
Library Preparation Based on Transposase Assisted RNA/DNA Hybrid Co-Tagmentation for Next-Generation Sequencing of Human Noroviruses


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. RNA Extraction and RT-qPCR Identification of Clinical Samples
2.2. Primer Sets Design
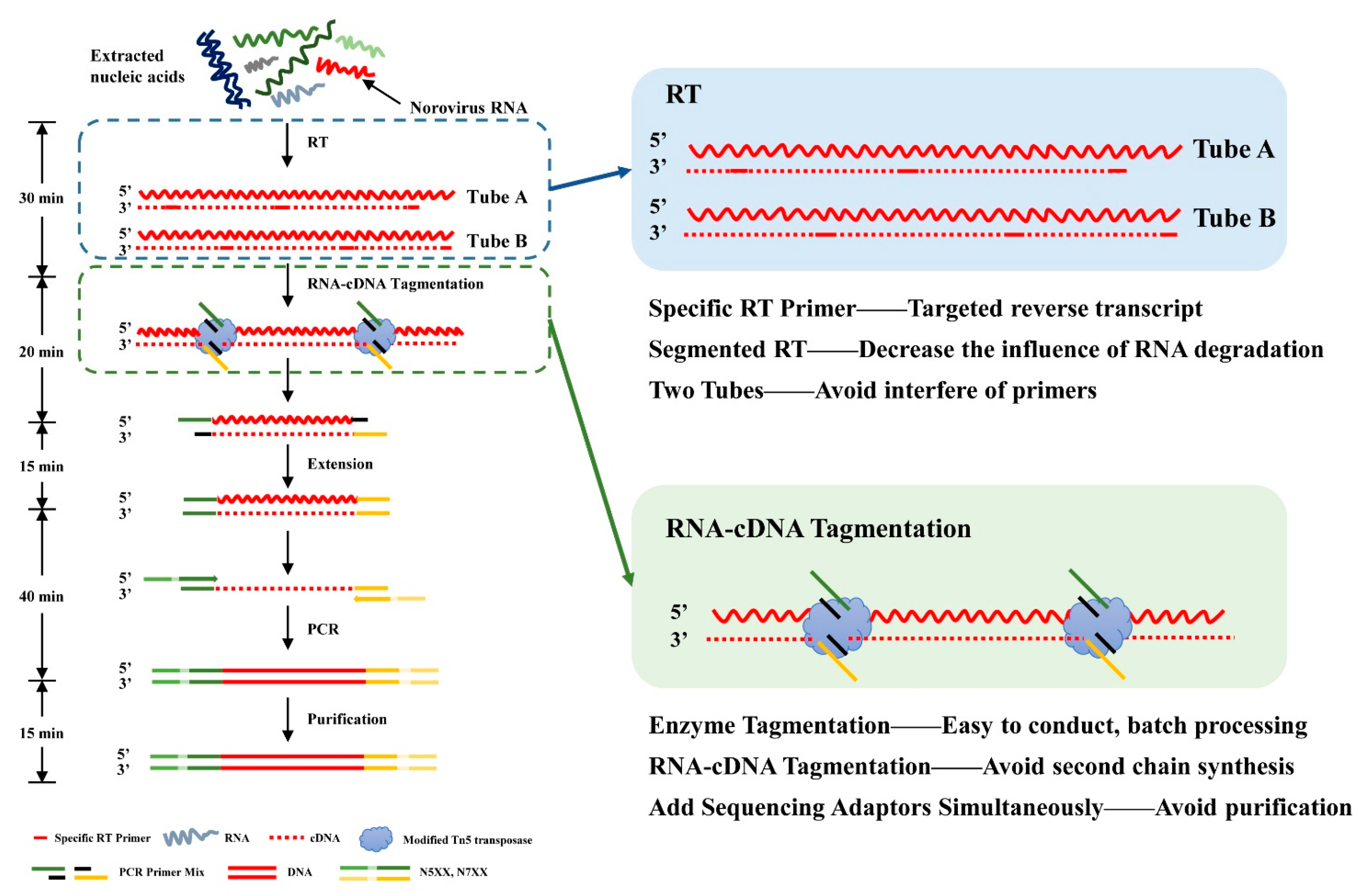
2.3. TRACE-Seq Library Preparation and Sequencing
2.4. Bioinformatics
3. Results
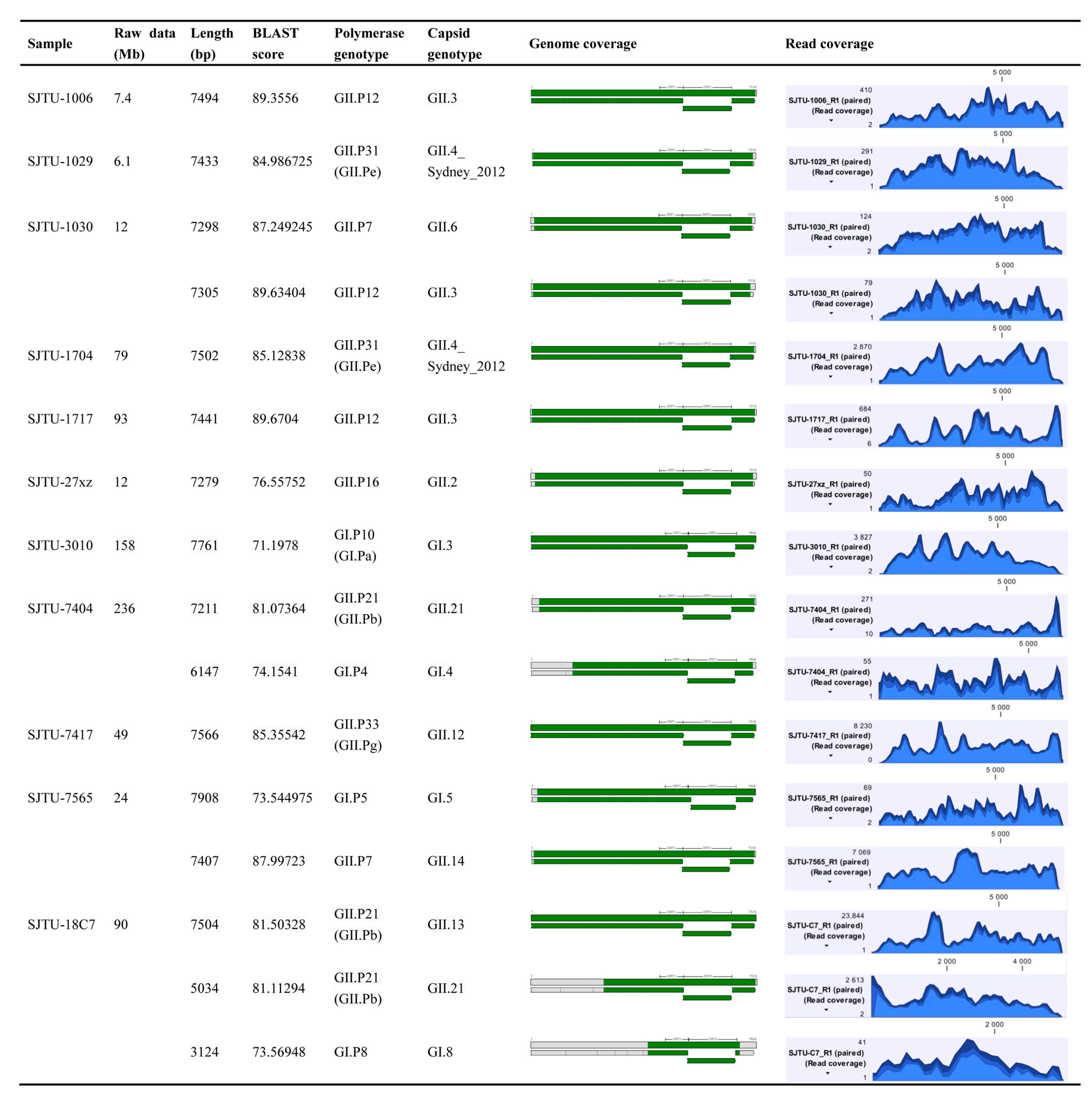
3.1. Sequencing Results
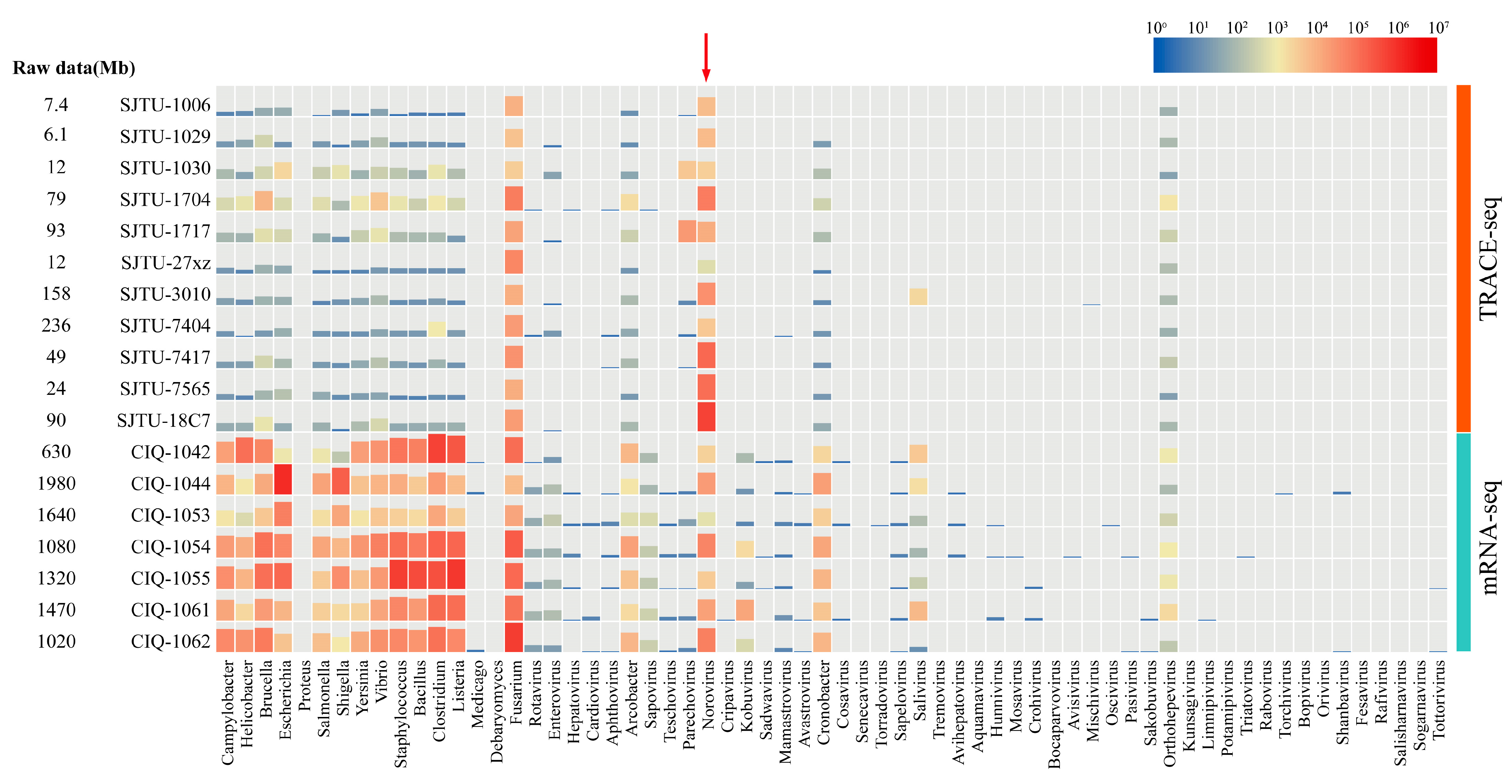
3.2. Effective Data Distribution
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cates, J.E.; Vinjé, J.; Parashar, U.; Hall, A.J. Recent advances in human norovirus research and implications for candidate vaccines. Expert Rev. Vaccines 2020, 19, 539–548. [Google Scholar] [CrossRef]
- Vinjé, J.; Estes, M.K.; Esteves, P.; Green, K.Y.; Katayama, K.; Knowles, N.J.; L’Homme, Y.; Martella, V.; Vennema, H.; White, P.A. ICTV Virus Taxonomy Profile: Caliciviridae. J. Gen. Virol. 2019, 100, 1469–1470. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, P.; de Graaf, M.; Parra, G.I.; Chan, M.C.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100, 1393–1406, Corrigendum in 2020, 101, 893. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Ando, T.; Fankhauser, R.L.; Beard, R.S.; Glass, R.I.; Monroe, S.S. Norovirus classification and proposed strain nomenclature. Virology 2006, 346, 312–323. [Google Scholar] [CrossRef]
- Estes, M.K.; Ettayebi, K.; Tenge, V.R.; Murakami, K.; Karandikar, U.; Lin, S.; Ayyar, B.V.; Cortes-Penfield, N.W.; Haga, K.; Neill, F.H.; et al. Human norovirus cultivation in nontransformed stem cell-derived human intestinal enteroid cultures: Success and challenges. Viruses 2019, 11, 638. [Google Scholar] [CrossRef] [PubMed]
- Gardy, J.L.; Loman, N.J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet. 2018, 19, 9–20. [Google Scholar] [CrossRef]
- Wilson, M.R.; Sample, H.A.; Zorn, K.C.; Arevalo, S.; Chiu, C.Y. Clinical metagenomic sequencing for diagnosis of meningitis and encephalitis. N. Engl. J. Med. 2019, 380, 2327–2340. [Google Scholar] [CrossRef]
- Cui, P.; Lin, Q.; Ding, F.; Xin, C.; Gong, W.; Zhang, L.; Geng, J.; Zhang, B.; Yu, X.; Yang, J.; et al. A comparison between ribo-minus RNA-sequencing and polyA-selected RNA-sequencing. Genomics 2010, 96, 259–265. [Google Scholar] [CrossRef]
- Gubler, U.; Hoffman, B.J. A simple and very efficient method for generating cDNA libraries. Gene 1983, 25, 263–269. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Endoh, D.; Mizutani, T.; Kirisawa, R.; Maki, Y.; Saito, H.; Kon, Y.; Morikawa, S.; Hayashi, M. Species-independent detection of RNA virus by representational difference analysis using non-ribosomal hexanucleotides for reverse transcription. Nucleic Acids Res. 2005, 33, e65. [Google Scholar] [CrossRef] [PubMed]
- Strubbia, S.; Schaeffer, J.; Besnard, A.; Wacrenier, C.; Le Mennec, C.; Garry, P.; Desdouits, M.; Le Guyader, F.S. Metagenomic to evaluate norovirus genomic diversity in oysters: Impact on hexamer selection and targeted capture-based enrichment. Int. J. Food Microbiol. 2020, 323, 108588. [Google Scholar] [CrossRef] [PubMed]
- Allen, U.D.; Hu, P.; Pereira, S.L.; Robinson, J.L.; Paton, T.A.; Beyene, J.; Khodai Booran, N.; Dipchand, A.; Hébert, D.; Ng, V. The genetic diversity of Epstein-Barr virus in the setting of transplantation relative to non-transplant settings: A feasibility study. Pediatr. Transplant. 2016, 20, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Calvet, G.; Aguiar, R.S.; Melo, A.S.; Sampaio, S.A.; De Filippis, I.; Fabri, A.; Araujo, E.S.; de Sequeira, P.C.; de Mendonça, M.C.; de Oliveira, L. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: A case study. Lancet Infect. Dis. 2016, 16, 653–660. [Google Scholar] [CrossRef]
- Desdouits, M.; de Graaf, M.; Strubbia, S.; Oude Munnink, B.B.; Kroneman, A.; Le Guyader, F.S.; Koopmans, M.P.G. Novel opportunities for NGS-based one health surveillance of foodborne viruses. One Health Outlook 2020, 2, 1–8. [Google Scholar] [CrossRef]
- Adey, A.; Morrison, H.G.; Asan; Xun, X.; Kitzman, J.O.; Turner, E.H.; Stackhouse, B.; MacKenzie, A.P.; Caruccio, N.C.; Zhang, X.; et al. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 2010, 11, R119. [Google Scholar] [CrossRef]
- Picelli, S.; Björklund, A.K.; Reinius, B.; Sagasser, S.; Winberg, G.; Sandberg, R. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 2014, 24, 2033–2040. [Google Scholar] [CrossRef]
- Di, L.; Fu, Y.; Sun, Y.; Li, J.; Liu, L.; Yao, J.; Wang, G.; Wu, Y.; Lao, K.; Lee, R.W.; et al. RNA sequencing by direct tagmentation of RNA/DNA hybrids. Proc. Natl. Acad. Sci. USA 2020, 117, 2886–2893. [Google Scholar] [CrossRef]
- Lu, B.; Dong, L.; Yi, D.; Zhang, M.; Zhu, C.; Li, X.; Yi, C. Transposase-assisted tagmentation of RNA/DNA hybrid duplexes. Elife 2020, 9, e54919. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, Z.; Wu, Q.; Tian, P.; Geng, H.; Xu, T.; Wang, D. Redesigned duplex RT-qPCR for the detection of GI and GII human noroviruses. Engineering 2020, 6, 442–448. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, D.; Zhang, Z.; Hewitt, J.; Li, X.; Hou, P.; Wang, D.; Wu, Q. Surveillance of human norovirus in oysters collected from production area in Shandong Province, China during 2017–2018. Food Control 2021, 121, 107649. [Google Scholar] [CrossRef]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinform. Oxf. Engl. 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.A.; Korobeynikov, A.; Lapidus, A.; Prjibelski, A.D.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. A J. Comput. Mol. Cell Biol. 2013, 20, 714–737. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.; Luo, R.; Sadakane, K.; Lam, T. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinform. Oxf. Engl. 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Kroneman, A.; Vennema, H.; Deforche, K.; Avoort, H.V.D.; Peñaranda, S.; Oberste, M.S.; Vinjé, J.; Koopmans, M. An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 2011, 51, 121–125. [Google Scholar] [CrossRef]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantara, L.C.; Vanden Eynden, E.; Vandamme, A.; et al. Genome Detective: An automated system for virus identification from high-throughput sequencing data. Bioinformatics 2019, 35, 871–873. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Labbé, R.G.; García, S. Foodborne viruses. In Guide to Foodborne Pathogens, 2nd ed.; Wiley Blackwell: Hoboken, NJ, USA, 2013; pp. 352–376. [Google Scholar]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Song, F.; Zhu, J.; Zhang, S.; Yang, Y.; Chen, T.; Tang, B. GSA: Genome Sequence Archive. Genom. Proteom. Bioinform. 2017, 15, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ma, L.; Abbasi, A.A.; Raza, R.Z.; Gao, F. Database resources of the National Genomics Data Center in 2020. Nucleic Acids Res. 2020, 48, D24–D33. [Google Scholar]
- Liu, D.; Zhang, Z.; Li, S.; Wu, Q.; Tian, P.; Zhang, Z.; Wang, D. Fingerprinting of human noroviruses co-infections in a possible foodborne outbreak by metagenomics. Int. J. Food Microbiol. 2020, 333, 108787. [Google Scholar] [CrossRef]
- Radford, A.D.; Chapman, D.; Dixon, L.; Chantrey, J.; Darby, A.C.; Hall, N. Application of next-generation sequencing technologies in virology. J. Gen. Virol. 2012, 93, 1853. [Google Scholar] [CrossRef]
- Brown, J.R.; Roy, S.; Ruis, C.; Romero, E.Y.; Shah, D.; Williams, R.; Breuer, J. Norovirus whole-genome sequencing by SureSelect target enrichment: A robust and sensitive method. J. Clin. Microbiol. 2016, 54, 2530–2537. [Google Scholar] [CrossRef]
- Deng, X.; Achari, A.; Federman, S.; Yu, G.; Somasekar, S.; Bártolo, I.; Yagi, S.; Mbala-Kingebeni, P.; Kapetshi, J.; Ahuka-Mundeke, S.; et al. Metagenomic sequencing with spiked primer enrichment for viral diagnostics and genomic surveillance. Nat. Microbiol. 2020, 5, 443–454. [Google Scholar] [CrossRef]
- Briese, T.; Kapoor, A.; Mishra, N.; Jain, K.; Kumar, A.; Jabado, O.J.; Lipkin, W.I. Virome capture sequencing enables sensitive viral diagnosis and comprehensive virome analysis. mBio 2015, 6, e1415–e1491. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Liu, D.; Wang, D.; Wu, Q. Library Preparation Based on Transposase Assisted RNA/DNA Hybrid Co-Tagmentation for Next-Generation Sequencing of Human Noroviruses. Viruses 2021, 13, 65. https://doi.org/10.3390/v13010065
Zhang Z, Liu D, Wang D, Wu Q. Library Preparation Based on Transposase Assisted RNA/DNA Hybrid Co-Tagmentation for Next-Generation Sequencing of Human Noroviruses. Viruses. 2021; 13(1):65. https://doi.org/10.3390/v13010065
Chicago/Turabian StyleZhang, Zilei, Danlei Liu, Dapeng Wang, and Qingping Wu. 2021. "Library Preparation Based on Transposase Assisted RNA/DNA Hybrid Co-Tagmentation for Next-Generation Sequencing of Human Noroviruses" Viruses 13, no. 1: 65. https://doi.org/10.3390/v13010065
APA StyleZhang, Z., Liu, D., Wang, D., & Wu, Q. (2021). Library Preparation Based on Transposase Assisted RNA/DNA Hybrid Co-Tagmentation for Next-Generation Sequencing of Human Noroviruses. Viruses, 13(1), 65. https://doi.org/10.3390/v13010065