K.; Britt, W.J.; Cobbs, C.S. High prevalence of human cytomegalovirus in prostatic intraepithelial neoplasia and prostatic carcinoma. J. Urol. 2003, 170, 998–1002. [Google Scholar] [CrossRef]
- Taher, C.; Taher, C.; Frisk, G.; Fuentes, S.; Religa, P.; Costa, H.; Assinger, A.; Vetvik, K.K.; Bukholm, I.R.; Yaiw, K.C.; et al. High prevalence of human cytomegalovirus in brain metastases of patients with primary breast and colorectal cancers. Transl. Oncol. 2014, 7, 732–740. [Google Scholar] [CrossRef]
- Chen, H.P.; Jiang, J.K.; Chen, C.Y.; Chou, T.Y.; Chen, Y.T.; Lin, S.F. Human cytomegalovirus preferentially infects the neoplastic epithelium of colorectal cancer: A quantitative and histological analysis. J. Clin. Virol. 2012, 54, 240–244. [Google Scholar] [CrossRef]
- Faivre, J.; Bouvier, A.; Bonithon-Kopp, C. Epidemiology and screening of colorectral cancer. Best Pract. Res. Clin. Gastroenterol. 2002, 16, 187–199. [Google Scholar] [CrossRef]
- Chen, H.P.; Jiang, J.K.; Lai, P.Y.; Chen, C.Y.; Chou, T.Y.; Chen, Y.C.; Chang, Y.T.; Lin, S.F.; Chan, C.H.; Yang, C.Y.; et al. Tumoral presence of human cytomegalovirus is associated with shorter disease-free survival in elderly patients with colorectal cancer and higher levels of intratumoral interleukin-17. Clin. Microbiol. Infect. 2014, 20, 664–671. [Google Scholar] [CrossRef]
- Chen, H.P.; Jiang, J.K.; Chen, C.Y.; Yang, C.Y.; Chen, Y.C.; Lin, C.H.; Chou, T.Y.; Cho, W.L.; Chan, Y.J. Identification of human cytomegalovirus in tumour tissues of colorectal cancer and its association with the outcome of non-elderly patients. J. Gen. Virol. 2016, 97, 2411–2420. [Google Scholar] [CrossRef][Green Version]
- Chen, H.P.; Jiang, J.K.; Lai, P.Y.; Teo, W.H.; Yang, C.Y.; Chou, T.Y.; Lin, C.H.; Chan, Y.J. Serological and viraemic status of human cytomegalovirus infection in patients with colorectal cancer is not correlated with viral replication and transcription in tumours. J. Gen. Virol. 2016, 91, 152–159. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cinatl, J., Jr.; Vogel, J.U.; Kotchetkov, R.; Doerr, H. Oncomodulatory signals by regulatory proteins encoded by human cytomegalovirus: A novel role for viral infection in tumor progression. FEMS Microbiol. Rev. 2004, 28, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Salvant, B.S.; Fortunato, E.A.; Spector, D.H. Cell cycle dysregulation by human cytomegalovirus: Influence of the cell cycle phase at the time of infection and effects on cyclin transcription. J. Virol. 1998, 72, 3729–3741. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Kotchetkov, R.; Vogel, J.-U.; Doerr, H.W.; Cinatl, J., Jr. Cytomegalovirus infection blocks apoptosis in cancer cells. Cell. Mol. Life Sci. 2004, 61, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Karki, K.; Haradhchandra, S.; Sage, S. Bortezomib targets Sp transcription factors in cancer cells. Mol. Pharmacol. 2018, 94, 1187–1196. [Google Scholar] [CrossRef]
- Hong, S.W.; Kim, S.M.; Jin, D.H.; Shin, J.S.; Yoon, D.H.; Kim, K.P.; Lee, J.L.; Heo, D.S.; Lee, J.S.; Kim, T.W. Bortezomib induces G2-M arrest in human colon cancer cells through ROS-inducible phosphorylation of ATM-CHK1. Int. J. Oncol. 2012, 41, 76–83. [Google Scholar] [CrossRef]
- Zimmermann, C.; Büscher, N.; Krauter, S.; Krämer, N.; Sehn, E.; Tenzer, S.; Plachter, B. The abundant tegument protein pUL25 of human cytomegalovirus prevents proteasomal degradation of pUL26 and supports its suppression of ISGylation. J. Virol. 2018, 92, e01180-18. [Google Scholar] [CrossRef]
- Biolatti, M.; Dell’Oste, V.; Pautasso, S.; Gugliesi, F.; von Einem, J.; Krapp, C.; Jakobsen, M.-R.; Borgogna, C.; Gargilio, M.; De Andrea, M.; et al. Human cytomegalovirus tegument protein pp65 (pUL83) dampens type I interferon production by inactivating the DNA sensor cGAS without affecting STING. J. Virol. 2018, 92, e01774-17. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, Y.E.; Stinski, M.F.; Ahn, J.H.; Song, Y.J. Human cytomegalovirus IE2 86 kDa protein induces STING degradation and inhibits cGAMP-mediated IFN-β induction. Front. Microbiol. 2017, 26, 1854. [Google Scholar] [CrossRef]
- Lee, S.H.; Kalejta, R.F.; Kerry, J.; Semmes, O.J.; O’Connor, C.M.; Khan, Z.; Garcia, B.A.; Shenk, T.; Murphy, E. BclAF1 restriction factor is neutralized by proteasomal degradation and microRNA repression during human cytomegalovirus infection. Proc. Natl. Acad. Sci. USA 2012, 109, 9575–9580. [Google Scholar] [CrossRef]
- Fennell, D.A.; Chacko, A.; Mutti, L. BCL-2 family regulation by the 20S proteasome inhibitor bortezomib. Oncogene 2008, 27, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Bartle, L.M.; Skaletskaya, A.; Poncet, D.; Zamzami, N.; Park, P.U.; Sharpe, J.; Youle, R.J.; Goldmacher, V.S. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc. Natl. Acad. Sci. USA 2004, 101, 7988–7993. [Google Scholar] [CrossRef] [PubMed]
- Brune, W.; Andoniou, C.E. Die another day: Inhibition of cell death pathways by cytomegalovirus. Viruses 2017, 9, 249. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, M.Z.; Casalegno-Garduno, R.; Sitnik, K.M.; Kasmapour, B.; Pulm, A.K.; Brizic, I.; Eiz-Vesper, B.; Moosmann, A.; Jonjic, S.; Mocarski, E.S.; et al. Cytomegalovirus inhibition of extrinsic apoptosis determines fitness and resistance to cytotoxic CD8 T cells. Proc. Natl. Acad. Sci. USA 2020, 22, 201914667. [Google Scholar] [CrossRef] [PubMed]
- Terhune, S.; Torigoi, E.; Moorman, N.; Silva, M.; Qian, Z.; Shenk, T.; Yu, D. Human cytomegalovirus UL38 protein blocks apoptosis. J. Virol. 2007, 81, 3109–3123. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. Pathogenesis of colorectal carcinoma and therapeutic implications: The roles of the ubiquitin-proteasome system and Cox-2. J. Cell. Mol. Med. 2007, 11, 252–285. [Google Scholar] [CrossRef]
- Boccadoro, M.; Morgan, G.; Cavenagh, J. Preclinical evaluation of the proteasome inhibitor bortezomib in cancer therapy. Cancer Cell Int. 2005, 5, 18. [Google Scholar] [CrossRef]
- Levin, A.; Minis, A.; Lalazar, G.; Rodriguez, J.; Steller, H. PSMD5 inactivation promotes 26S proteasome assembly during colorectal tumor progression. Cancer Res. 2018, 78, 3458–3468. [Google Scholar] [CrossRef]
- Bonvini, P.; Zorzi, E.; Basso, G.; Rosolen, A. Bortezomib-mediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in CD-30+ anaplastic large cell lymphomas. Leukemia 2007, 21, 838–842. [Google Scholar] [CrossRef]
- Roy, S.S.; Kirma, N.B.; Santhamma, B.; Tekmal, R.R.; Agyin, J.K. Effects of a novel proteasome inhibitor BU-32 on multiple myeloma cells. Cancer Chemother. Pharmacol. 2014, 73, 1263–1271. [Google Scholar] [CrossRef]
- Adams, J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell 2004, 5, 417–421. [Google Scholar] [CrossRef]
- Sampaio, K.L.; Cavignac, Y.; Stierhof, Y.D.; Sinzger, C. Human cytomegalovirus labeled with green fluorescent protein for live analysis of intracellular Particle Movements. J. Virol. 2005, 79, 2754–2767. [Google Scholar] [CrossRef] [PubMed]
- Smuda, C.; Bogner, E.; Radsak, K. The human cytomegalovirus glycoprotein B gene (ORF UL55) is expressed early in the infectious cycle. J. Gen. Virol. 1997, 78, 1981–1992. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bhat, M.; Toledo-Velasquez, D.; Wang, L.; Malanga, C.J.; Ma, J.K.; Rojanasakul, Y. Regulation of tight junction permeability by calcium mediators and cell cytoskeleton in rabbit tracheal epithelium. Pharm. Res. 1993, 10, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Deli, M.A. Potential use of tight junction modulators to reversibly open membranous barriers and improve drug delivery. Biochim. Biohys. Acta 2008, 1788, 892–910. [Google Scholar] [CrossRef]
- Turk, S.R.; Shipman, C., Jr.; Nassiri, R.; Genzlinger, G.; Krawczyk, S.H.; Townsend, L.B.; Drach, J.C. Pyrrolo[2,3-d]pyrimidine nucleosides as inhibitors of human cytomegalovirus. Antimicrob. Agents Chemother. 1987, 31, 544–550. [Google Scholar] [CrossRef]
- Hofmann, H.; Sindre, H.; Stamminger, T. Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx. J. Virol. 2002, 76, 5769–5783. [Google Scholar] [CrossRef]
- Jarvis, M.A.; Wang, C.; Meyers, H.L.; Smith, P.P.; Corless, C.L.; Henderson, G.J.; Vieira, J.; Britt, W.J.; Nelson, J.A. Human cytomegalovirus infection of Caco-2 cells occurs at the basolateral membrane and is differentiation state dependent. J. Virol. 1999, 73, 4552–4560. [Google Scholar] [CrossRef]
- Esclatine, A.; Lemullois, M.; Servin, A.L.; Quero, A.-M.; Geniteau-Legendre, M. Human cytomegalovirus infects Caco-2 intestinal epithelial cells basolaterally regardless of the differentiation state. J. Virol. 2000, 74, 513–517. [Google Scholar] [CrossRef]
- Ryckman, B.J.; Jarvis, M.A.; Drummond, D.D.; Nelson, J.A.; Johnson, D.C. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 2006, 80, 710–722. [Google Scholar] [CrossRef]
- Liu, X.; Li, Y.; Peng, S.; Yu, X.; Li, W.; Shi, F.; Luo, X.; Tang, M.; Tan, Z.; Bode, A.M.; et al. Epstein-Barr virus encoded latent membrane protein 1 suppresses necroptosis through targeting RIPK1/3 ubiquitination. Cell Death Dis. 2018, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Endlich, F.; Bermejo, G.A.; Norris, K.L.; Youle, R.J.; Tjandra, N. Structural mechanism of Bax inhibition by cytomegalovirus protein vMIA. Proc. Natl. Acad. Sci. USA 2012, 109, 20901–20906. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.T.; Chau, K.-Y.; Schapira, H.V. The cytomegalovirus protein pUL37x1 targets mitochondria to mediate neuroprotection. Sci. Rep. 2016, 6, 31373. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Omoto, S.; Harris, P.A.; Finger, J.N.; Bertin, J.; Gough, P.J.; Kaiser, W.J.; Mocarski, E.S. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 2015, 17, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Omoto, S.; Guo, H.; Talekar, G.R.; Roback, L.; Kaiser, W.J.; Mocarski, E.S. Suppression of RIP3-dependent necroptosis by human cytomegalovirus. J. Biol. Chem. 2015, 290, 11635–11648. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, F.; Mengarelli, A.; Giannotti, F.; Tendas, A.; Anacierico, B.; Porrini, R.; Picardi, A.; Cerchiara, E.; Dentamaro, T.; Chierichini, A.; et al. High incidence of post-transplant cytomegalovirus reactivations in myeloma patients undergoing autologous stem cell transplantation after treatment with bortezomib-based regimens: A survey from the Rome transplant network. Transpl. Infect. Dis. 2014, 16, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Kaya, A.H.; Tekgunduz, E.; Akpinar, S.; Batgi, H.; Bekdemir, F.; Kayikci, O.; Namdarogl, S.; Ulu, B.U.; Dal, M.S.; Cakar, M.K.; et al. Is cytomegalovirus surveillance necessary for patients with low reactivation risk in an autologous hematopoietic cell transplantation setting? Transpl. Proc. 2017, 49, 1911–1915. [Google Scholar] [CrossRef]
- Sharpley, F.A.; De-Silva, D.; Mahmood, S.; Sachchithanantham, S.; Ramsay, I.; Mingo, A.G.; Worthington, S.; Hughes, D.; Mehta, A.; Kyriakou, C.; et al. Cytomegalovirus reactivation after bortezomib treatment for multiple myeloma and light chain amyloidosis. Eur. J. Haematol. 2020, 104, 230–235. [Google Scholar] [CrossRef]
- Li, J.; Li, Y.; Huang, B.; Zheng, D.; Chen, M.; Zhou, Z. Drug-induced modulation of T lymphocytes as a potential mechanism of susceptibility to infections in patients with multiple myeloma during bortezomib therapy. Cell Biochem. Biophys. 2015, 71, 457–464. [Google Scholar] [CrossRef]
- Neuzillet, C.; Tijeras-Rabbaland, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Cavaco, A.C.M.; Rezaei, M.; Caliandro, M.F.; Lima, A.M.; Stehling, M.; Dhayat, A.; Haier, J.; Brakebusch, C.; Eble, J.A. The interaction between laminin-332 and alpha3beta1 integrin determines differentiation and maintenance of CAFs, and supports invasion of pancreatic duct adenocarcinoma cells. Cancers 2018, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Madar, S.; Goldstein, I.; Rotter, V. ‘Cancer associated fibroblasts’—More than meets the eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Template | Time Post Infection (h) | ||
|---|---|---|---|
| 36 | 96 | 168 | |
| Inf. Caco-2 | 1.724 × 103 | 3.273 × 103 | 4.305 × 103 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Härtel, H.; Theiß, J.; Abdelaziz, M.O.; Raftery, M.J.; Pecher, G.; Bogner, E. HCMV-Mediated Interference of Bortezomib-Induced Apoptosis in Colon Carcinoma Cell Line Caco-2. Viruses 2021, 13, 83. https://doi.org/10.3390/v13010083
Härtel H, Theiß J, Abdelaziz MO, Raftery MJ, Pecher G, Bogner E. HCMV-Mediated Interference of Bortezomib-Induced Apoptosis in Colon Carcinoma Cell Line Caco-2. Viruses. 2021; 13(1):83. https://doi.org/10.3390/v13010083
Chicago/Turabian StyleHärtel, Heike, Janine Theiß, Mohammed O. Abdelaziz, Martin J. Raftery, Gabriele Pecher, and Elke Bogner. 2021. "HCMV-Mediated Interference of Bortezomib-Induced Apoptosis in Colon Carcinoma Cell Line Caco-2" Viruses 13, no. 1: 83. https://doi.org/10.3390/v13010083
APA StyleHärtel, H., Theiß, J., Abdelaziz, M. O., Raftery, M. J., Pecher, G., & Bogner, E. (2021). HCMV-Mediated Interference of Bortezomib-Induced Apoptosis in Colon Carcinoma Cell Line Caco-2. Viruses, 13(1), 83. https://doi.org/10.3390/v13010083