
Computational Analysis of SARS-CoV-2 and SARS-Like Coronavirus Diversity in Human, Bat and Pangolin Populations


Abstract
1. Background
2. Results
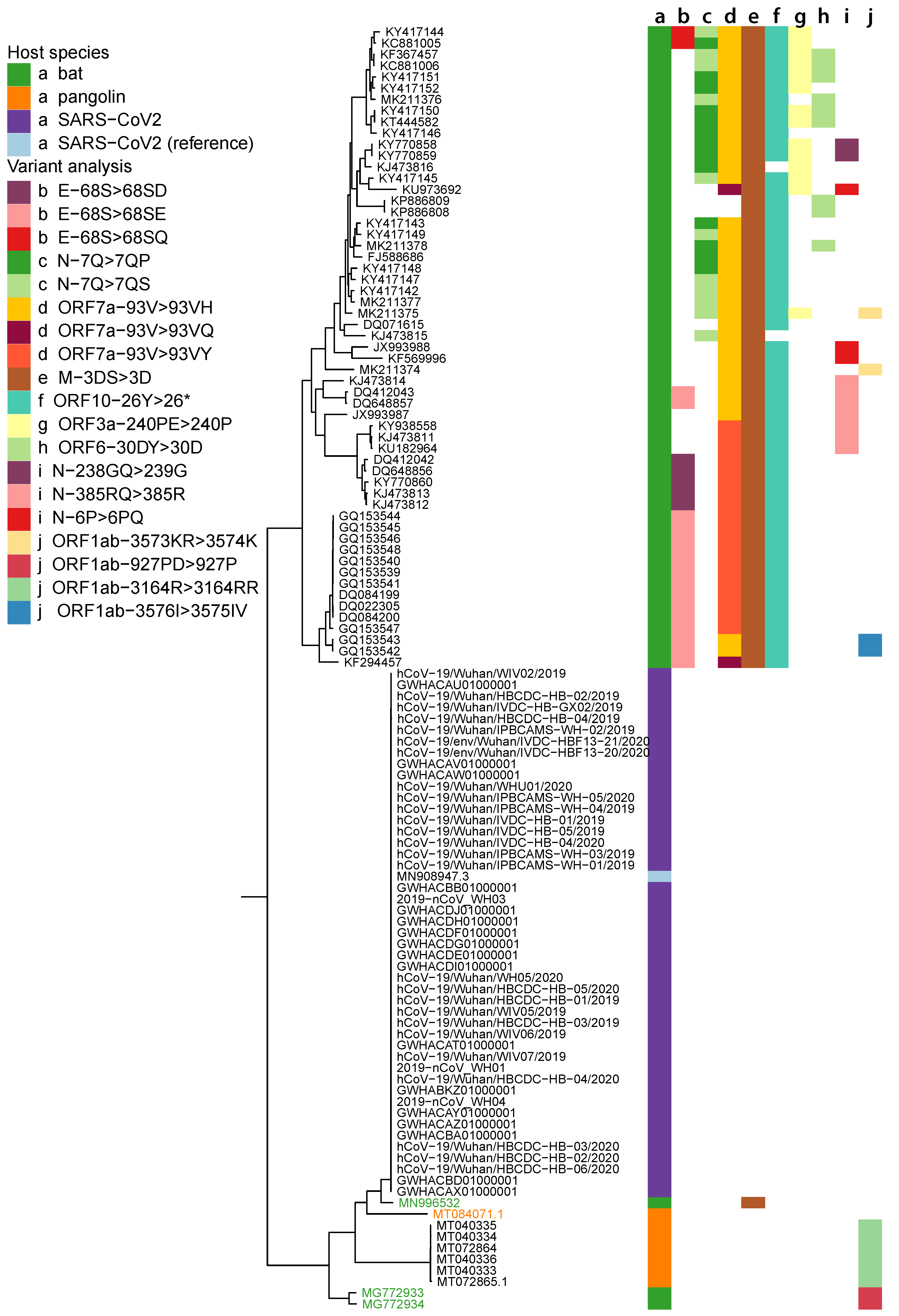
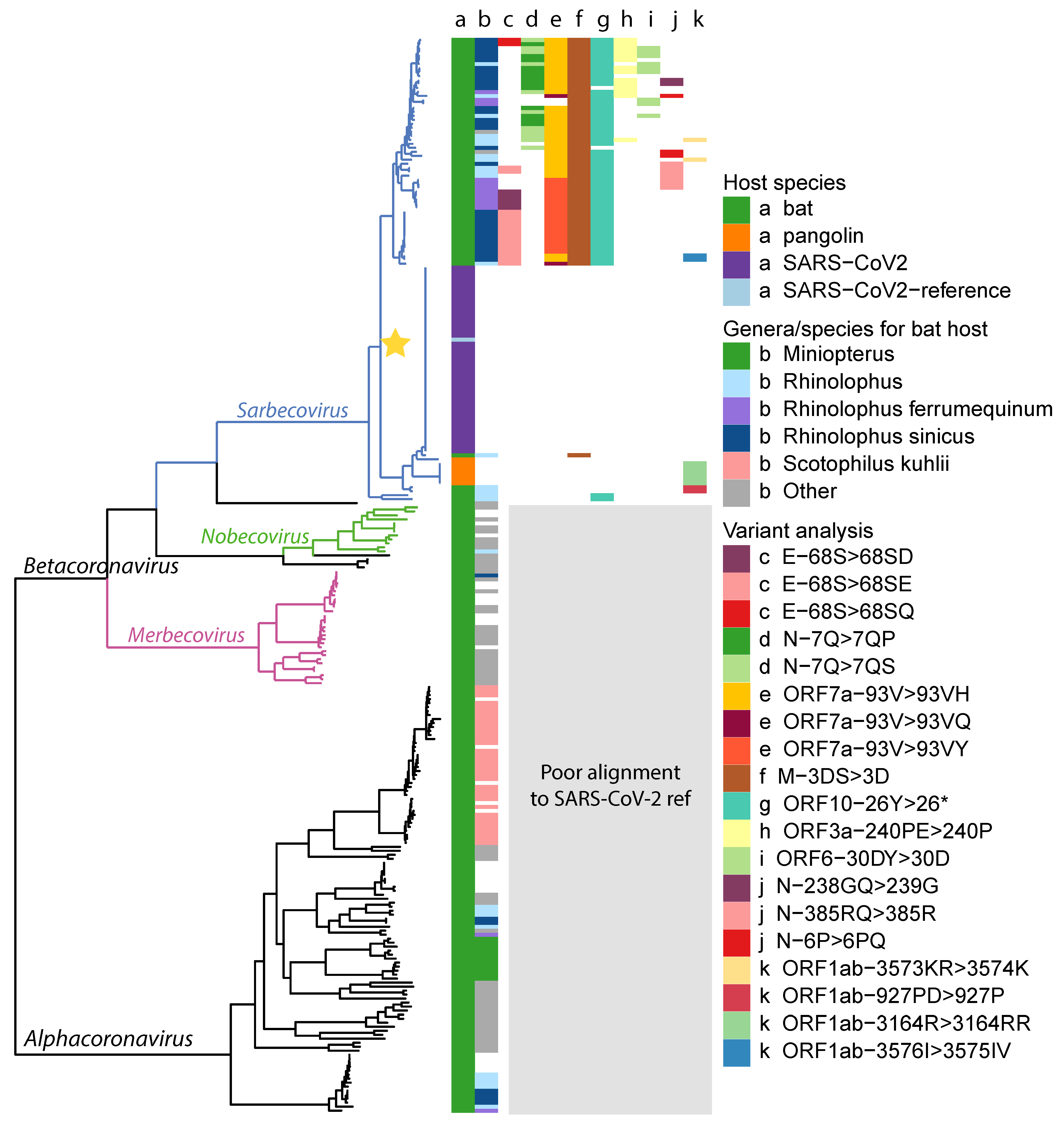
2.1. Data Collection and Phylogenetic Analysis
2.2. Gene Identification
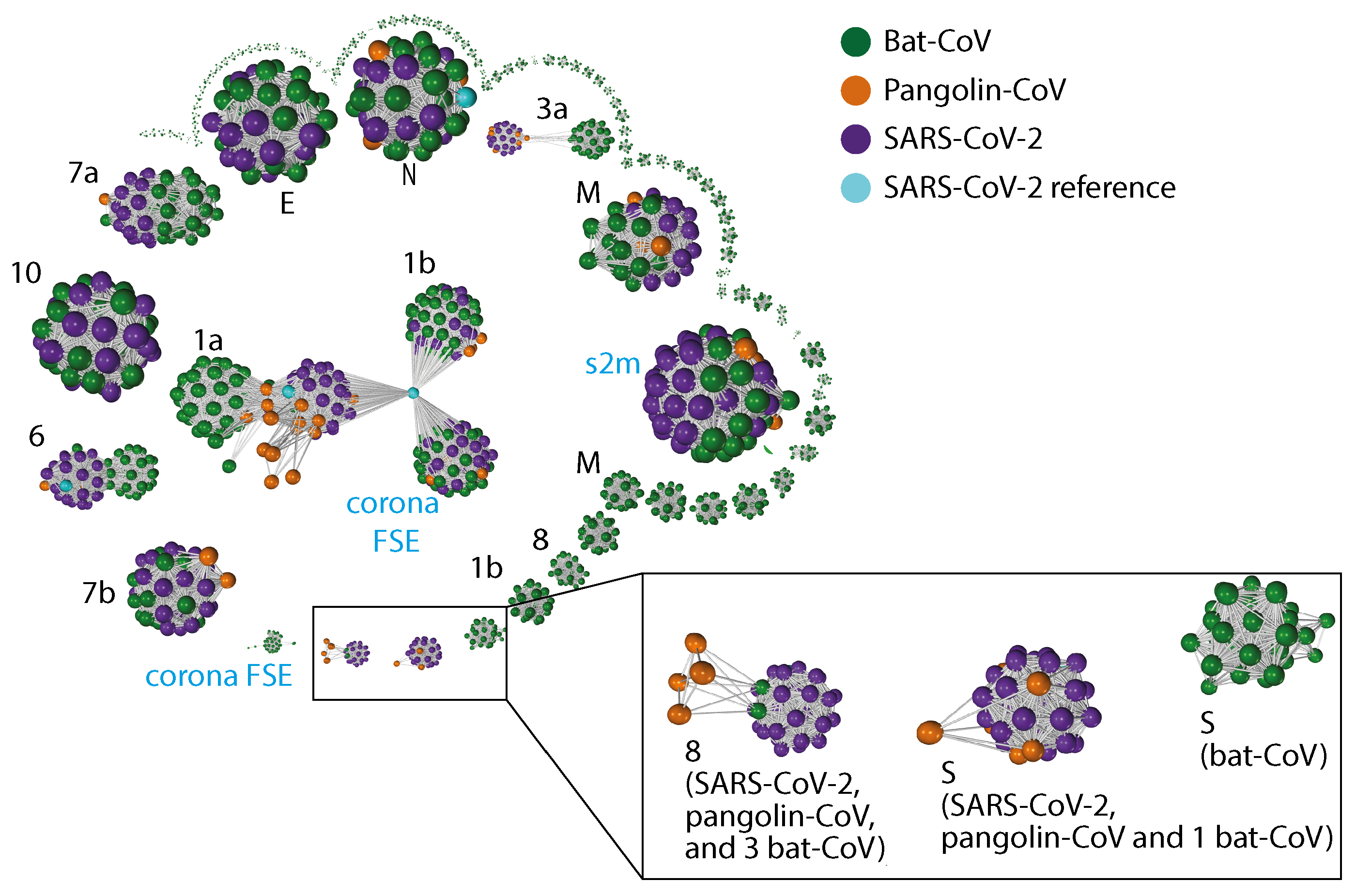
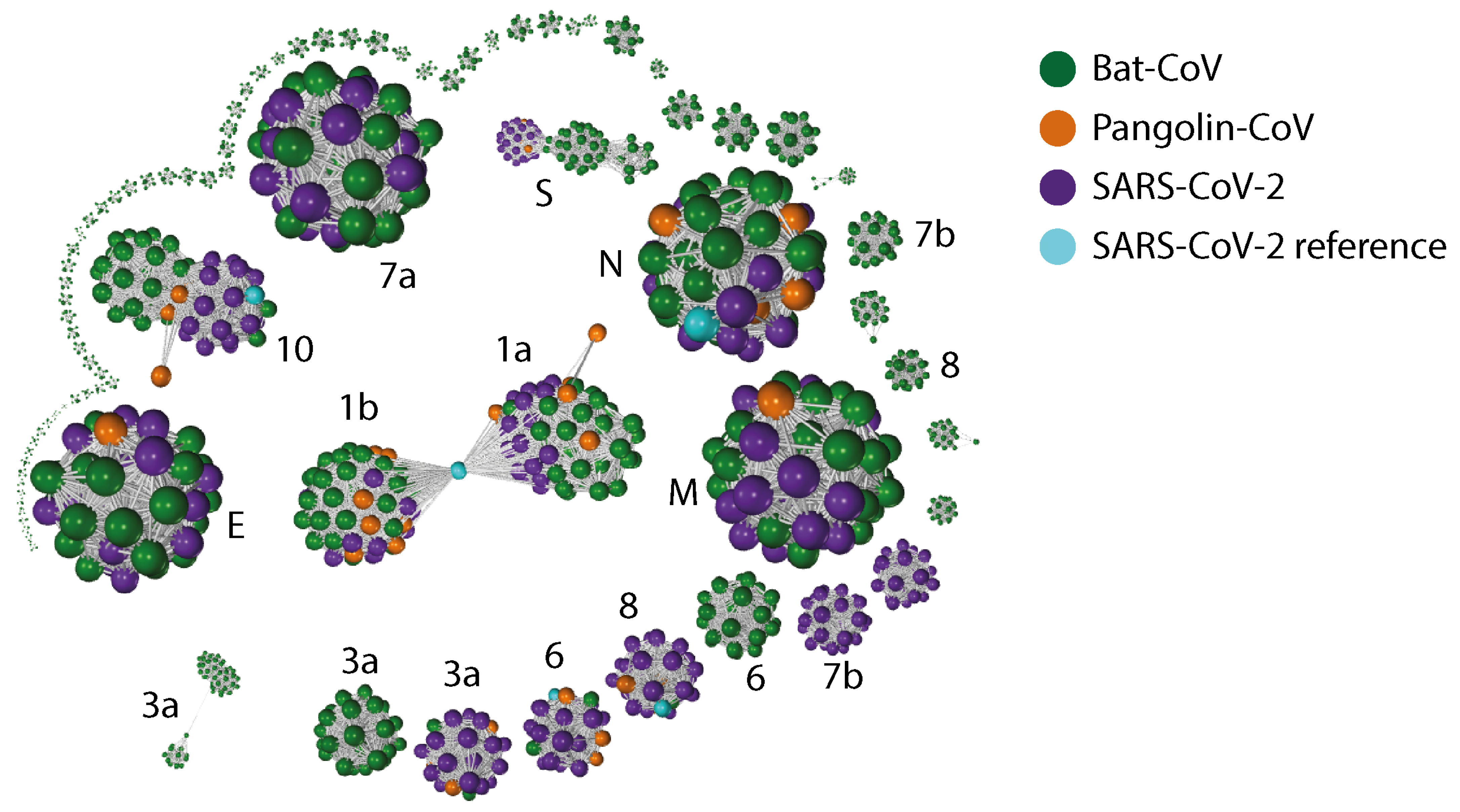
2.3. Gene Relationship Network Graph
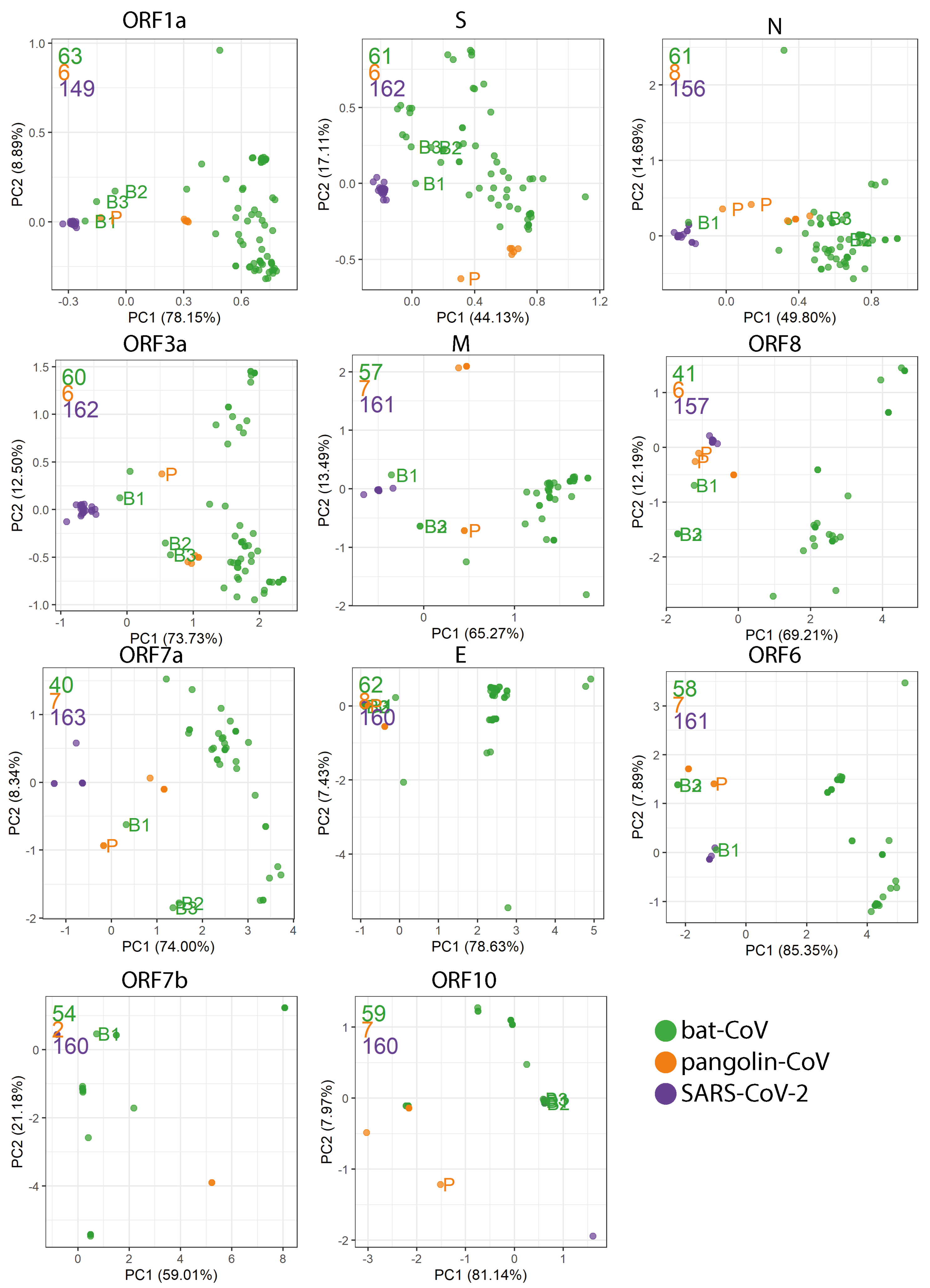
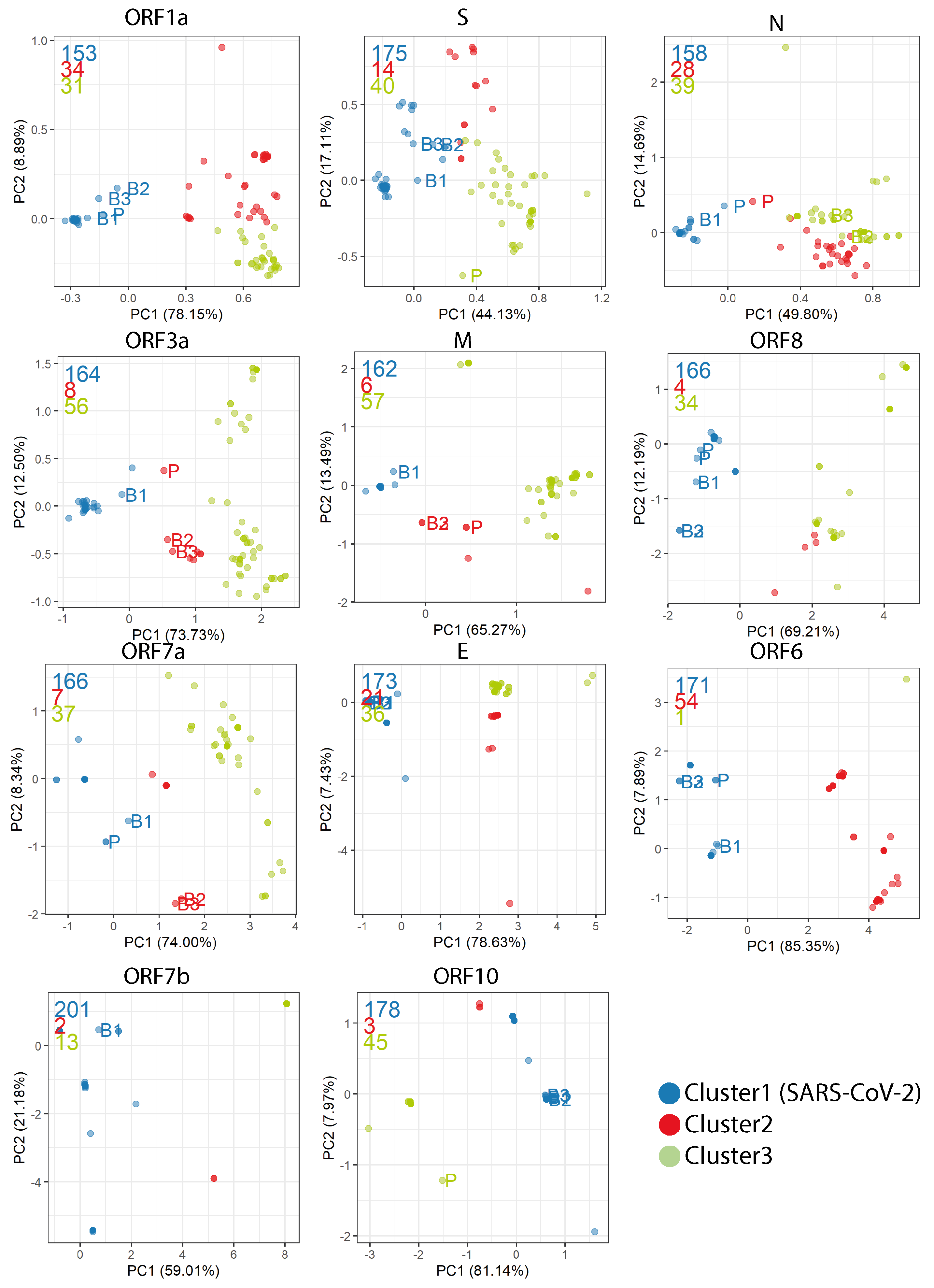
2.4. Codon Usage Bias
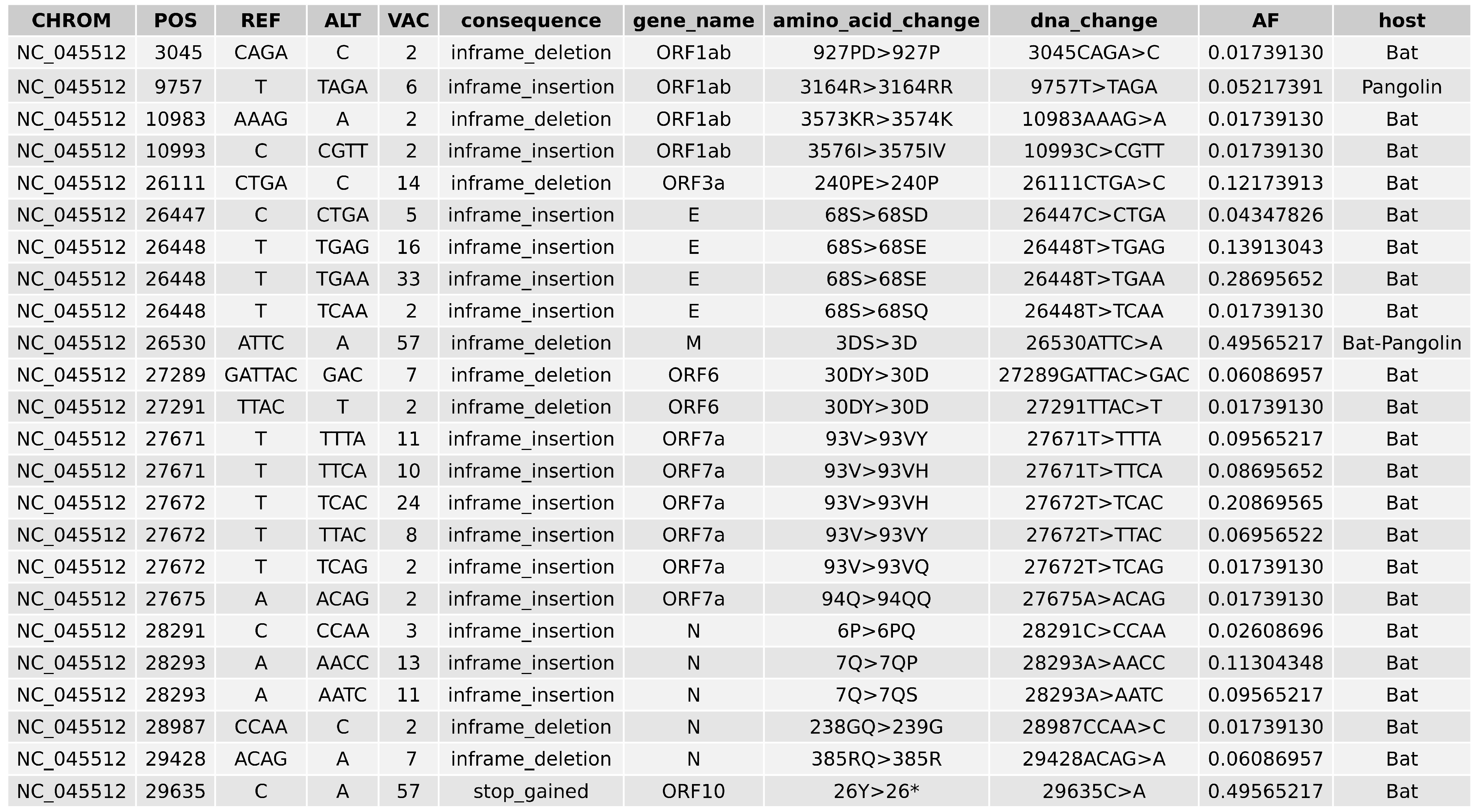
2.5. Variant Analysis
- ORF1ab gene at position 9757 (NC045512_9757 T > TAGA 3164R > 3164RR) of all pangolin-Cov genomes which represents an extra Arginine.
- E gene at position 26448 (NC045512_26448 T > TGAA 68S > 68SE) in 33 bat-Cov genomes which caused an addition of Glutamine.
- ORF7a gene at position 27672 (NC045512_27672 T > TCAC 93V > 93VH) in 24 bat-Cov genomes by addition of an Histamine.
- N gene at position 28293 (NC405512z_28293 A > AACC 7Q > 7QP) in 13 bat-Cov genomes by addition of a Proline.
3. Discussion
4. Methods
4.1. Genomes
4.2. Genome Annotation
4.3. Phylogenetic Trees
4.4. Gene Relationship Network Graph
4.5. Codon Usage
4.6. Variant Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE2 | Angiotensin Converting Enzyme |
| BLAST | Basic Local Alignment Search Tool |
| CoV | Coronavirus |
| DB | DataBiology |
| E | Envelope |
| M | Membrane |
| MERS-CoV | Middle East Respiratory Syndrome Coronavirus |
| N | Nucleocapsid |
| NCBI | National Center for Biotechnology Information |
| ORF | Open reading frame |
| PCA | Principle component analysis |
| PC | Principle component |
| PROKKA | Rapid Prokaryotic Genome Annotation |
| RaTG13 | SARSr-Ra-BatCoV-RaTG13 |
| RBD | Receptor binding domain |
| RSCU | Relative synonymous codon usage |
| SARS | Severe Acute Respiratory Syndrome |
| S | Spike glycoprotein |
| ViPR | Virus Pathogen Resource |
Appendix A. Phylogenetic Tree

Appendix B. Genome Annotation Identified by Source

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host–Dataset | No. Genomes | No. Seq Matching Genes | No. by (PROKKA) | No. by (BLAST) |
|---|---|---|---|---|
| SARS-CoV-2 WI | 46 | 681 | 591 | 90 |
| SARS-CoV-2 GI | 117 | 1736 | 1495 | 241 |
| SARS-CoV-2 EWR | 1 | 12 | N/A | N/A |
| Bat | 215 | 2427 | 2365 | 62 |
| Pangolin | 7 | 97 | 95 | 2 |
Appendix C. Gene–Gene Network Graph Using Amino Acid Sequences

Appendix D. PCA Plots Based on the RSCU for Each Gene

Appendix E. Synonymous Codon Ratios

Appendix F. Combined Variant Analysis

References
- Patz, J.A.; Graczyk, T.K.; Geller, N.; Vittor, A.Y. Effects of environmental change on emerging parasitic diseases. Int. J. Parasitol. 2000, 30, 1395–1405. [Google Scholar] [CrossRef]
- Madhav, N.; Oppenheim, B.; Gallivan, M.; Mulembakani, P.; Rubin, E.; Wolfe, N. Pandemics: Risks, Impacts, and Mitigation; The International Bank for Reconstruction and Development/The World Bank: Washington, DC, USA, 2017. [Google Scholar]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536. [Google Scholar] [CrossRef]
- Weiss, S.R. Forty years with coronaviruses. J. Exp. Med. 2020, 217, e20200537. [Google Scholar] [CrossRef] [PubMed]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nat. Rev. Microbiol. 2009, 7, 439–450. [Google Scholar] [CrossRef]
- Amer, H.; Alqahtani, A.S.; Alzoman, H.; Aljerian, N.; Memish, Z.A. Unusual presentation of Middle East respiratory syndrome coronavirus leading to a large outbreak in Riyadh during 2017. Am. J. Infect. Control 2018, 46, 1022–1025. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.S. The SARS epidemic in Hong Kong: What lessons have we learned? J. R. Soc. Med. 2003, 96, 374–378. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Lian, X.; Su, X.; Wu, W.; Marraro, G.A.; Zeng, Y. From SARS and MERS to COVID-19: A brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir. Res. 2020, 21, 1–14. [Google Scholar] [CrossRef]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.Y.; Perry, B.; Castoe, T.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Holmes, E.C. A Genomic Perspective on the Origin and Emergence of SARS-CoV-2. Cell 2020, 181, 223–227. [Google Scholar] [CrossRef]
- Jitobaom, K.; Phakaratsakul, S.; Sirihongthong, T.; Chotewutmontri, S.; Suriyaphol, P.; Suptawiwat, O.; Auewarakul, P. Codon usage similarity between viral and some host genes suggests a codon-specific translational regulation. Heliyon 2020, 6, e03915. [Google Scholar] [CrossRef]
- Kumar, N.; Kulkarni, D.D.; Lee, B.; Kaushik, R.; Bhatia, S.; Sood, R.; Pateriya, A.K.; Bhat, S.; Singh, V.P. Evolution of codon usage bias in Henipaviruses is governed by natural selection and is host-specific. Viruses 2018, 10, 604. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wu, P.; Deng, S.; Zhang, H.; Hou, Y.; Hu, Z.; Zhang, J.; Chen, X.; Yang, J.R. Dissimilation of synonymous codon usage bias in virus–host coevolution due to translational selection. Nat. Ecol. Evol. 2020, 4, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Elbe, S.; Buckland-Merrett, G.; Falkename, T.; Thistoo, A. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Challenges 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Baranov, P.V.; Henderson, C.M.; Anderson, C.B.; Gesteland, R.F.; Atkins, J.F.; Howard, M.T. Programmed ribosomal frameshifting in decoding the SARS-CoV genome. Virology 2005, 332, 498–510. [Google Scholar] [CrossRef]
- Robertson, M.P.; Igel, H.; Baertsch, R.; Haussler, D.; Ares, M., Jr.; Scott, W.G. The structure of a rigorously conserved RNA element within the SARS virus genome. PLoS Biol. 2004, 3, e5. [Google Scholar] [CrossRef]
- Tengs, T.; Jonassen, C.M. Distribution and evolutionary history of the mobile genetic element s2m in coronaviruses. Diseases 2016, 4, 27. [Google Scholar] [CrossRef]
- Tengs, T.; Delwiche, C.F.; Jonassen, C.M. A mobile genetic element in the SARS-CoV-2 genome is shared with multiple insect species. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lau, S.K.; Luk, H.K.; Wong, A.C.; Li, K.S.; Zhu, L.; He, Z.; Fung, J.; Chan, T.T.; Fung, K.S.; Woo, P.C. Possible bat origin of severe acute respiratory syndrome coronavirus 2. Emerg. Infect. Dis. 2020, 26, 1542. [Google Scholar] [CrossRef] [PubMed]
- Malaiyan, J.; Arumugam, S.; Mohan, K.; Radhakrishnan, G.G. An update on origin of SARS-CoV-2: Despite closest identity, bat (RaTG13) and Pangolin derived Coronaviruses varied in the critical binding site and O-linked glycan residues. J. Med Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Kulcsar, K.; Misra, V.; Frieman, M.; Mossman, K. Bats and coronaviruses. Viruses 2019, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Yi, H. 2019 novel coronavirus is undergoing active recombination. Clin. Infect. Dis. 2020, 71, 884–887. [Google Scholar] [CrossRef]
- Lopes, L.R.; de Mattos Cardillo, G.; Paiva, P.B. Molecular evolution and phylogenetic analysis of SARS-CoV-2 and hosts ACE2 protein suggest Malayan pangolin as intermediary host. Braz. J. Microbiol. 2020, 51, 1593–1599. [Google Scholar] [CrossRef]
- Fahmi, M.; Kubota, Y.; Ito, M. Nonstructural proteins NS7b and NS8 are likely to be phylogenetically associated with evolution of 2019-nCoV. Infect. Genet. Evol. 2020, 81, 104272. [Google Scholar] [CrossRef]
- Liu, P.; Jiang, J.Z.; Wan, X.F.; Hua, Y.; Li, L.; Zhou, J.; Wang, X.; Hou, F.; Chen, J.; Zou, J.; et al. Are pangolins the intermediate host of the 2019 novel coronavirus (SARS-CoV-2)? PLoS Pathog. 2020, 16, e1008421. [Google Scholar] [CrossRef]
- Xiao, K.; Zhai, J.; Feng, Y.; Zhou, N.; Zhang, X.; Zou, J.J.; Li, N.; Guo, Y.; Li, X.; Shen, X.; et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature 2020, 583, 286–289. [Google Scholar] [CrossRef]
- Ceraolo, C.; Giorgi, F.M. Genomic variance of the 2019-nCoV coronavirus. J. Med Virol. 2020, 92, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F. Evolutionary dynamics of the SARS-CoV-2 ORF8 accessory gene. Infect. Genet. Evol. 2020, 85, 104525. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Liao, C.H.; Wang, Q.; Tan, Y.J.; Luo, R.; Qiu, Y.; Ge, X.Y. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020, 286, 198074. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.H.; Fung, T.S.; Fang, S.; Huang, M.; Le, M.T.; Liu, D.X. Accessory proteins 8b and 8ab of severe acute respiratory syndrome coronavirus suppress the interferon signaling pathway by mediating ubiquitin-dependent rapid degradation of interferon regulatory factor 3. Virology 2018, 515, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Hachim, A.; Kavian, N.; Cohen, C.A.; Chin, A.W.; Chu, D.K.; Mok, C.K.; Tsang, O.T.; Yeung, Y.C.; Perera, R.A.; Poon, L.L.; et al. ORF8 and ORF3b Antibodies Are Accurate Serological Markers of Early and Late SARS-CoV-2 Infection. Nat. Immunol. 2020, 21, 1293–1301. [Google Scholar] [CrossRef]
- Lau, S.K.; Feng, Y.; Chen, H.; Luk, H.K.; Yang, W.H.; Li, K.S.; Zhang, Y.Z.; Huang, Y.; Song, Z.Z.; Chow, W.N.; et al. Severe acute respiratory syndrome (SARS) coronavirus ORF8 protein is acquired from SARS-related coronavirus from greater horseshoe bats through recombination. J. Virol. 2015, 89, 10532–10547. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, Q.; Zhang, Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr. Biol. 2020, 30, 1346–1351.e2. [Google Scholar] [CrossRef]
- Wrobel, A.G.; Benton, D.J.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 2020, 27, 763–767. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Baranowski, E.; Ruiz-Jarabo, C.M.; Domingo, E. Evolution of cell recognition by viruses. Science 2001, 292, 1102–1105. [Google Scholar] [CrossRef]
- Baranowski, E.; Ruiz-Jarabo, C.M.; Pariente, N.; Verdaguer, N.; Domingo, E. Evolution of cell recognition by viruses: A source of biological novelty with medical implications. Adv. Virus Res. 2003, 62, 19. [Google Scholar] [PubMed]
- Wu, K.; Chen, L.; Peng, G.; Zhou, W.; Pennell, C.A.; Mansky, L.M.; Geraghty, R.J.; Li, F. A virus-binding hot spot on human angiotensin-converting enzyme 2 is critical for binding of two different coronaviruses. J. Virol. 2011, 85, 5331–5337. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.M.; Morales, A.C.; Ho, A.T.; Mordstein, C.; Mühlhausen, S.; Watson, S.; Cano, L.; Young, B.; Kudla, G.; Hurst, L.D. Evidence for strong mutation bias towards, and selection against, U content in SARS-CoV-2: Implications for vaccine design. Mol. Biol. Evol. 2020, msaa188. [Google Scholar] [CrossRef]
- Gu, H.; Chu, D.K.; Peiris, M.; Poon, L.L. Multivariate analyses of codon usage of SARS-CoV-2 and other betacoronaviruses. Virus Evol. 2020, 6, veaa032. [Google Scholar] [CrossRef]
- Nambou, K.; Anakpa, M. Deciphering the co-adaptation of codon usage between respiratory coronaviruses and their human host uncovers candidate therapeutics for COVID-19. Infect. Genet. Evol. 2020, 85, 104471. [Google Scholar] [CrossRef]
- Alonso, A.M.; Diambra, L. SARS-CoV-2 Codon Usage Bias Downregulates Host Expressed Genes with Similar Codon Usage. Front. Cell Dev. Biol. 2020, 8, 831. [Google Scholar] [CrossRef]
- Digard, P.; Lee, H.M.; Sharp, C.; Grey, F.; Gaunt, E. Intra-genome variability in the dinucleotide composition of SARS-CoV-2. Virus Evol. 2020, 6, veaa057. [Google Scholar] [CrossRef]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef]
- Smith, E.C.; Sexton, N.R.; Denison, M.R. Thinking outside the triangle: Replication fidelity of the largest RNA viruses. Annu. Rev. Virol. 2014, 1, 111–132. [Google Scholar] [CrossRef]
- Hassan, S.S.; Attrish, D.; Ghosh, S.; Choudhury, P.P.; Uversky, V.N.; Uhal, B.D.; Lundstrom, K.; Rezaei, N.; Aljabali, A.A.; Seyran, M.; et al. Notable sequence homology of the ORF10 protein introspects the architecture of SARS-COV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The architecture of SARS-CoV-2 transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Jia, P.; Fang, B.; Zhao, Z. Differential expression of viral transcripts from single-cell RNA sequencing of moderate and severe COVID-19 patients and its implications for case severity. Front. Microbiol. 2020, 11, 2568. [Google Scholar] [CrossRef]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 1–22. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Álvarez, E.; Almazán, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. J. Virol. 2007, 81, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Teoh, K.T.; Siu, Y.L.; Chan, W.L.; Schlüter, M.A.; Liu, C.J.; Peiris, J.M.; Bruzzone, R.; Margolis, B.; Nal, B. The SARS coronavirus E protein interacts with PALS1 and alters tight junction formation and epithelial morphogenesis. Mol. Biol. Cell 2010, 21, 3838–3852. [Google Scholar] [CrossRef]
- Hassan, S.S.; Choudhury, P.P.; Roy, B. SARS-CoV2 envelope protein: Non-synonymous mutations and its consequences. Genomics 2020. [Google Scholar] [CrossRef]
- Taylor, J.K.; Coleman, C.M.; Postel, S.; Sisk, J.M.; Bernbaum, J.G.; Venkataraman, T.; Sundberg, E.J.; Frieman, M.B. Severe acute respiratory syndrome coronavirus ORF7a inhibits bone marrow stromal antigen 2 virion tethering through a novel mechanism of glycosylation interference. J. Virol. 2015, 89, 11820–11833. [Google Scholar] [CrossRef]
- Cong, Y.; Ulasli, M.; Schepers, H.; Mauthe, M.; V’kovski, P.; Kriegenburg, F.; Thiel, V.; de Haan, C.A.; Reggiori, F. Nucleocapsid protein recruitment to replication-transcription complexes plays a crucial role in coronaviral life cycle. J. Virol. 2020, 94, e01925-19. [Google Scholar] [CrossRef]
- Weber, S.; Ramirez, C.; Doerfler, W. Signal hotspot mutations in SARS-CoV-2 genomes evolve as the virus spreads and actively replicates in different parts of the world. Virus Res. 2020, 289, 198170. [Google Scholar] [CrossRef]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef] [PubMed]
- Coordinators, N.R. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2018, 46, D8. [Google Scholar] [CrossRef] [PubMed]
- Dinman, J.D. Programmed–1 Ribosomal Frameshifting in SARS Coronavirus. In Molecular Biology of the SARS-Coronavirus; Springer: Berlin/Heidelberg, Germany, 2010; pp. 63–72. [Google Scholar]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Revell, L.J. phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Yu, G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef]
- Freeman, T.; Horsewell, S.; Patir, A.; Harling-Lee, J.; Regan, T.; Shih, B.B.; Prendergast, J.; Hume, D.A.; Angus, T. Graphia: A platform for the graph-based visualisation and analysis of complex data. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sharp, P.M.; Tuohy, T.M.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef]
- Danecek, P.; McCarthy, S.A. BCFtools/csq: Haplotype-aware variant consequences. Bioinformatics 2017, 33, 2037–2039. [Google Scholar] [CrossRef]




| Dataset | Min. | Median | Mean | Max. | Sample Count |
|---|---|---|---|---|---|
| Wuhan | 7 | 11 | 11 | 13 | 46 |
| Charite | 9 | 11 | 11 | 12 | 117 |
| Bat | 2 | 9 | 9 | 12 | 215 |
| Pangolin | 10 | 11 | 12 | 17 | 7 |
| Host–Source | No. Genomes | Database |
|---|---|---|
| SARS-CoV-2 Wuhan isolates | 20 | https://doi.org/10.1101/2020.10.22.328864 |
| SARS-CoV-2 Wuhan isolates | 26 | GISAID-Charite [14] |
| SARS-CoV-2 German isolates | 117 | GISAID-Charite [14] |
| SARS-CoV-2 Ensembl Wuhan Reference | 1 | Ensembl [63] |
| Bat | 139 | https://doi.org/10.1101/2020.10.22.328864 |
| Bat | 76 | ViPR [62] |
| Pangolin | 5 | https://doi.org/10.1101/2020.10.22.328864 |
| Pangolin | 2 | NCBI [64] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimonaco, N.J.; Salavati, M.; Shih, B.B. Computational Analysis of SARS-CoV-2 and SARS-Like Coronavirus Diversity in Human, Bat and Pangolin Populations. Viruses 2021, 13, 49. https://doi.org/10.3390/v13010049
Dimonaco NJ, Salavati M, Shih BB. Computational Analysis of SARS-CoV-2 and SARS-Like Coronavirus Diversity in Human, Bat and Pangolin Populations. Viruses. 2021; 13(1):49. https://doi.org/10.3390/v13010049
Chicago/Turabian StyleDimonaco, Nicholas J., Mazdak Salavati, and Barbara B. Shih. 2021. "Computational Analysis of SARS-CoV-2 and SARS-Like Coronavirus Diversity in Human, Bat and Pangolin Populations" Viruses 13, no. 1: 49. https://doi.org/10.3390/v13010049
APA StyleDimonaco, N. J., Salavati, M., & Shih, B. B. (2021). Computational Analysis of SARS-CoV-2 and SARS-Like Coronavirus Diversity in Human, Bat and Pangolin Populations. Viruses, 13(1), 49. https://doi.org/10.3390/v13010049