
Interface between Bats and Pigs in Heavy Pig Production

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ecological Interface between Bats and Pigs
2.2. Presence of Viruses in Bats and Swines from Piggeries
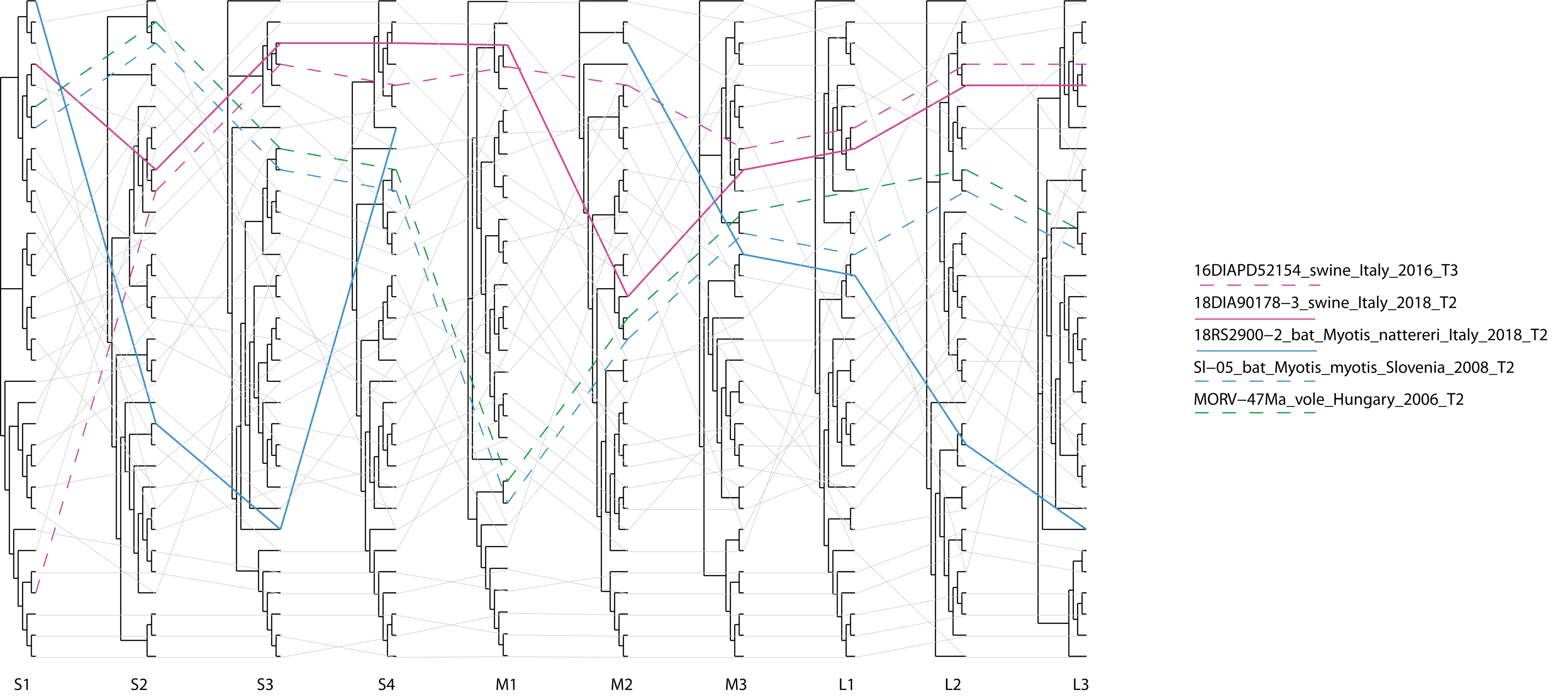
2.3. Phylogenetic Analyses
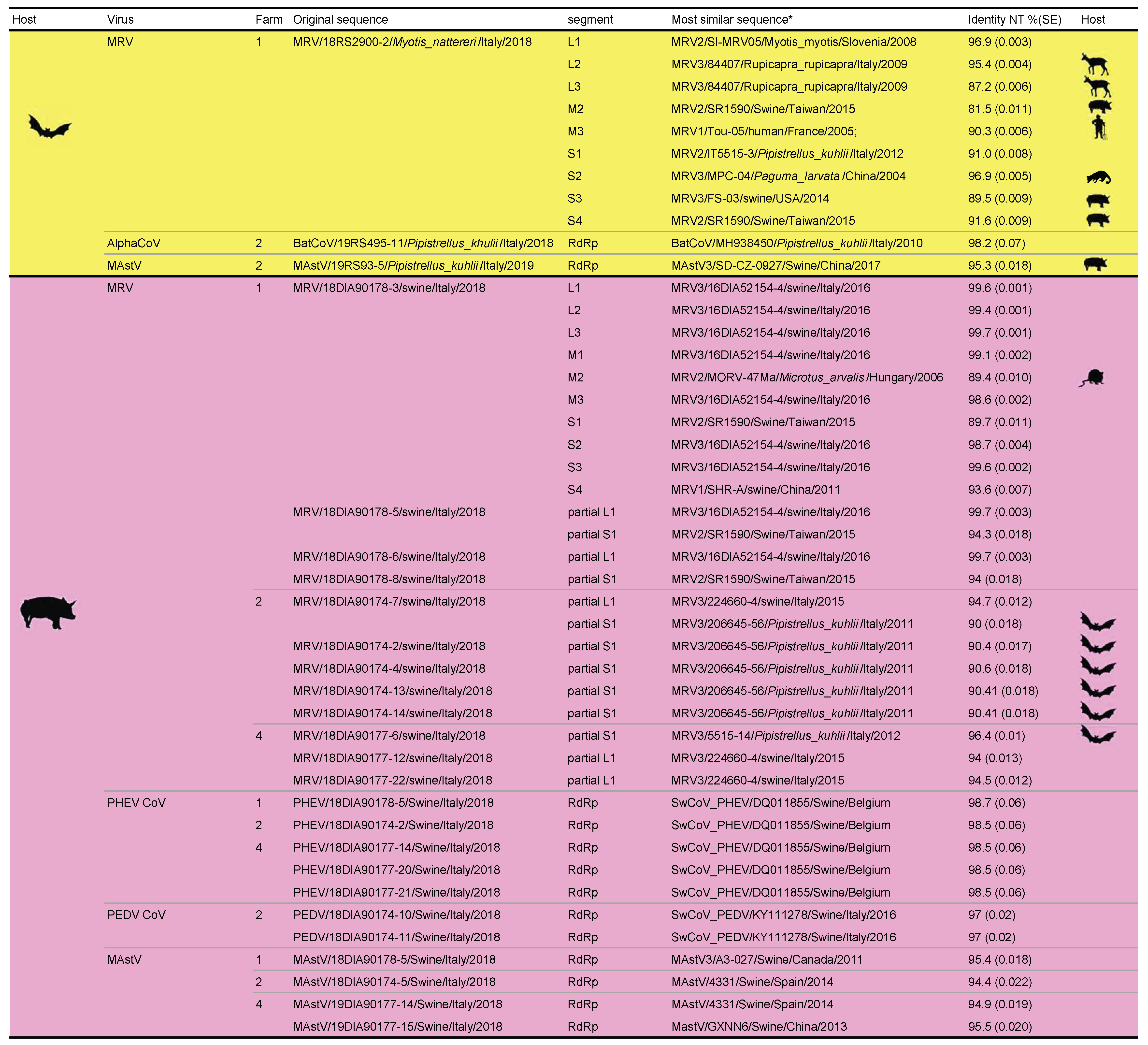
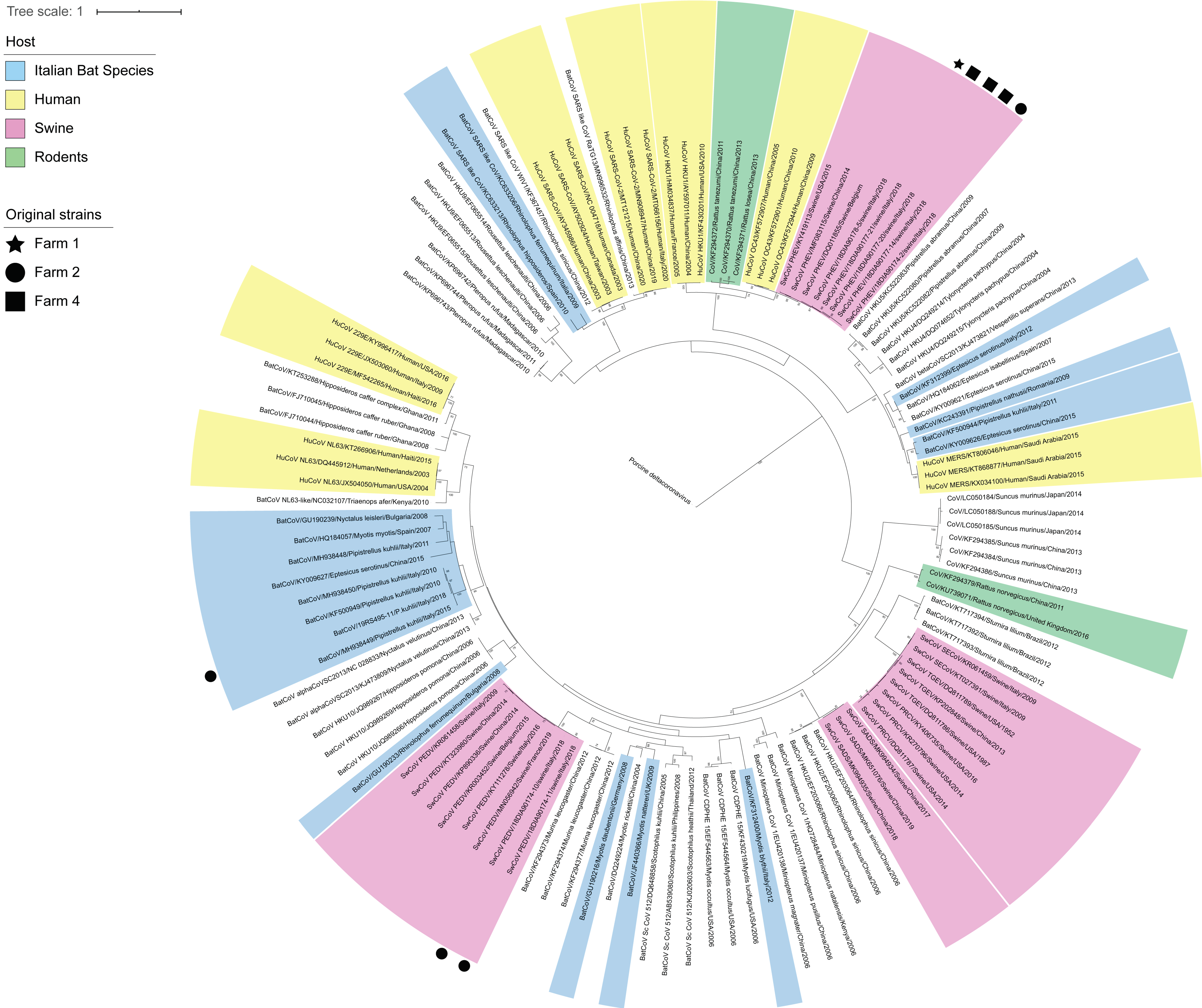
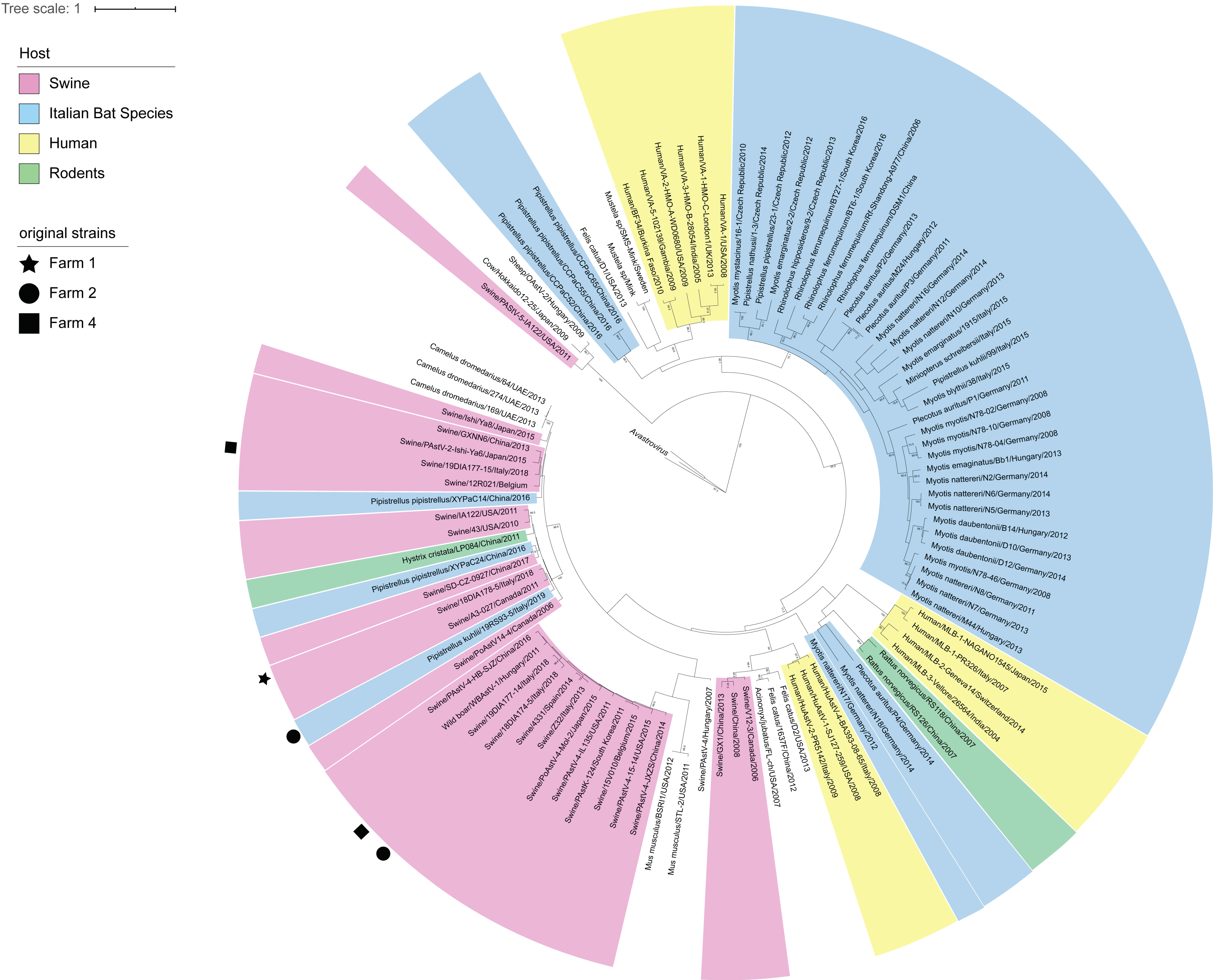
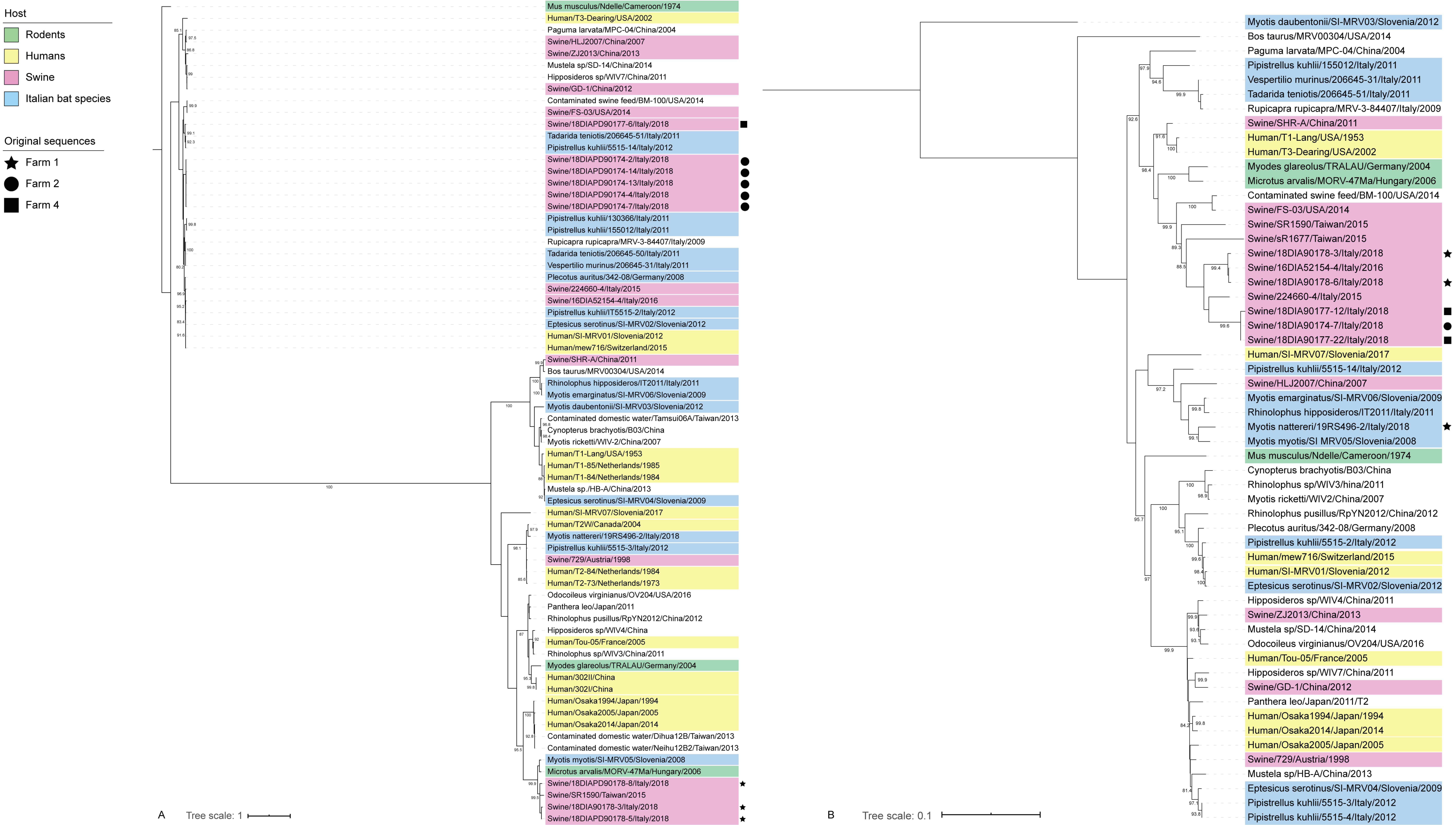
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Plowright, R.K.; Foley, P.; Field, H.E.; Dobson, A.P.; Foley, J.E.; Eby, P.; Daszak, P. Urban habituation, ecological connectivity and epidemic dampening: The emergence of Hendra virus from flying foxes (Pteropus spp.). Proc. R. Soc. B Biol. Sci. 2011, 278, 3703–3712. [Google Scholar] [CrossRef] [PubMed]
- Daszak, P.; Olival, K.J.; Li, H. A strategy to prevent future epidemics similar to the 2019-nCoV outbreak. Biosaf. Health 2020, 2, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Kock, R.A.; Begovoeva, M.; Ansumana, R.; Suluku, R. Searching for the source of Ebola: The elusive factors driving its spillover into humans during the West African outbreak of 2013–2016. Rev. Sci. Tech. 2019, 38, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Rulli, M.C.; D’Odorico, P.; Galli, N.; Hayman, D.T.S. Land Use Change and Coronavirus Emergence Risk. medRxiv 2020. [Google Scholar] [CrossRef]
- Ancillotto, L.; Santini, L.; Ranc, N.; Maiorano, L.; Russo, D. Extraordinary range expansion in a common bat: The potential roles of climate change and urbanisation. Sci. Nat. 2016, 103, 15. [Google Scholar] [CrossRef]
- Plowright, R.K.; Eby, P.; Hudson, P.J.; Smith, I.L.; Westcott, D.; Bryden, W.L.; Middleton, D.; Reid, P.A.; McFarlane, R.A.; Martin, G.; et al. Ecological dynamics of emerging bat virus spillover. Proc. R. Soc. B 2015, 282, 20142124. [Google Scholar] [CrossRef]
- Becker, D.J.; Czirják, G.; Volokhov, D.V.; Bentz, A.B.; Carrera, J.E.; Camus, M.S.; Navara, K.J.; Chizhikov, V.E.; Fenton, M.B.; Simmons, N.B.; et al. Livestock abundance predicts vampire bat demography, immune profiles and bacterial infection risk. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170089. [Google Scholar] [CrossRef]
- Kessler, M.K.; Becker, D.J.; Peel, A.J.; Justice, N.V.; Lunn, T.; Crowley, D.E.; Jones, D.N.; Eby, P.; Cecilia, A.S. Changing resource landscapes and spillover of henipaviruses. Ann. N. Y. Acad. Sci. 2018, 1429, 78–99. [Google Scholar] [CrossRef]
- Pulliam, J.R.C.; Epstein, J.H.; Dushoff, J.; Rahman, S.A.; Bunning, M.; Jamaluddin, A.A.; Hyatt, A.D.; Field, H.E.; Dobson, A.P.; Daszak, P. Agricultural intensification, priming for persistence and the emergence of Nipah virus: A lethal bat-borne zoonosis. J. R. Soc. Interface 2012, 9, 89–101. [Google Scholar] [CrossRef]
- Johnson, N.; Aréchiga-Ceballos, N.; Aguilar-Setien, A. Vampire bat rabies: Ecology, epidemiology and control. Viruses 2014, 6, 1911–1928. [Google Scholar] [CrossRef] [PubMed]
- Parrish, C.R.; Holmes, E.C.; Morens, D.M.; Park, E.; Burke, D.S.; Calisher, C.H.; Laughlin, C.A.; Saif, L.J.; Daszak, P. Cross-Species Virus Transmission and the Emergence of New Epidemic Diseases Cross-Species Virus Transmission and the Emergence of New Epidemic Diseases. Microbiol. Mol. Biol. Rev. 2008, 72, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.L.N.; Leach, M.; Waldman, L.; MacGregor, H.; Fooks, A.R.; Jones, K.E.; Restif, O.; Dechmann, D.; Hayman, D.T.S.; Baker, K.S.; et al. A framework for the study of zoonotic disease emergence and its drivers: Spillover of bat pathogens as a case study. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2881–2892. [Google Scholar] [CrossRef] [PubMed]
- FAO Livestock Production. World Agriculture:Towards 2015/2030; Food and Agriculture Organization of the United Nations (FAO): Rome, Italy, 2003; pp. 158–176. [Google Scholar]
- Borkenhagen, L.K.; Fieldhouse, J.K.; Gray, G.C.; Zemke, J.; Bailey, E.S.; Zhang, D.; Choi, J.Y. The continual threat of influenza virus infections at the human–animal interface. Evol. Med. Public Health 2018, 2018, 192–198. [Google Scholar]
- Chua, K.B.; Bellini, W.; Rota, P.A.; Harcourt, B.H.; Tamin, A.; Lam, S.K.; Kslazek, T.G.; Rollin, P.E.; Zaki, S.R.; Shieh, W.J.; et al. Nipah virus: A recently emergent deadly paramyxovirus. Science 2000, 288, 1432–1435. [Google Scholar] [CrossRef]
- Peiris, J.S.M.; Poon, L.L.M.; Guan, Y. Emergence of a novel swine-origin influenza A virus (S-OIV) H1N1 virus in humans. J. Clin. Virol. 2009, 45, 169–173. [Google Scholar] [CrossRef]
- Moratelli, R.; Calisher, C.H. Bats and zoonotic viruses: Can we confidently link bats with emerging deadly viruses? Mem. Inst. Oswaldo Cruz 2015, 110, 1–22. [Google Scholar] [CrossRef]
- Wang, L.-F.; Anderson, D.E. Viruses in bats and potential spillover to animals and humans. Curr. Opin. Virol. 2019, 34, 79–89. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Luo, Y.; Guo, H.; Anderson, D.E.; Zhang, Y.-W.; Zhou, L.; Lan, T.; Wu, Z.-X.; Li, J.; Tong, Y.-G.; Chen, J.-W.; et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 2018, 556, 255–258. [Google Scholar]
- Kohl, C.; Kurth, A. European bats as carriers of viruses with zoonotic potential. Viruses 2014, 6, 3110–3128. [Google Scholar] [CrossRef] [PubMed]
- Shipley, R.; Wright, E.; Selden, D.; Wu, G.; Aegerter, J.; Fooks, A.R.; Banyard, A.C. Bats and Viruses: Emergence of Novel Lyssaviruses and Association of Bats with Viral Zoonoses in the EU. Trop. Med. Infect. Dis. 2019, 4, 31. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Li, X.; Lau, S.; Woo, P. Global Epidemiology of Bat Coronaviruses. Viruses 2019, 11, 174. [Google Scholar] [CrossRef] [PubMed]
- Afelt, A.; Lacroix, A.; Zawadzka-Pawlewska, U.; Pokojski, W.; Buchy, P.; Frutos, R. Distribution of bat-borne viruses and environment patterns. Infect. Genet. Evol. 2018, 58, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; dos Reis, V.P.; Balkema-Buschmann, A. Bat astroviruses: Towards understanding the transmission dynamics of a neglected virus family. Viruses 2017, 9, 34. [Google Scholar] [CrossRef]
- De Benedictis, P.; Marciano, S.; Scaravelli, D.; Priori, P.; Zecchin, B.; Capua, I.; Monne, I.; Cattoli, G. Alpha and lineage C betaCoV infections in Italian bats. Virus Genes 2014, 48, 366–371. [Google Scholar] [CrossRef]
- Lelli, D.; Moreno, A.; Lavazza, A.; Bresaola, M.; Canelli, E.; Boniotti, M.B.; Cordioli, P. Identification of Mammalian Orthoreovirus Type 3 in Italian Bats. Zoonoses Public Health 2013, 60, 84–92. [Google Scholar] [CrossRef]
- Wohlgemuth, N.; Honce, R.; Schultz-Cherry, S. Astrovirus evolution and emergence. Infect. Genet. Evol. 2019, 69, 30–37. [Google Scholar] [CrossRef]
- Amoroso, M.G.; Russo, D.; Lanave, G.; Cistrone, L.; Pratelli, A.; Martella, V.; Galiero, G.; Decaro, N.; Fusco, G. Detection and phylogenetic characterization of astroviruses in insectivorous bats from Central-Southern Italy. Zoonoses Public Health 2018, 65, 702–710. [Google Scholar] [CrossRef]
- Bosi, P.; Russo, V. The production of the heavy pig for high quality processed products. Ital. J. Anim. Sci. 2010, 3, 309–321. [Google Scholar] [CrossRef]
- Fenton, M.B.; Grinnel, A.D.; Popper, A.N.; Fay, R.R. Bat Bioacoustics; Springer: New York, NY, USA, 2016; ISBN 9781493935253. [Google Scholar]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Decaro, N.; Campolo, M.; Desario, C.; Ricci, D.; Camero, M.; Lorusso, E.; Elia, G.; Lavazza, A.; Martella, V.; Buonavoglia, C. Virological and molecular characterization of a mammalian orthoreovirus type 3 strain isolated from a dog in Italy. Vet. Microbiol. 2005, 109, 19–27. [Google Scholar] [CrossRef] [PubMed]
- De Souza Luna, L.K.; Heiser, V.; Regamey, N.; Panning, M.; Drexler, J.F.; Mulangu, S.; Poon, L.; Baumgarte, S.; Haijema, B.J.; Kaiser, L.; et al. Generic detection of coronaviruses and differentiation at the prototype strain level by reverse transcription-PCR and nonfluorescent low-density microarray. J. Clin. Microbiol. 2007, 45, 1049–1052. [Google Scholar] [CrossRef]
- Chu, D.K.W.; Poon, L.L.M.; Guan, Y.; Peiris, J.S.M. Novel astroviruses in insectivorous bats. J. Virol. 2008, 82, 9107–9114. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Gloza-Rausch, F.; Glende, J.; Corman, V.M.; Muth, D.; Goettsche, M.; Seebens, A.; Niedrig, M.; Pfefferle, S.; Yordanov, S.; et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J. Virol. 2010, 84, 11336–11349. [Google Scholar] [CrossRef]
- Cavicchio, L.; Tassoni, L.; Zamperin, G.; Campalto, M. Unexpected Genetic Diversity of Two Novel Swine MRVs in Italy. Viruses 2020, 12, 574. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, 465–469. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Leopardi, S.; Terregino, C.; De Benedictis, P. Silent circulation of coronaviruses in pigs. Vet. Rec. 2020, 186, 323. [Google Scholar] [CrossRef] [PubMed]
- Toffoli, R.; Culasso, P.; Locatelli, A.G.; Giraudo, L. Bats of Alpi Marittime Nature Park (North West Italy) and Site of Community Importance IT1160056: Distribution and status. Nat. Hist. Sci. 2016, 3, 3. [Google Scholar] [CrossRef][Green Version]
- Ancillotto, L.; Ariano, A.; Nardone, V.; Budinski, I.; Rydell, J.; Russo, D. Effects of free-ranging cattle and landscape complexity on bat foraging: Implications for bat conservation and livestock management. Agric. Ecosyst. Environ. 2017, 241, 54–61. [Google Scholar] [CrossRef]
- Afelt, A.; Devaux, C.; Serra-Cobo, J.; Frutos, R. Bats, Bat-Borne Viruses, and Environmental Changes. In Bats; Mikkola, H., Ed.; InTechOpen: London, UK, 2018; pp. 113–132. [Google Scholar]
- Leopardi, S.; Holmes, E.C.; Gastaldelli, M.; Tassoni, L.; Priori, P.; Scaravelli, D.; Zamperin, G.; De Benedictis, P. Interplay between co-divergence and cross-species transmission in the evolutionary history of bat coronaviruses. Infect. Genet. Evol. 2018, 58, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, A.T.; Fooks, A.R.; Hayman, D.T.S.; Horton, D.L.; Müller, T.; Plowright, R.; Peel, A.J.; Bowen, R.; Wood, J.L.N.; Mills, J.; et al. Deciphering serology to understand the ecology of infectious diseases in wildlife. Ecohealth 2013, 10, 298–313. [Google Scholar] [CrossRef]
- Kohl, C.; Lesnik, R.; Brinkmann, A.; Ebinger, A.; Radonić, A.; Nitsche, A.; Mühldorfer, K.; Wibbelt, G.; Kurth, A. Isolation and characterization of three mammalian orthoreoviruses from European bats. PLoS ONE 2012, 7, e43106. [Google Scholar] [CrossRef]
- Nagli, T.; Rihtari, D.; Hostnik, P.; Koren, S.; Kutnjak, D.; Steyer, A. Identification of novel reassortant mammalian orthoreoviruses from bats in Slovenia. BMC Vet. Res. 2018, 14, 1–10. [Google Scholar] [CrossRef]
- Backhans, A.; Fellström, C. Rodents on pig and chicken farms—A potential threat to human and animal health. Infect. Ecol. Epidemiol. 2012, 2, 17093. [Google Scholar] [CrossRef]
- Dhingra, M.S.; Artois, J.; Dellicour, S.; Lemey, P.; Dauphin, G.; Von Dobschuetz, S.; Van Boeckel, T.P.; Castellan, D.M.; Morzaria, S.; Gilbert, M. Geographical and Historical Patterns in the Emergences of Novel Highly Pathogenic Avian Influenza (HPAI) H5 and H7 Viruses in Poultry. Front. Vet. Sci. 2018, 5, 1–14. [Google Scholar] [CrossRef]
- Greger, M. The human/animal interface: Emergence and resurgence of zoonotic infectious diseases. Crit. Rev. Microbiol. 2007, 33, 243–299. [Google Scholar] [CrossRef]
- Field, H.; Young, P.; Yob, J.M.; Mills, J.; Hall, L.; Mackenzie, J. The natural history of Hendra and Nipah viruses. Microbes Infect. 2001, 3, 307–314. [Google Scholar] [CrossRef]
- Hermann, L.; Embree, J.; Hazelton, P.; Wells, B.; Coombs, K. Reovirus Type 2 Isolated from Cerebrospinal Fluid. Pediatr. Infect. Dis. J. 2004, 23, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Joffrin, L.; Dietrich, M.; Mavingui, P.; Lebarbenchon, C. Bat pathogens hit the road: But which one? PLoS Pathog. 2018, 14, e1007134. [Google Scholar] [CrossRef] [PubMed]
- Conzade, R.; Grant, R.; Malik, M.R.; Elkholy, A.; Elhakim, M.; Samhouri, D.; Ben Embarek, P.K.; Van Kerkhove, M.D. Reported direct and indirect contact with dromedary camels among laboratory-confirmed MERS-CoV cases. Viruses 2018, 10, 425. [Google Scholar] [CrossRef]
- Ithete, N.L.; Stoffberg, S.; Corman, V.M.; Cottontail, V.M.; Richards, L.R.; Schoeman, M.C.; Drosten, C.; Drexler, J.F.; Preiser, W. Close relative of human middle east respiratory syndrome coronavirus in bat, South Africa. Emerg. Infect. Dis. 2013, 19, 1697–1699. [Google Scholar] [CrossRef]
- Qin, P.; Li, H.; Wang, J.W.; Wang, B.; Xie, R.H.; Xu, H.; Zhao, L.Y.; Li, L.; Pan, Y.; Song, Y.; et al. Genetic and pathogenic characterization of a novel reassortant mammalian orthoreovirus 3 (MRV3) from a diarrheic piglet and seroepidemiological survey of MRV3 in diarrheic pigs from east China. Vet. Microbiol. 2017, 208, 126–136. [Google Scholar] [CrossRef]
- Mora-Díaz, J.C.; Magtoto, R.; Houston, E.; Baum, D.; Carrillo-Ávila, J.A.; Temeeyasen, G.; Zimmerman, J.; Piñeyro, P.; Giménez-Lirola, L. Detecting and Monitoring Porcine Hemagglutinating Encephalomyelitis Virus, an Underresearched Betacoronavirus. mSphere 2020, 5, 1–8. [Google Scholar] [CrossRef]
- Rickli, C.I. Detection of Potentially Commensal Viruses and Associated Bacteria in Pigs by Metagenomic Analysis. Inaugural Dissertation, University of Zurich Vetsuisse faculty, Rämistrasse, Zurich, 2020. [Google Scholar]
- De Benedictis, P.; Schultz-Cherry, S.; Burnham, A.; Cattoli, G. Astrovirus infections in humans and animals—Molecular biology, genetic diversity, and interspecies transmissions. Infect. Genet. Evol. 2011, 11, 1529–1544. [Google Scholar] [CrossRef]
- Donato, C.; Vijaykrishna, D. The broad host range and genetic diversity of mammalian and avian astroviruses. Viruses 2017, 9, 102. [Google Scholar] [CrossRef]
- Leopardi, S.; Blake, D.; Puechmaille, S.J. White-Nose Syndrome fungus introduced from Europe to North America. Curr. Biol. 2015, 25, R217–R219. [Google Scholar] [CrossRef]
- Bernard, R.F.; Reichard, J.D.; Coleman, J.T.H.; Blackwood, J.C.; Verant, M.L.; Segers, J.L.; Lorch, J.M.; White, J.; Moore, M.S.; Russell, A.L.; et al. Identifying research needs to inform white-nose syndrome management decisions. Conserv. Sci. Pract. 2020, 2, 1–17. [Google Scholar]
- Olival, K.J.; Cryan, P.M.; Amman, B.R.; Baric, R.S.; Blehert, D.S.; Brook, C.E.; Calisher, C.H.; Castle, K.T.; Coleman, J.T.H.; Daszak, P.; et al. Possibility for reverse zoonotic transmission of sars-cov-2 to free-ranging wildlife: A case study of bats. PLoS Pathog. 2020, 16, 1–19. [Google Scholar] [CrossRef] [PubMed]
- IUCN SSC Bat Specialist Group; Nuñez, G.B.; Cunningham, A.; Moise, E.; Fils, B.; Frick, W.; Islam, N.; Jolliffe, T.; Kading, R.; Kepel, A.; et al. BSG Recommended Strategy for Researchers to Reduce the Risk of Transmission of SARS-CoV-2 from Humans to Bats. Available online: https://www.iucnbsg.org/bsg-publications.html (accessed on 21 December 2020).
- IUCN The IUCN Red List of Threatened Species Version 2020-3. Available online: www.iucnredlist.org (accessed on 21 December 2020).
- Park, K.J. Mitigating the impacts of agriculture on biodiversity: Bats and their potential role as bioindicators. Mamm. Biol. 2015, 80, 191–204. [Google Scholar] [CrossRef]
- Meerburg, B.G.; Vermeer, H.M.; Kijlstra, A. Controlling risks of pathogen transmission by flies on organic pig farms: A review. Outlook Agric. 2007, 36, 193–197. [Google Scholar] [CrossRef]
- Sarfraz, M.; Keddie, A.B.; Dosdall, L.M. Biological control of the diamondback moth, Plutella xylostella: A review. Biocontrol Sci. Technol. 2005, 15, 763–789. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Farm n. | Bat Activity (Passes/Night) | Feeding Activity (%) | Social Activity (%) | Species Richness (n) |
|---|---|---|---|---|
| 1 | 142 | 17.6 | 4.2 | 9 |
| 2 | 67 | 28.4 | 4.5 | 9 |
| 3 | 83 | 72.3 | 22.9 | 5 |
| 4 | 93 | 14 | 1.1 | 6 |
| 96.25 mean/farm | 33.1 mean/farm | 8.2 mean/farm | 13 species in total |
| Farm n. | Species | Activity (p/h) | Occurrence (%) | Feeding Activity (%) | Social Activity (%) | Presumed/Expected Activity |
|---|---|---|---|---|---|---|
| 1 | H. savii | 14.8 | 21.13 | 0 | 0 | hP; R |
| P. kuhlii | 16.6 | 23.24 | 54.6 | 15.2 | hP; hFa; R | |
| P. pipistrellus | 19.0 | 26.76 | 2.6 | 0 | hP; oF; R | |
| E. serotinus | 6.8 | 9.86 | 14.3 | 7.1 | oP; hFa; R | |
| N. leisleri | 8.5 | 11.97 | 23.5 | 0 | oP; Fa; R | |
| M. emarginatus | 0.5 | 0.70 | 0 | 0 | rP; R | |
| M. mystacinus | 0.2 | 0.70 | 0 | 0 | rP | |
| M. nattereri | 1.1 | 2.11 | 0 | 0 | rP; R * | |
| M. myotis/blythii | 0.2 | 3.52 | 0 | 0 | rP | |
| 2 | H. savii | 4.6 | 17.39 | 83.3 | 16.7 | oP; hFa |
| P. kuhlii | 5.4 | 20.29 | 21.4 | 0 | oP; Fa; R * | |
| P. pipistrellus | 6.5 | 24.64 | 23.5 | 0 | oP; Fa | |
| E. serotinus | 0.8 | 2.90 | 50 | 50 | rP; hFa | |
| N. leisleri | 0.2 | 13.04 | 11.1 | 0 | oP; Fa | |
| Nyctalus sp. | 0.2 | 1.45 | 0 | 0 | rP | |
| M. daubentonii | 0.9 | 2.90 | 0 | 0 | rP | |
| M. nattereri | 4.5 | 11.59 | 0 | 0 | oP | |
| R. ferrumequinum | 1.6 | 5.80 | 0 | 0 | oP | |
| 3 | H. savii | 1.9 | 7.23 | 100 | 16.7 | oP; hFa; R |
| P. kuhlii | 4.5 | 14.46 | 0 | 0 | oP | |
| P. pipistrellus | 3.7 | 69.88 | 93.1 | 31.0 | hP; hFa; R | |
| N. leisleri | 2 | 6.02 | 0 | 0 | oP | |
| R. hipposideros | 0.6 | 2.41 | 0 | 0 | rP; R | |
| 4 | H. savii | 10.3 | 31.18 | 0 | 0 | hP |
| P. kuhlii | 6.4 | 19.35 | 5.5 | 0 | oP; oF | |
| P. pipistrellus | 13.3 | 39.78 | 32.4 | 2.7 | hP; hFa | |
| T. teniotis | 0.8 | 2.15 | 0 | 0 | rP | |
| M. emarginatus | 1.9 | 6.45 | 0 | 0 | oP | |
| Myotis sp. | 0.3 | 1.08 | 0 | 0 | rP | |
| Distribution (% farms) | Mean/farm | |||||
| 50 | E. serotinus | 3.8 | 6.4 | 32.1 | 28.6 | P; F; R |
| 100 | H. savii | 8 | 19.2 | 45.8 | 8.3 | P; F; R |
| 25 | M. daubentonii | 0.9 | 2.9 | 0 | 0 | P |
| 50 | M. emarginatus | 1.2 | 3.6 | 0 | 0 | P; R |
| 25 | M. myotis/blythii | 0.2 | 3.5 | 0 | 0 | P |
| 25 | M. mystacinus | 0.2 | 0.7 | 0 | 0 | P |
| 50 | M. nattereri | 2.8 | 6.9 | 0 | 0 | P |
| 75 | N. leisleri | 3.6 | 10.3 | 11.5 | 0 | P; F; R |
| 100 | P. kuhlii | 8.2 | 19.3 | 20.4 | 3.8 | P; F; R |
| 100 | P. pipistrellus | 10.6 | 40.3 | 37.9 | 8.4 | P; F; R |
| 25 | R. ferrumequinum | 1.6 | 5.8 | 0 | 0 | P |
| 25 | R. hipposideros | 0.6 | 2.4 | 0 | 0 | P; R |
| 25 | T. teniotis | 0.8 | 2.2 | 0 | 0 | P |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leopardi, S.; Priori, P.; Zecchin, B.; Zamperin, G.; Milani, A.; Tonon, F.; Giorgiutti, M.; Beato, M.S.; De Benedictis, P. Interface between Bats and Pigs in Heavy Pig Production. Viruses 2021, 13, 4. https://doi.org/10.3390/v13010004
Leopardi S, Priori P, Zecchin B, Zamperin G, Milani A, Tonon F, Giorgiutti M, Beato MS, De Benedictis P. Interface between Bats and Pigs in Heavy Pig Production. Viruses. 2021; 13(1):4. https://doi.org/10.3390/v13010004
Chicago/Turabian StyleLeopardi, Stefania, Pamela Priori, Barbara Zecchin, Gianpiero Zamperin, Adelaide Milani, Francesco Tonon, Mirco Giorgiutti, Maria Serena Beato, and Paola De Benedictis. 2021. "Interface between Bats and Pigs in Heavy Pig Production" Viruses 13, no. 1: 4. https://doi.org/10.3390/v13010004
APA StyleLeopardi, S., Priori, P., Zecchin, B., Zamperin, G., Milani, A., Tonon, F., Giorgiutti, M., Beato, M. S., & De Benedictis, P. (2021). Interface between Bats and Pigs in Heavy Pig Production. Viruses, 13(1), 4. https://doi.org/10.3390/v13010004