
Therapy Implications of Hepatitis C Virus Genetic Diversity



Abstract
1. Introduction
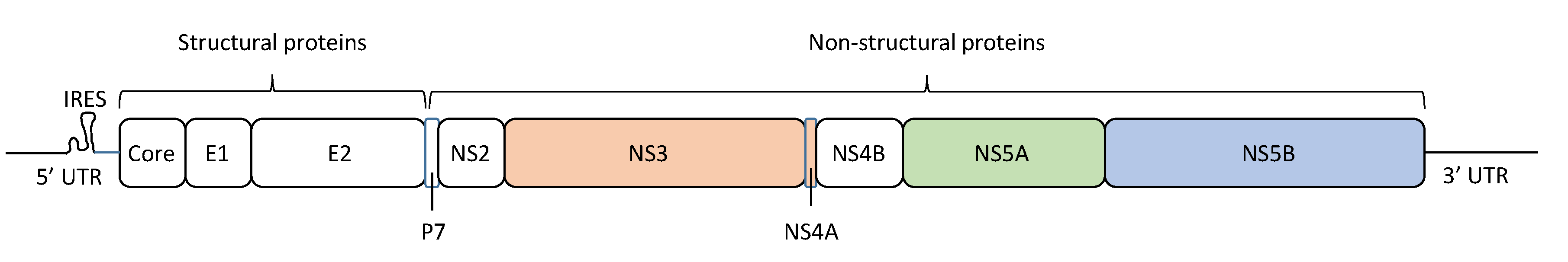
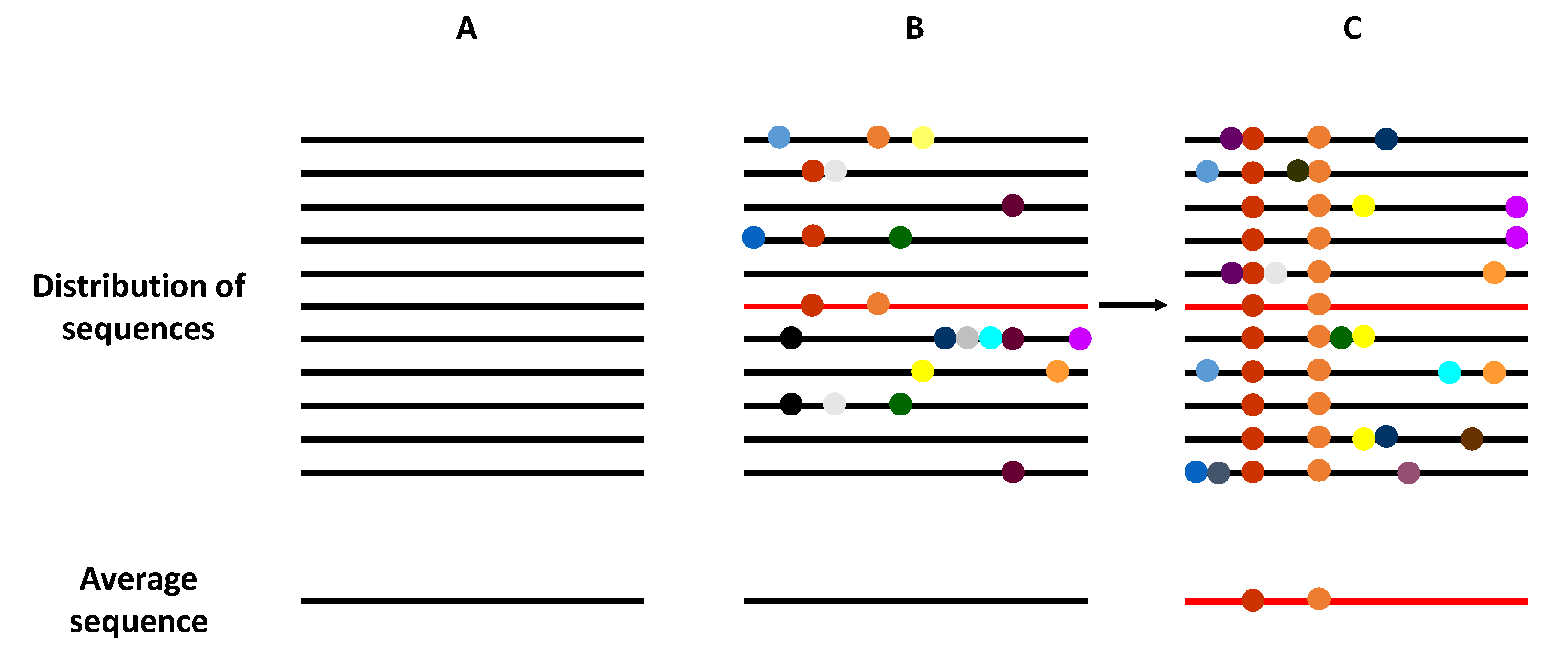
2. HCV Quasispecies and Diversity
3. Implications of HCV Diversity in Pathogenesis and Transmission
4. HCV Therapy
5. DAA Resistance
6. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Simmonds, P. Genetic diversity and evolution of hepatitis C virus—15 years on. J. Gen. Virol. 2004, 85, 3173–3188. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Simmonds, P.; Gerold, G.; Qaisar, N.; Jain, K.; Henriquez, J.A.; Firth, C.; Hirschberg, D.L.; Rice, C.M.; Shields, S.; et al. Characterization of a canine homolog of hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 11608–11613. [Google Scholar] [CrossRef] [PubMed]
- Burbelo, P.D.; Dubovi, E.J.; Simmonds, P.; Medina, J.L.; Henriquez, J.A.; Mishra, N.; Wagner, J.; Tokarz, R.; Cullen, J.M.; Iadarola, M.J.; et al. Serology-Enabled Discovery of Genetically Diverse Hepaciviruses in a New Host. J. Virol. 2012, 86, 6171–6178. [Google Scholar] [CrossRef] [PubMed]
- Feinstone, S.M.; Kapikian, A.Z.; Purcell, R.H.; Alter, H.J.; Holland, P.V. Transfusion-Associated Hepatitis Not Due to Viral Hepatitis Type A or B. N. Engl. J. Med. 1975, 292, 767–770. [Google Scholar] [CrossRef]
- Kolykhalov, A.A.; Agapov, E.V.; Blight, K.J.; Mihalik, K.; Feinstone, S.M.; Rice, C.M. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 1997, 277, 570–574. [Google Scholar] [CrossRef]
- Kuo, G.; Choo, Q.L.; Alter, H.J.; Gitnick, G.L.; Redeker, A.G.; Purcell, R.H.; Miyamura, T.; Dienstag, J.L.; Alter, M.J.; Stevens, C.E.; et al. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science 1989, 244, 362–364. [Google Scholar] [CrossRef]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef]
- Grebely, J.; Dore, G.J.; Kim, A.Y.; Lloyd, A.; Shoukry, N.H.; Prins, M.; Page, K. Genetics of spontaneous clearance of hepatitis C virus infection: A complex topic with much to learn. Hepatology 2014, 60, 2127–2128. [Google Scholar] [CrossRef]
- Westbrook, R.H.; Dusheiko, G. Natural history of hepatitis C. J. Hepatol. 2014, 61, S58–S68. [Google Scholar] [CrossRef]
- World Health Organization. Global Hepatitis Report 2017. Available online: http://apps.who.int/iris/bitstream/10665/255016/1/9789241565455-eng.pdf?ua=1 (accessed on 10 March 2017).
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef]
- Catanese, M.T.; Uryu, K.; Kopp, M.; Edwards, T.J.; Andrus, L.; Rice, W.J.; Silvestry, M.; Kuhn, R.J.; Rice, C.M. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. USA 2013, 110, 9505–9510. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F. Hepatitis C Virus Proteins: From Structure to Function. In Current Topics in Microbiology and Immunology; Curr Top Microbiol Immunol, Springer: New York, NY, USA, 2013; Volume 369, pp. 113–142. [Google Scholar]
- Tabata, K.; Neufeldt, C.J.; Bartenschlager, R. Hepatitis C virus replication. Cold Spring Harb. Perspect. Med. 2020, 10, a037093. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V.; Körner, F.; Koch, J.O.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Kräusslich, H.G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Más, A.; López-Galíndez, C.; Cacho, I.; Gómez, J.; Martínez, M.A. Unfinished stories on viral quasispecies and darwinian views of evolution. J. Mol. Biol. 2010, 397, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Perales, C. Viral quasispecies. PLoS Genet. 2019, 15. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M. Selforganization of matter and the evolution of biological macromolecules. Naturwissenschaften 1971, 58, 465–523. [Google Scholar] [CrossRef]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef]
- Ogata, N.; Alter, H.J.; Miller, R.H.; Purcell, R.H. Nucleotide sequence and mutation rate of the H strain of hepatitis C virus. Proc. Natl. Acad. Sci. USA 1991, 88, 3392–3396. [Google Scholar] [CrossRef]
- Franco, S.; Parera, M.; Aparicio, E.; Clotet, B.; Martinez, M.A. Genetic and catalytic efficiency structure of an HCV protease quasispecies. Hepatology 2007, 45, 899–910. [Google Scholar] [CrossRef]
- Martinez, M.A.; Nevot, M.; Jordan-Paiz, A.; Franco, S. Similarities between Human Immunodeficiency Virus Type 1 and Hepatitis C Virus Genetic and Phenotypic Protease Quasispecies Diversity. J. Virol. 2015, 89, 9758–9764. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martell, M.; Esteban, J.I.; Quer, J.; Vargas, V.; Esteban, R.; Guardia, J.; Gómez, J. Dynamic behavior of hepatitis C virus quasispecies in patients undergoing orthotopic liver transplantation. J. Virol. 1994, 68, 3425–3436. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, J.M.; González-Candelas, F.; Moya, A.; Sanjuán, R. Effect of Ribavirin on the Mutation Rate and Spectrum of Hepatitis C Virus In Vivo. J. Virol. 2009, 83, 5760–5764. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, R.M.; Li, H.; Wang, S.; Stoddard, M.B.; Learn, G.H.; Korber, B.T.; Bhattacharya, T.; Guedj, J.; Parrish, E.H.; Hahn, B.H.; et al. Quantifying the Diversification of Hepatitis C Virus (HCV) during Primary Infection: Estimates of the In Vivo Mutation Rate. PLoS Pathog. 2012, 8, e1002881. [Google Scholar] [CrossRef] [PubMed]
- Borgia, S.M.; Hedskog, C.; Parhy, B.; Hyland, R.H.; Stamm, L.M.; Brainard, D.M.; Subramanian, M.G.; McHutchison, J.G.; Mo, H.; Svarovskaia, E.; et al. Identification of a novel hepatitis C virus genotype from Punjab, India: Expanding classification of hepatitis C virus into 8 genotypes. J. Infect. Dis. 2018, 218, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Blach, S.; Zeuzem, S.; Manns, M.; Altraif, I.; Duberg, A.S.; Muljono, D.H.; Waked, I.; Alavian, S.M.; Lee, M.H.; Negro, F.; et al. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: A modelling study. Lancet Gastroenterol. Hepatol. 2017, 2, 161–176. [Google Scholar] [CrossRef]
- Galli, A.; Bukh, J. Comparative analysis of the molecular mechanisms of recombination in hepatitis C virus. Trends Microbiol. 2014, 22, 354–364. [Google Scholar] [CrossRef]
- Farci, P.; Shimoda, A.; Coiana, A.; Diaz, G.; Peddis, G.; Melpolder, J.C.; Strazzera, A.; Chien, D.Y.; Munoz, S.J.; Balestrieri, A.; et al. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science 2000, 288, 339–344. [Google Scholar] [CrossRef]
- Geller, R.; Estada, Ú.; Peris, J.B.; Andreu, I.; Bou, J.V.; Garijo, R.; Cuevas, J.M.; Sabariegos, R.; Mas, A.; Sanjuán, R. Highly heterogeneous mutation rates in the hepatitis C virus genome. Nat. Microbiol. 2016, 1, 16045. [Google Scholar] [CrossRef]
- Raghwani, J.; Rose, R.; Sheridan, I.; Lemey, P.; Suchard, M.A.; Santantonio, T.; Farci, P.; Klenerman, P.; Pybus, O.G. Exceptional Heterogeneity in Viral Evolutionary Dynamics Characterises Chronic Hepatitis C Virus Infection. PLoS Pathog. 2016, 12, e1005894. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.R.; Tanaka, Y.; Takebe, Y.; Magiorkinis, G.; Buskell, Z.; Seeff, L.; Alter, H.J.; Pybus, O.G. Evolutionary analysis of hepatitis C virus gene sequences from 1953. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 2013.0168. [Google Scholar] [CrossRef] [PubMed]
- Farci, P.; Wollenberg, K.; Diaz, G.; Engle, R.E.; Lai, M.E.; Klenerman, P.; Purcell, R.H.; Pybus, O.G.; Alter, H.J. Profibrogenic chemokines and viral evolution predict rapid progression of hepatitis C to cirrhosis. Proc. Natl. Acad. Sci. USA 2012, 109, 14562–14567. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Kosakovsky Pond, S.L.; Drummond, A.J.; Pybus, O.G.; Shapiro, B.; Barroso, H.; Taveira, N.; Rambaut, A. Synonymous substitution rates predict HIV disease progression as a result of underlying replication dynamics. PLoS Comput. Biol. 2007, 3, 0282–0292. [Google Scholar] [CrossRef]
- Alter, H.J.; Farci, P.; Bukh, J.; Purcell, R.H. Reflections on the History of HCV: A Posthumous Examination. Clin. Liver Dis. 2020, 15, S64–S71. [Google Scholar] [CrossRef]
- Fafi-Kremer, S.; Fofana, I.; Soulier, E.; Carolla, P.; Meuleman, P.; Leroux-Roels, G.; Patel, A.H.; Cosset, F.L.; Pessaux, P.; Doffoël, M.; et al. Viral entry and escape from antibody-mediated neutralization influence hepatitis C virus reinfection in liver transplantation. J. Exp. Med. 2010, 207, 2019–2031. [Google Scholar] [CrossRef]
- Harouaka, D.; Engle, R.E.; Wollenberg, K.; Diaz, G.; Tice, A.B.; Zamboni, F.; Govindarajan, S.; Alter, H.; Kleiner, D.E.; Farci, P. Diminished viral replication and compartmentalization of hepatitis C virus in hepatocellular carcinoma tissue. Proc. Natl. Acad. Sci. USA 2016, 113, 1375–1380. [Google Scholar] [CrossRef]
- Ramirez, S.; Perez-del-Pulgar, S.; Carrion, J.A.; Costa, J.; Gonzalez, P.; Massaguer, A.; Fondevila, C.; Garcia-Valdecasas, J.C.; Navasa, M.; Forns, X. Hepatitis C Virus Compartmentalization and Infection Recurrence after Liver Transplantation. Am. J. Transplant. 2009, 9, 1591–1601. [Google Scholar] [CrossRef]
- Hedegaard, D.L.; Tully, D.C.; Rowe, I.A.; Reynolds, G.M.; Bean, D.J.; Hu, K.; Davis, C.; Wilhelm, A.; Ogilvie, C.B.; Power, K.A.; et al. High resolution sequencing of hepatitis C virus reveals limited intra-hepatic compartmentalization in end-stage liver disease. J. Hepatol. 2017, 66, 28–38. [Google Scholar] [CrossRef]
- Li, H.; Stoddard, M.B.; Wang, S.; Blair, L.M.; Giorgi, E.E.; Parrish, E.H.; Learn, G.H.; Hraber, P.; Goepfert, P.A.; Saag, M.S.; et al. Elucidation of Hepatitis C Virus Transmission and Early Diversification by Single Genome Sequencing. PLoS Pathog. 2012, 8, e1002880. [Google Scholar] [CrossRef]
- Li, H.; Stoddard, M.B.; Wang, S.; Giorgi, E.E.; Blair, L.M.; Learn, G.H.; Hahn, B.H.; Alter, H.J.; Busch, M.P.; Fierer, D.S.; et al. Single-Genome Sequencing of Hepatitis C Virus in Donor-Recipient Pairs Distinguishes Modes and Models of Virus Transmission and Early Diversification. J. Virol. 2016, 90, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Abayasingam, A.; Leung, P.; Eltahla, A.; Bull, R.A.; Luciani, F.; Grebely, J.; Dore, G.J.; Applegate, T.; Page, K.; Bruneau, J.; et al. Genomic characterization of hepatitis C virus transmitted founder variants with deep sequencing. Infect. Genet. Evol. 2019, 71, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, C.; Leung, P.; Lloyd, A.R.; Bull, R.A.; Luciani, F.; Grebely, J.; Dore, G.J.; Applegate, T.; Page, K.; Bruneau, J.; et al. Genomic variability of within-host hepatitis C variants in acute infection. J. Viral Hepat. 2019, 26, 476–484. [Google Scholar] [CrossRef]
- Bukh, J. The history of hepatitis C virus (HCV): Basic research reveals unique features in phylogeny, evolution and the viral life cycle with new perspectives for epidemic control. J. Hepatol. 2016, 65, S2–S21. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.D.; Salinas, E.; Grakoui, A. Immune system control of hepatitis C virus infection. Curr. Opin. Virol. 2021, 46, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Farci, P.; Alter, H.J.; Govindarajan, S.; Wong, D.C.; Engle, R.; Lesniewski, R.R.; Mushahwar, I.K.; Desai, S.M.; Miller, R.H.; Ogata, N.; et al. Lack of protective immunity against reinfection with hepatitis C virus. Science 1992, 258, 135–140. [Google Scholar] [CrossRef]
- Bassett, S.E.; Guerra, B.; Brasky, K.; Miskovsky, E.; Houghton, M.; Klimpel, G.R.; Lanford, R.E. Protective immune response to hepatitis C virus in chimpanzees rechallenged following clearance of primary infection. Hepatology 2001, 33, 1479–1487. [Google Scholar] [CrossRef]
- Franco, S.; Tural, C.; Nevot, M.; Moltó, J.; Rockstroh, J.K.; Clotet, B.; Martinez, M.A. Detection of a sexually transmitted hepatitis C virus protease inhibitor-resistance variant in a human immunodeficiency virus-infected homosexual man. Gastroenterology 2014, 147, 599–601. [Google Scholar] [CrossRef]
- Wrensch, F.; Ligat, G.; Heydmann, L.; Schuster, C.; Zeisel, M.B.; Pessaux, P.; Habersetzer, F.; King, B.J.; Tarr, A.W.; Ball, J.K.; et al. Interferon-Induced Transmembrane Proteins Mediate Viral Evasion in Acute and Chronic Hepatitis C Virus Infection. Hepatology 2019, 70, 1506–1520. [Google Scholar] [CrossRef]
- Grakoui, A.; Shoukry, N.H.; Woollard, D.J.; Han, J.H.; Hanson, H.L.; Ghrayeb, J.; Murthy, K.K.; Rice, C.M.; Walker, C.M. HCV Persistence and Immune Evasion in the Absence of Memory T Cell Help. Science 2003, 302, 659–662. [Google Scholar] [CrossRef]
- Cox, A.L.; Mosbruger, T.; Lauer, G.M.; Pardoll, D.; Thomas, D.L.; Ray, S.C. Comprehensive analyses of CD8+ T cell responses during longitudinal study of acute human hepatitis C. Hepatology 2005, 42, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Neumann-Haefelin, C.; Timm, J.; Spangenberg, H.C.; Wischniowski, N.; Nazarova, N.; Kersting, N.; Roggendorf, M.; Allen, T.M.; Blum, H.E.; Thimme, R. Virological and immunological determinants of intrahepatic virus-specific CD8+ T-cell failure in chronic hepatitis C virus infection. Hepatology 2008, 47, 1824–1836. [Google Scholar] [CrossRef] [PubMed]
- Neumann-Haefelin, C.; McKiernan, S.; Ward, S.; Viazov, S.; Spangenberg, H.C.; Killinger, T.; Baumert, T.F.; Nazarova, N.; Sheridan, I.; Pybus, O.; et al. Dominant influence of an HLA-B27 restricted CD8+ T cell response in mediating HCV clearance and evolution. Hepatology 2006, 43, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Fitzmaurice, K.; Petrovic, D.; Ramamurthy, N.; Simmons, R.; Merani, S.; Gaudieri, S.; Sims, S.; Dempsey, E.; Freitas, E.; Lea, S.; et al. Molecular footprints reveal the impact of the protective HLA-A*03 allele in hepatitis C virus infection. Gut 2011, 60, 1563–1571. [Google Scholar] [CrossRef]
- Thimme, R. T cell immunity to hepatitis C virus: Lessons for a prophylactic vaccine. J. Hepatol. 2021, 74, 220–229. [Google Scholar] [CrossRef]
- Cornberg, M.; Tacke, F.; Karlsen, T.H. Clinical Practice Guidelines of the European Association for the study of the Liver – Advancing methodology but preserving practicability. J. Hepatol. 2019, 70, 5–7. [Google Scholar] [CrossRef]
- Li, G.; De Clercq, E. Current therapy for chronic hepatitis C: The role of direct-acting antivirals. Antiviral Res. 2017, 142, 83–122. [Google Scholar] [CrossRef]
- Ansaldi, F.; Orsi, A.; Sticchi, L.; Bruzzone, B.; Icardi, G. Hepatitis C virus in the new era: Perspectives in epidemiology, prevention, diagnostics and predictors of response to therapy. World J. Gastroenterol. 2014, 20, 9633–9652. [Google Scholar] [CrossRef]
- Jacobson, I.M.; McHutchison, J.G.; Dusheiko, G.; Di Bisceglie, A.M.; Reddy, K.R.; Bzowej, N.H.; Marcellin, P.; Muir, A.J.; Ferenci, P.; Flisiak, R.; et al. Telaprevir for Previously Untreated Chronic Hepatitis C Virus Infection. N. Engl. J. Med. 2011, 364, 2405–2416. [Google Scholar] [CrossRef]
- Poordad, F.; McCone, J.; Bacon, B.R.; Bruno, S.; Manns, M.P.; Sulkowski, M.S.; Jacobson, I.M.; Reddy, K.R.; Goodman, Z.D.; Boparai, N.; et al. Boceprevir for Untreated Chronic HCV Genotype 1 Infection. N. Engl. J. Med. 2011, 364, 1195–1206. [Google Scholar] [CrossRef]
- Manns, M.; Marcellin, P.; Poordad, F.; De Araujo, E.S.A.; Buti, M.; Horsmans, Y.; Janczewska, E.; Villamil, F.; Scott, J.; Peeters, M.; et al. Simeprevir with pegylated interferon alfa 2a or 2b plus ribavirin in treatment-naive patients with chronic hepatitis C virus genotype 1 infection (QUEST-2): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2014, 384, 414–426. [Google Scholar] [CrossRef]
- Lawitz, E.; Mangia, A.; Wyles, D.; Rodriguez-Torres, M.; Hassanein, T.; Gordon, S.C.; Schultz, M.; Davis, M.N.; Kayali, Z.; Reddy, K.R.; et al. Sofosbuvir for Previously Untreated Chronic Hepatitis C Infection. N. Engl. J. Med. 2013, 368, 1878–1887. [Google Scholar] [CrossRef]
- Jacobson, I.M.; Gordon, S.C.; Kowdley, K.V.; Yoshida, E.M.; Rodriguez-Torres, M.; Sulkowski, M.S.; Shiffman, M.L.; Lawitz, E.; Everson, G.; Bennett, M.; et al. Sofosbuvir for Hepatitis C Genotype 2 or 3 in Patients without Treatment Options. N. Engl. J. Med. 2013, 368, 1867–1877. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V.; Gordon, S.C.; Reddy, K.R.; Rossaro, L.; Bernstein, D.E.; Lawitz, E.; Shiffman, M.L.; Schiff, E.; Ghalib, R.; Ryan, M.; et al. Ledipasvir and Sofosbuvir for 8 or 12 Weeks for Chronic HCV without Cirrhosis. N. Engl. J. Med. 2014, 370, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Afdhal, N.; Zeuzem, S.; Kwo, P.; Chojkier, M.; Gitlin, N.; Puoti, M.; Romero-Gomez, M.; Zarski, J.-P.; Agarwal, K.; Buggisch, P.; et al. Ledipasvir and Sofosbuvir for Untreated HCV Genotype 1 Infection. N. Engl. J. Med. 2014, 370, 1889–1898. [Google Scholar] [CrossRef]
- Afdhal, N.; Reddy, K.R.; Nelson, D.R.; Lawitz, E.; Gordon, S.C.; Schiff, E.; Nahass, R.; Ghalib, R.; Gitlin, N.; Herring, R.; et al. Ledipasvir and Sofosbuvir for Previously Treated HCV Genotype 1 Infection. N. Engl. J. Med. 2014, 370, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.; Sulkowski, M.S.; Ghalib, R.; Rodriguez-Torres, M.; Younossi, Z.M.; Corregidor, A.; Dejesus, E.; Pearlman, B.; Rabinovitz, M.; Gitlin, N.; et al. Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non-responders to pegylated interferon and ribavirin and treatment-naive patients: The COSMOS randomised study. Lancet 2014, 384, 1756–1765. [Google Scholar] [CrossRef]
- Poordad, F.; Hezode, C.; Trinh, R.; Kowdley, K.V.; Zeuzem, S.; Agarwal, K.; Shiffman, M.L.; Wedemeyer, H.; Berg, T.; Yoshida, E.M.; et al. ABT-450/r–Ombitasvir and Dasabuvir with Ribavirin for Hepatitis C with Cirrhosis. N. Engl. J. Med. 2014, 370, 1973–1982. [Google Scholar] [CrossRef]
- Feld, J.J.; Kowdley, K.V.; Coakley, E.; Sigal, S.; Nelson, D.R.; Crawford, D.; Weiland, O.; Aguilar, H.; Xiong, J.; Pilot-Matias, T.; et al. Treatment of HCV with ABT-450/r–Ombitasvir and Dasabuvir with Ribavirin. N. Engl. J. Med. 2014, 370, 1594–1603. [Google Scholar] [CrossRef]
- Li, D.K.; Chung, R.T. Overview of direct-acting antiviral drugs and drug resistance of hepatitis C virus. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2019; Volume 1911, pp. 3–32. [Google Scholar]
- Lazarus, J.V.; Roel, E.; Elsharkawy, A.M. Hepatitis c virus epidemiology and the impact of interferon-free hepatitis c virus therapy. Cold Spring Harb. Perspect. Med. 2020, 10, a036913. [Google Scholar] [CrossRef]
- Wang, G.; Dyatkina, N.; Prhavc, M.; Williams, C.; Serebryany, V.; Hu, Y.; Huang, Y.; Wu, X.; Chen, T.; Huang, W.; et al. Synthesis and Anti-HCV Activity of Sugar-Modified Guanosine Analogues: Discovery of AL-611 as an HCV NS5B Polymerase Inhibitor for the Treatment of Chronic Hepatitis C. J. Med. Chem. 2020, 63, 10380–10395. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Dyatkina, N.; Prhavc, M.; Williams, C.; Serebryany, V.; Hu, Y.; Huang, Y.; Wan, J.; Wu, X.; Deval, J.; et al. Synthesis and Anti-HCV Activities of 4′-Fluoro-2′-Substituted Uridine Triphosphates and Nucleotide Prodrugs: Discovery of 4′-Fluoro-2′-C-methyluridine 5′-Phosphoramidate Prodrug (AL-335) for the Treatment of Hepatitis C Infection. J. Med. Chem. 2019, 62, 4555–4570. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.Y.; Shotwell, J.B.; Miller, J.; Price, D.J.; Maynard, A.; Voitenleitner, C.; Mathis, A.; Williams, S.; Pouliot, J.J.; Creech, K.; et al. Design of N-Benzoxaborole Benzofuran GSK8175—Optimization of Human Pharmacokinetics Inspired by Metabolites of a Failed Clinical HCV Inhibitor. J. Med. Chem. 2019, 62, 3254–3267. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Kang, D.; Liu, X. Resurrecting the Condemned: Identification of N-Benzoxaborole Benzofuran GSK8175 as a Clinical Candidate with Reduced Metabolic Liability. J. Med. Chem. 2019, 62, 3251–3253. [Google Scholar] [CrossRef] [PubMed]
- Ramdas, V.; Talwar, R.; Banerjee, M.; Joshi, A.A.; Das, A.K.; Walke, D.S.; Borhade, P.; Dhayagude, U.; Loriya, R.; Gote, G.; et al. Discovery and Characterization of Potent Pan-Genotypic HCV NS5A Inhibitors Containing Novel Tricyclic Central Core Leading to Clinical Candidate. J. Med. Chem. 2019, 62, 10563–10582. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Gai, K.; Qin, H.; Wang, J.; Liu, X.; Cao, Y.; Lu, Q.; Lu, D.; Chen, D.; Shen, H.; et al. Discovery of a Silicon-Containing Pan-Genotype Hepatitis C Virus NS5A Inhibitor. J. Med. Chem. 2020, 63, 5312–5323. [Google Scholar] [CrossRef]
- Kazmierski, W.M.; Baskaran, S.; Walker, J.T.; Miriyala, N.; Meesala, R.; Beesu, M.; Adjabeng, G.; Grimes, R.M.; Hamatake, R.; Leivers, M.R.; et al. GSK2818713, a Novel Biphenylene Scaffold-Based Hepatitis C NS5A Replication Complex Inhibitor with Broad Genotype Coverage. J. Med. Chem. 2020, 63, 4155–4170. [Google Scholar] [CrossRef]
- Jiang, X.; Tan, J.; Wang, Y.; Chen, J.; Li, J.; Li, J.; Jiang, Z.; Quan, Y.; Jin, J.; Li, Y.; et al. 2-((4-Arylpiperazin-1-yl)methyl)benzonitrile Derivatives as Orally Available Inhibitors of Hepatitis C Virus with a Novel Mechanism of Action. J. Med. Chem. 2020, 63, 5972–5989. [Google Scholar] [CrossRef]
- Perales, C. Quasispecies dynamics and clinical significance of hepatitis C virus (HCV) antiviral resistance. Int. J. Antimicrob. Agents 2020, 56, 105562. [Google Scholar] [CrossRef]
- Dietz, J.; Susser, S.; Vermehren, J.; Peiffer, K.H.; Grammatikos, G.; Berger, A.; Ferenci, P.; Buti, M.; Müllhaupt, B.; Hunyady, B.; et al. Patterns of Resistance-Associated Substitutions in Patients With Chronic HCV Infection Following Treatment With Direct-Acting Antivirals. Gastroenterology 2018, 154, 976–988. [Google Scholar] [CrossRef]
- Pawlotsky, J.M. Hepatitis C Virus Resistance to Direct-Acting Antiviral Drugs in Interferon-Free Regimens. Gastroenterology 2016, 151, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. J. Hepatol. 2016, 64, 486–504. [Google Scholar] [CrossRef] [PubMed]
- Childs, K.; Davis, C.; Cannon, M.; Montague, S.; Filipe, A.; Tong, L.; Simmonds, P.; Smith, D.; Thomson, E.C.; Dusheiko, G.; et al. Suboptimal SVR rates in African patients with atypical genotype 1 subtypes: Implications for global elimination of hepatitis C. J. Hepatol. 2019, 71, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, J.M.; Pham, L.V.; Mikkelsen, L.S.; Ghanem, L.; Ramirez, S.; Scheel, T.K.H.; Carlsen, T.H.R.; Bukh, J. Efficacy of NS5A Inhibitors Against Hepatitis C Virus Genotypes 1–7 and Escape Variants. Gastroenterology 2018, 154, 1435–1448. [Google Scholar] [CrossRef]
- Pawlotsky, J.M.; Negro, F.; Aghemo, A.; Berenguer, M.; Dalgard, O.; Dusheiko, G.; Marra, F.; Puoti, M.; Wedemeyer, H. EASL recommendations on treatment of hepatitis C: Final update of the series. J. Hepatol. 2020, 73, 1170–1218. [Google Scholar] [CrossRef]
- Aparicio-Puerta, E.; Lebrón, R.; Rueda, A.; Gómez-Martín, C.; Giannoukakos, S.; Jaspez, D.; Medina, J.M.; Zubkovic, A.; Jurak, I.; Fromm, B.; et al. SRNAbench and sRNAtoolbox 2019: Intuitive fast small RNA profiling and differential expression. Nucleic Acids Res. 2019, 47, W530–W535. [Google Scholar] [CrossRef]
- Chevaliez, S.; Rodriguez, C.; Pawlotsky, J.M. New virologic tools for management of chronic hepatitis B and C. Gastroenterology 2012, 142, 1303–1313. [Google Scholar] [CrossRef]
- Jensen, S.B.; Fahnøe, U.; Pham, L.V.; Serre, S.B.N.; Tang, Q.; Ghanem, L.; Pedersen, M.S.; Ramirez, S.; Humes, D.; Pihl, A.F.; et al. Evolutionary Pathways to Persistence of Highly Fit and Resistant Hepatitis C Virus Protease Inhibitor Escape Variants. Hepatology 2019, 70, 771–787. [Google Scholar] [CrossRef]
- Bourlière, M.; Gordon, S.C.; Flamm, S.L.; Cooper, C.L.; Ramji, A.; Tong, M.; Ravendhran, N.; Vierling, J.M.; Tran, T.T.; Pianko, S.; et al. Sofosbuvir, Velpatasvir, and Voxilaprevir for Previously Treated HCV Infection. N. Engl. J. Med. 2017, 376, 2134–2146. [Google Scholar] [CrossRef]
- Poordad, F.; Pol, S.; Asatryan, A.; Buti, M.; Shaw, D.; Hézode, C.; Felizarta, F.; Reindollar, R.W.; Gordon, S.C.; Pianko, S.; et al. Glecaprevir/Pibrentasvir in patients with hepatitis C virus genotype 1 or 4 and past direct-acting antiviral treatment failure. Hepatology 2018, 67, 1253–1260. [Google Scholar] [CrossRef]
- Nguyen, D.; Smith, D.; Vaughan-Jackson, A.; Magri, A.; Barnes, E.; Simmonds, P. Efficacy of NS5A inhibitors against unusual and potentially difficult-to-treat HCV subtypes commonly found in sub-Saharan Africa and South East Asia. J. Hepatol. 2020, 73, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, A.; Gimson, A.; Agarwal, K.; Aldersley, M.; Bathgate, A.; MacDonald, D.; McPherson, S.; Mutimer, D.; Gelson, W. Liver transplant listing for hepatitis C-associated cirrhosis and hepatocellular carcinoma has fallen in the United Kingdom since the introduction of direct-acting antiviral therapy. J. Viral Hepat. 2019, 26, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Carrat, F.; Fontaine, H.; Dorival, C.; Simony, M.; Diallo, A.; Hezode, C.; De Ledinghen, V.; Larrey, D.; Haour, G.; Bronowicki, J.P.; et al. Clinical outcomes in patients with chronic hepatitis C after direct-acting antiviral treatment: A prospective cohort study. Lancet 2019, 393, 1453–1464. [Google Scholar] [CrossRef]
- Pawlotsky, J.M. Retreatment of Hepatitis C Virus-Infected Patients with Direct-Acting Antiviral Failures. Semin. Liver Dis. 2019, 39, 354–368. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| DAA Virus Target | DAA Name |
|---|---|
| NS3/4A inhibitor | Telaprevir a |
| Boceprevir a | |
| Semiprevir | |
| Paritaprevir | |
| Grazoprevir | |
| Voxilaprevir | |
| Glecaprevir | |
| NS3/4A booster | Ritonavir |
| NS5A inhibitor | Ledivaspir |
| Ombitasvir | |
| Daclatasvir | |
| Elbasvir | |
| Velpatasvir | |
| Pibrentasvir | |
| NS5B inhibitor | Sofosbuvir |
| Dasabuvir |
| Genome Region Drug Class a | Amino Acid Position | Genotype | ||||||
|---|---|---|---|---|---|---|---|---|
| 1a | 1b | 2 | 3 | 4 | 5 | 6 | ||
| NS3 Protease Inhibitors: Semiprevir, Paritaprevir, Grazoprevir, Voxilaprevir and Glecaprevir b | ||||||||
| 36 | V36A/C/F/G/L/M | V36A/C/G/L/M | V36I | |||||
| 41 | Q41R | Q41R | Q41K | Q41R | Q41K/R | |||
| 43 | F43I/L/S/V | F43I/S/V | F43V | |||||
| 54 | T54A/S | T54A/C/G/S | ||||||
| 55 | V55I | V55A | V55A/I | |||||
| 56 | Y56H | Y56H/L/F | Y56H/F | Y56H | Y56H | Y56H | ||
| 80 | Q80K/L/R | Q80H/K/L/R | Q80K/R | Q80R | L80K/Q | |||
| 122 | S122G/N/R | S122A/D/G/I/N/R/T | S122T | |||||
| 155 | R155G/I/K/M/Q/S/T/V/W | R155C/G/I/K/L/Q/M/S/T/W | R155K | R155C/K | R155K | |||
| 156 | A156G/P/S/T/V | A156G/P/S/T/V | A156L/M/T/V | A156G/P/T/V | A156G/H/K/L/S/T/V | A156T/V | A156T/V | |
| 158 | V158I | V158I | ||||||
| 166 | A166S/T/Y | |||||||
| 168 | D168A/C/E/F/G/H/I/K/L/N/Q/R/T/V/Y | D168A/C/E/F/G/H/I/K/L/N/Q/T/V/Y | D168A/E/F/G/H/N/S/T/V/Y | Q168H/K/L/R | D168A/E/G/H/T/V | D168A/E/H/K/R/V/Y | D168A/E/G/H/V/Y | |
| 170 | I/V170T/V | I/V170A/L/T | I170V | |||||
| 175 | M175L | |||||||
| NS5A Inhibitors: Ledivaspir, Ombitasvir, Daclatasvir, Elbasvir, Velpatasvir and Pibrentasvir | ||||||||
| 24 | K24E/QR/T | Q24K | T24A/S | S24F | ||||
| 26 | K26E | |||||||
| 28 | M28A/G/S/T/V | L28A/M/T | L/F28C/S | M28T/K | L28M/S/T/V | L28I | F/L28A/I/L/M/T/V | |
| 29 | 29 P29R | P29S, del29 | P29S | |||||
| 30 | Q30C/D/E/G/H/K/L/N/R/T/Y, del30 | R30G/H/P/Q/S | L30H/S | A30D/E/K/S | L30F/G/H/R/S | Q30H | R30E/H/N/S | |
| 31 | L31I/F/M/P/V | L31F/I/M/V/W | L31I/M/V | L31F/I/M/P/V | M/L31I/V | L31F/I/V | L31I/M/V | |
| 32 | P32L/S, del32 | P32F/L/S, del32 | P32L | P32A/L/Q/R/S | ||||
| 38 | S38F | |||||||
| 58 | H58C/D/L/P/R | P58A/D/L/S/R/T | T58A/P/S | T58A/G/H/N/S | ||||
| 62 | Q/E62D | S62L | ||||||
| 92 | A92K/T | A92E/K/T/V | C92R/S/T/W | E92K | E92T | |||
| 93 | Y93C/F/H/L/N/R/S/T/W | Y93C/H/N/R/S/T | Y93F/N/H | Y93H/N/S | Y93C/H/N/S/R/W | T93A/H/N/S | ||
| NS5B Non-nucleoside Inhibitors: Dasabuvir | ||||||||
| 314 | L314H | |||||||
| 316 | C316Y | C316H/N/Y/W | ||||||
| 368 | S368T | |||||||
| 395 | A395G | |||||||
| 411 | ||||||||
| 414 | M414I/T/V | M414I/T/V | ||||||
| 445 | C445F/Y | |||||||
| 446 | E446K/Q | |||||||
| 448 | Y448C/H | Y448C/H | ||||||
| 553 | A553T/V | A553V | ||||||
| 554 | G554S | G554S | ||||||
| 555 | Y555H | |||||||
| 556 | S556G/R | S556G/R | ||||||
| 557 | G557R | |||||||
| 558 | G558R | G558R | ||||||
| 559 | D559G/N | D559G/N | ||||||
| 561 | Y561H/N | |||||||
| 565 | S565F | |||||||
| NS5B Nucleotide analogue Inhibitors: Sofosbuvir | ||||||||
| 150 | A150V | |||||||
| 159 | L159F | L159F | L159F | L159F | ||||
| 206 | K206E | |||||||
| 282 | S282G/R/T | S282G/R/T | S282G/R/T | S282G/R/T | S282C/G/R/T | S282G/R/T | S282G/R/T | |
| 316 | C316H/R | C316F/H/N | ||||||
| 320 | L320I/F/V | |||||||
| 321 | V321A | V321I | V321A | V321A | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez, M.A.; Franco, S. Therapy Implications of Hepatitis C Virus Genetic Diversity. Viruses 2021, 13, 41. https://doi.org/10.3390/v13010041
Martinez MA, Franco S. Therapy Implications of Hepatitis C Virus Genetic Diversity. Viruses. 2021; 13(1):41. https://doi.org/10.3390/v13010041
Chicago/Turabian StyleMartinez, Miguel Angel, and Sandra Franco. 2021. "Therapy Implications of Hepatitis C Virus Genetic Diversity" Viruses 13, no. 1: 41. https://doi.org/10.3390/v13010041
APA StyleMartinez, M. A., & Franco, S. (2021). Therapy Implications of Hepatitis C Virus Genetic Diversity. Viruses, 13(1), 41. https://doi.org/10.3390/v13010041