
Respiratory Epithelial Cells Respond to Lactobacillus plantarum but Provide No Cross-Protection against Virus-Induced Inflammation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. MLE-12 Cells
2.3. Generation of Mouse Tracheal Epithelial Cell (mTEC) Cultures
2.4. Preparation of Virus Stocks and Titration by qPCR
2.5. Preparation of Lactobacillus plantarum (Lp)
2.6. Evaluation of Lp-Mediated Protection against Inf A Infection In Vivo
2.7. Responses of MLE-12 Cells to Administration of Lp and Pattern Recognition Receptor (PRR) Agonists
2.8. Treatment of mTECs with Lp and PRR Ligands
2.9. Infection of MLE-12 Cells with Inf A
2.10. Infection of mTECs with Inf A
2.11. Quantitative Evaluation of Virus Recovery by qRT-PCR
2.12. Lp Priming Prior to Infection with Inf A
2.13. Determination of Cytokine Concentrations in MLE-12 Supernatants and mTEC Basal Medium by ELISA
2.14. Flow Cytometry
2.15. Statistical Evaluation
3. Results and Discussion
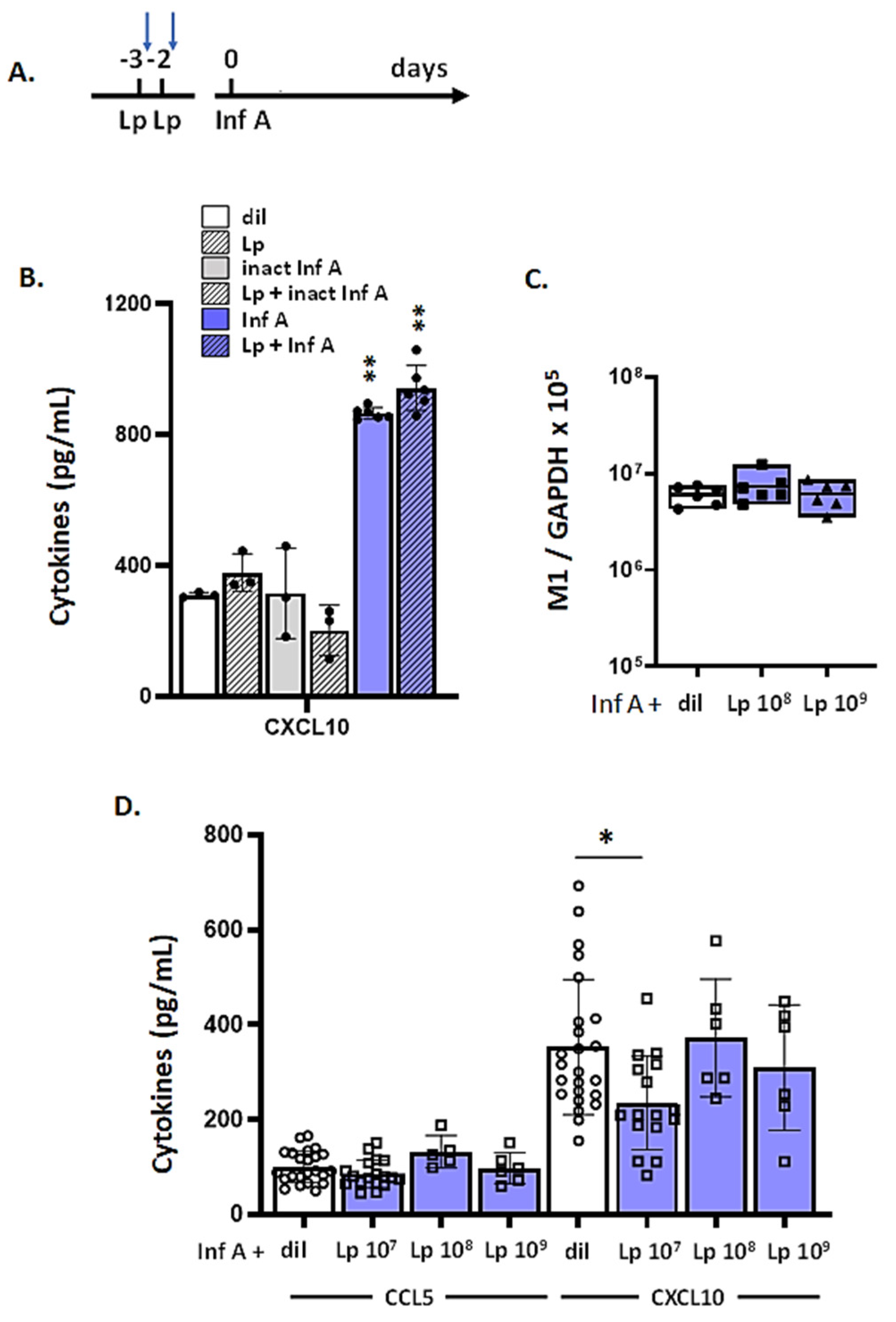
3.1. Lactobacillus plantarum (Lp) Administered Directly to the Respiratory Tract Protects Mice against Inflammation and Weight Loss in Response to Infection with Inf A
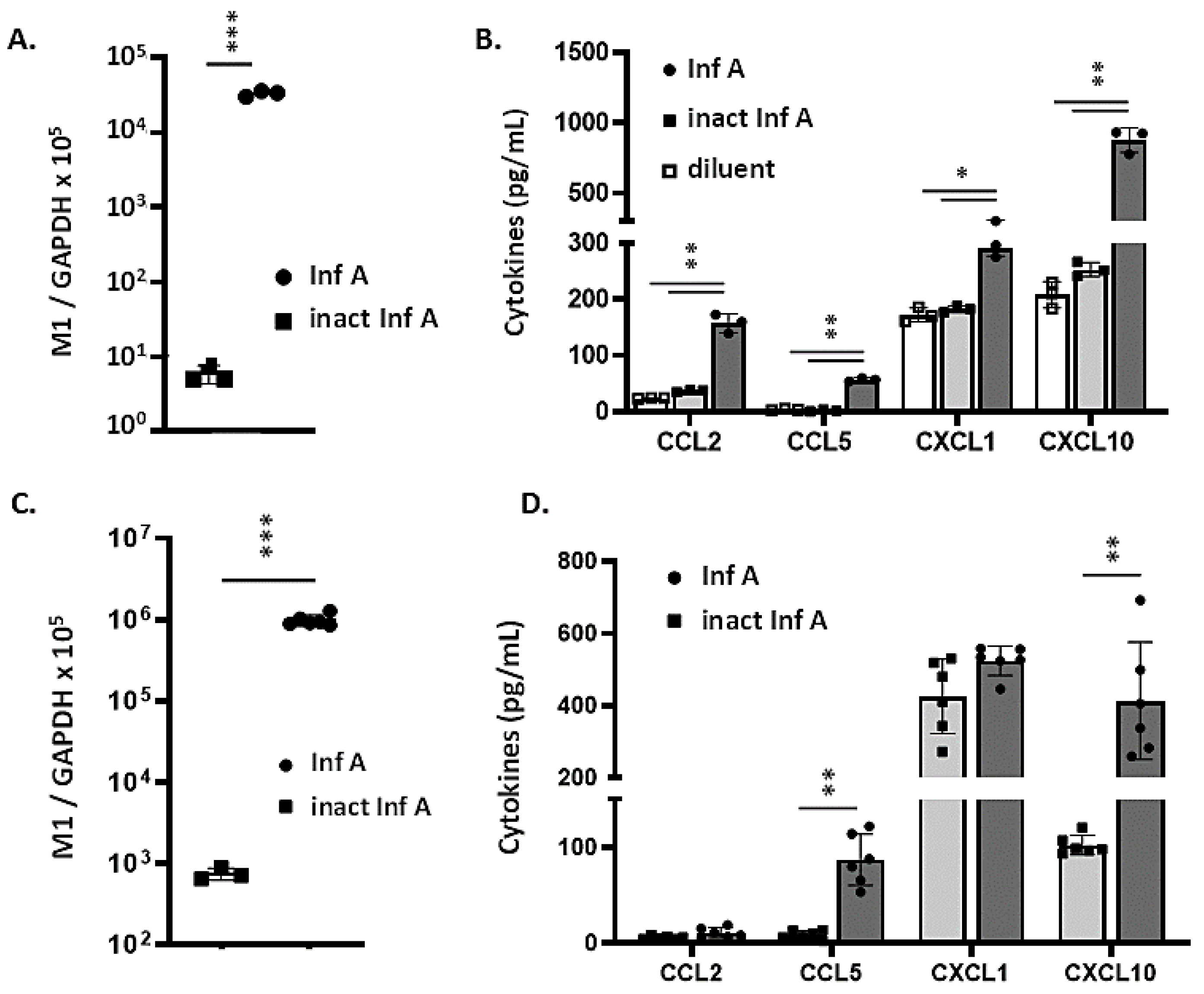
3.2. MLE-12 Cells and mTECs Support Replication of Inf A and Produce Proinflammatory Cytokines in Response to Virus Infection
3.3. Detection of Critical Pattern Recognition Receptors (PRRs) in MLE-12 Cells and mTECs by Flow Cytometry
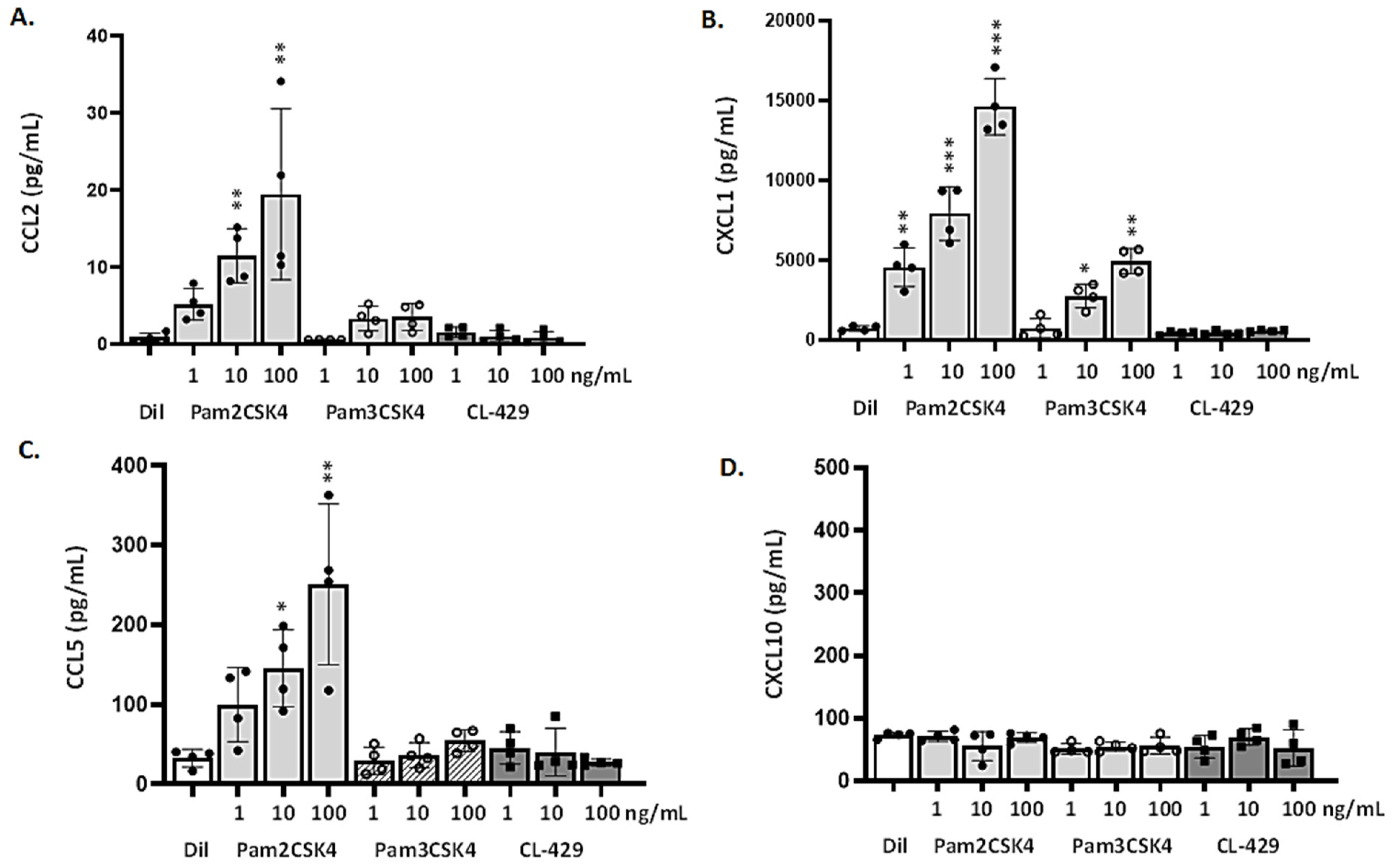
3.4. MLE-12 Cells Respond to Lp and TLR2 Agonists
3.5. Differential Responses of mTECs to TLR2 Ligands and CL-429
3.6. mTECs from Wild-Type (WT) but Not Tlr2−/− Mice Respond to Lp
3.7. Lp Does Not Elicit Cross-Protection against Virus-Induced Inflammation
3.8. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hall, C.B.; Weinberg, G.A.; Iwane, M.K.; Blumkin, A.K.; Edwards, K.M.; Staat, M.A.; Auinger, P.; Griffin, M.R.; Poehling, K.A.; Erdman, D.; et al. The burden of respiratory syncytial virus infection in young children. N. Engl. J. Med. 2009, 360, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Belongia, E.A.; King, J.P.; Kieke, B.A.; Pluta, J.; Al-Hilli, A.; Meece, J.K.; Shinde, V. Clinical features, severity, and incidence of RSV illness during 12 consecutive seaons in a community cohort of adults 60 years old. Open Forum Infect. Dis. 2018, 5, ofy316. [Google Scholar] [CrossRef] [PubMed]
- Rahil, Z.; Leylek, R.; Schürch, C.M.; Chen, H.; Bjornson-Hooper, Z.; Christensen, S.R.; Gherardini, P.F.; Bhate, S.S.; Spitzer, M.H.; Fragiadakis, G.K.; et al. Landscape of coordinated immune responses to H1N1 challenge in humans. J. Clin. Investig. 2020, 130, 5800–5816. [Google Scholar] [CrossRef] [PubMed]
- Andrew, M.K.; Shinde, V.; Hatchette, T.F.; Ambrose, A.; Boivin, G.; Bowie, W.; Chit, A.; Dos Santos, G.; Elsherif, M.; Green, K.; et al. Influenza vaccine effectiveness against influenza-related hospitalization during a season with mixed outbreaks of four influenza viruses: A test-negative case-control study in adults in Canada. BMC Infect. Dis. 2017, 17, 805. [Google Scholar] [CrossRef]
- Wang, F.; Cao, J.; Yu, Y.; Ding, J.; Eshak, E.S.; Liu, K.; Mubarik, S.; Shi, F.; Wen, H.; Zeng, Z.; et al. OUP accepted manuscript. Int. J. Epidemiol. 2020, in press. [Google Scholar]
- Smatti, M.K.; Al-Sarraj, Y.; Albagha, O.; Yassine, H.M. Host Genetic Variants Potentially Associated with SARS-Cov-2: A Multi-Population Analysis. Front Genet. 2020, 11, 578523. [Google Scholar] [CrossRef]
- Sebina, I.; Phipps, S. The Contribution of Neutrophils to the Pathogenesis of RSV Bronchiolitis. Viruses 2020, 12, 808. [Google Scholar] [CrossRef]
- Nuriev, R.; Johansson, C. Chemokine regulation during respiratory syncytial virus infection. F1000Reserch 2019, 8, 1837. [Google Scholar] [CrossRef]
- Gu, Y.; Hsu, A.C.-Y.; Pang, Z.; Pan, H.; Zuo, X.; Wang, G.; Zheng, J.; Wang, F. Role of the Innate Cytokine Storm Induced by the Influenza A Virus. Viral Immunol. 2019, 32, 244–251. [Google Scholar] [CrossRef]
- Betakova, T.; Kostrabova, A.; Lachova, V.; Turianova, L. Cytokines induced during influenza virus infection. Curr. Pharm. Des. 2017, 23, 2616–2622. [Google Scholar] [CrossRef]
- Miyazawa, M. Immunopathogenesis of SARS-CoV-2-induced pneumonia: Lessons from influenza virus infection. Inflamm. Regen. 2020, 40, 39. [Google Scholar] [CrossRef] [PubMed]
- Heimfarth, L.; Serafini, M.R.; Martins-Filho, P.R.; Quintans, J.D.S.S.; Quintans-Júnior, L.J. Drug repurposing and cytokine management in response to COVID-19: A review. Int. Immunopharmacol. 2020, 88, 106947. [Google Scholar] [CrossRef] [PubMed]
- Dyer, K.D.; Garcia-Crespo, K.E.; Glineur, S.; Domachowske, J.B.; Rosenberg, H.F. The pneumonia virus of mice (PVM) model of acute respiratory virus infection. Viruses 2012, 4, 3494–3510. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, H.F.; Bonville, C.A.; Easton, A.J.; Domachowske, J.B. The pneumonia virus of mice infection model for severe respiratory syncytial virus infection: Identifying novel targets for therapeutic intervention. Pharmacol. Ther. 2005, 105, 1–6. [Google Scholar] [CrossRef]
- Bem, R.A.; Domachowske, J.B.; Rosenberg, H.F. Animal models of human respiratory syncytial virus disease. Am. J. Physiol. Cell. Mol. Physiol. 2011, 301, L148–L156. [Google Scholar] [CrossRef]
- Öner, D.; Drysdale, S.B.; McPherson, C.; Lin, G.L.; Janet, S.; Broad, J.; Pollard, A.J.; Aerssens, J.; Nair, H.; Campbell, H.; et al. Biomarkers for Disease Severity in Children Infected with Respiratory Syncytial Virus: A Systematic Literature Review. J. Infect. Dis. 2020, 222, S648–S657. [Google Scholar] [CrossRef]
- Walsh, K.B.; Teijaro, J.R.; Brock, L.G.; Fremgen, D.M.; Collins, P.L.; Rosen, H.; Oldstone, M.B. Animal model of respiratory syncytial virus: CD8+ T cells cause a cytokine storm that is chemically tractable by spingosine-1-phophate 1 receptor agonist therapy. J. Virol. 2014, 88, 6281–6293. [Google Scholar] [CrossRef]
- Bonville, C.A.; Easton, A.J.; Rosenberg, H.F.; Domachowske, J.B. Altered pathogenesis of severe pneumovirus infection in response to combined antiviral and specific immunomodulatory agents. J. Virol. 2003, 77, 1237–1244. [Google Scholar] [CrossRef]
- Bonville, C.A.; Lau, V.K.; DeLeon, J.M.; Gao, J.L.; Easton, A.J.; Rosenberg, H.F.; Domachowske, J.B. Functional Antagonism of Chemokine Receptor CCR1 Reduces Mortality in Acute Pneumovirus Infection In Vivo. J. Virol. 2004, 78, 7894–7899. [Google Scholar] [CrossRef]
- Bondue, B.; Vosters, O.; De Nadai, P.; Glineur, S.; De Henau, O.; Luangsay, S.; Van Gool, F.; Communi, D.; De Vuyst, P.; Desmecht, D.; et al. ChemR23 Dampens Lung Inflammation and Enhances Anti-viral Immunity in a Mouse Model of Acute Viral Pneumonia. PLoS Pathog. 2011, 7, e1002358. [Google Scholar] [CrossRef]
- Gabryszewski, S.J.; Bachar, O.; Dyer, K.D.; Percopo, C.M.; Killoran, K.E.; Domachowske, J.B.; Rosenberg, H.F. Lactobacillus-Mediated Priming of the Respiratory Mucosa Protects against Lethal Pneumovirus Infection. J. Immunol. 2011, 186, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Percopo, C.M.; Dyer, K.D.; Garcia-Crespo, K.E.; Gabryszewski, S.J.; Shaffer, A.L.; Domachowske, J.B.; Rosenberg, H.F. B cells are not essential for Lactobacillus-mediated protection against lethal pneumovirus infection. J. Immunol. 2014, 192, 5265–5272. [Google Scholar] [CrossRef] [PubMed]
- Percopo, C.M.; Rice, T.A.; Brenner, T.A.; Dyer, K.D.; Luo, J.L.; Kanakabandi, K.; Sturdevant, D.E.; Porcella, S.F.; Domachowske, J.B.; Keicher, J.D.; et al. Immunobiotic Lactobacillus administered post-exposure averts the lethal sequelae of respiratory virus infection. Antivir. Res. 2015, 121, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Rice, T.A.; Brenner, T.A.; Percopo, C.M.; Ma, M.; Keicher, J.D.; Domachowske, J.B.; Rosenberg, H.F. Signaling via pattern recognition receptors NOD2 and TLR2 contributes to immunomodulatory control of lethal pneumovirus infection. Antivir. Res. 2016, 132, 131–140. [Google Scholar] [CrossRef][Green Version]
- Percopo, C.M.; Ma, M.; Rosenberg, H.F. Administration of immunobiology Lactobacillus plantarum delays but does not prevent lethal pneumovirus infection in Rag1-/-mice. J. Leukoc. Biol. 2017, 102, 905–913. [Google Scholar] [CrossRef]
- Percopo, C.M.; Ma, M.; Brenner, T.; Krumholz, J.O.; Break, T.J.; Laky, K.; Rosenberg, H.F. Critical Adverse Impact of IL-6 in Acute Pneumovirus Infection. J. Immunol. 2019, 202, 871–882. [Google Scholar] [CrossRef]
- Tonetti, F.R.; Islam, A.; Vizoso-Pinto, M.G.; Takahashi, H.; Kitazawa, H.; Villena, J. Nasal priming with immunobiotic lactobacilli improves the adaptive immune response against influenza virus. Int. Immunopharmacol. 2020, 78, 106115. [Google Scholar] [CrossRef]
- Tomosada, Y.; Chiba, E.; Zelaya, H.; Takahashi, T.; Tsukida, K.; Kitazawa, H.; Alvarez, S.; Villena, J. Nasally administered Lactobacillus rhamnosus strains differentially modulate respiratory antiviral immune responses and induce protection against respiratory syncytial virus infection. BMC Immunol. 2013, 14, 40. [Google Scholar] [CrossRef]
- Jung, Y.-J.; Lee, Y.-T.; Ngo, V.; Cho, Y.-H.; Ko, E.-J.; Hong, S.-M.; Kim, K.-H.; Jang, J.-H.; Oh, J.-S.; Park, M.-K.; et al. Heat-killed Lactobacillus casei confers broad protection against influenza A virus primary infection and develops heterosubtypic immunity against future secondary infection. Sci. Rep. 2017, 7, 17360. [Google Scholar] [CrossRef]
- Mahooti, M.; Miri, S.M.; Abdolalipour, E.; Ghaemi, A. The immunomodulatory effects of probiotics on respiratory viral infections: A hint for COVID-19 treatment? Microb. Pathog. 2020, 148, 104452. [Google Scholar] [CrossRef]
- Park, M.-K.; Ngo, V.; Kwon, Y.-M.; Lee, Y.-T.; Yoo, S.; Cho, Y.-H.; Hong, S.-M.; Hwang, H.S.; Ko, E.-J.; Jung, Y.-J.; et al. Lactobacillus plantarum DK119 as a Probiotic Confers Protection against Influenza Virus by Modulating Innate Immunity. PLoS ONE 2013, 8, e75368. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Salazar, C.; Shilts, M.H.; Tovchigrechko, A.; Schobel, S.; Chappell, J.D.; Larkin, E.K.; Gebretsadik, T.; Halpin, R.A.; Nelson, K.E.; Moore, M.L.; et al. Nasopharyngeal Lactobacillus is associated with a reduced risk of childhood wheezing illnesses following acute respiratory syncytial virus infection in infancy. J. Allergy Clin. Immunol. 2018, 142, 1447–1456.e9. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Crespo, K.E.; Chan, C.C.; Gabryszewski, S.J.; Percopo, C.M.; Rigaux, P.; Dyer, K.D.; Domachowske, J.B.; Rosenberg, H.F. Lactobacillus priming of the respiratory tract: Heterologous immunity and protection against lethal pneumovirus infection. Antivir. Res. 2013, 97, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef]
- Wikenheiser, K.A.; Vorbroker, D.K.; Rice, W.R.; Clark, J.C.; Bachurski, C.J.; Oie, H.K.; Whitsett, J.A. Production of immortalized distal respiratory epithelial cell lines from surfactant protein C/simian virus 40 large tumor antigen transgenic mice. Proc. Natl. Acad. Sci. USA 1993, 90, 11029–11033. [Google Scholar] [CrossRef]
- You, Y.; Richer, E.J.; Huang, T.; Brody, S.L. Growth and differentiation of mouse tracheal epithelial cells: Selection of a proliferative population. Am. J. Physiol. Cell. Mol. Physiol. 2002, 283, L1315–L1321. [Google Scholar] [CrossRef]
- You, Y.; Brody, S.L. Culture and Differentiation of Mouse Tracheal Epithelial Cells. Bioinform. MicroRNA Res. 2012, 945, 123–143. [Google Scholar] [CrossRef]
- Percopo, C.M.; Ma, M.; Mai, E.; Redes, J.L.; Kraemer, L.S.; Minai, M.; Moore, I.N.; Druey, K.M.; Rosenberg, H.F. Alternaria alternata Accelerates Loss of Alveolar Macrophages and Promotes Lethal Influenza A Infection. Viruses 2020, 12, 946. [Google Scholar] [CrossRef]
- Brown, E.G.; Bailly, J.E. Genetic analysis of mouse-adapted influenza A virus identifies roles for the NA, PB1, and PB2 genes in virulence. Virus Res. 1999, 61, 63–76. [Google Scholar] [CrossRef]
- Ma, M.; Redes, J.L.; Percopo, C.M.; Druey, K.M.; Rosenberg, H.F. Alternaria alternata challenge at the nasal mucosa results in eosinophilic inflammation and increased susceptibility to influenza virus infection. Clin. Exp. Allergy 2018, 48, 691–702. [Google Scholar] [CrossRef]
- Rosenberger, C.M.; Podyminogin, R.L.; Askovich, P.S.; Navarro, G.; Kaiser, S.M.; Sanders, C.J.; McClaren, J.L.; Tam, V.C.; Dash, P.; Noonan, J.G.; et al. Characterization of innate responses to influenza virus infection in a novel lung type I epithelial cell model. J. Gen. Virol. 2014, 95, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.K.-W.; Smee, D.F.; Wong, M.-H.; Nayak, D.P. Mutations in Influenza Virus M1 CCHH, the Putative Zinc Finger Motif, Cause Attenuation in Mice and Protect Mice against Lethal Influenza Virus Infection. J. Virol. 2006, 80, 5697–5707. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Maemura, T.; Fukuyama, S.; Sugita, Y.; Lopes, T.J.S.; Nakao, T.; Noda, T.; Kawaoka, Y. Lung-Derived Exosomal miR-483-3p Regulates the Innate Immune Response to Influenza Virus Infection. J. Infect. Dis. 2018, 217, 1372–1382. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, T.; Cai, D.; Hu, Z.; Chen, J.; Liao, H.; Zhi, L.; Wei, H.; Zhang, Z.; Qiu, Y.; et al. Cytokine storm intervention in the early stages of COVID-19 pneumonia. Cytokine Growth Factor Rev. 2020, 53, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.G.; Becher, A.; Tönnies, M.; Holland, G.; Knepper, J.; Bauer, T.T.; Schneider, P.; Neudecker, J.; Rückert, J.C.; Szymanski, K.; et al. Influenza A Viruses Target Type II Pneumocytes in the Human Lung. J. Infect. Dis. 2012, 206, 1685–1694. [Google Scholar] [CrossRef]
- Ibricevic, A.; Pekosz, A.; Walter, M.J.; Newby, C.; Battaile, J.T.; Brown, E.G.; Holtzman, M.J.; Brody, S.L. Influenza Virus Receptor Specificity and Cell Tropism in Mouse and Human Airway Epithelial Cells. J. Virol. 2006, 80, 7469–7480. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, Y.-H.; Yang, Z.-Q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell. Mol. Immunol. 2016, 13, 3–10. [Google Scholar] [CrossRef]
- Hartshorn, K.L. Innate Immunity and Influenza A Virus Pathogenesis: Lessons for COVID-19. Front. Cell. Infect. Microbiol. 2020, 10, 563850. [Google Scholar] [CrossRef]
- Guo, X.-Z.J.; Thomas, P.G. New fronts emerge in the influenza cytokine storm. Semin. Immunopathol. 2017, 39, 541–550. [Google Scholar] [CrossRef]
- Van Reeth, K. Cytokines in the pathogenesis of influenza. Vet. Microbiol. 2000, 74, 109–116. [Google Scholar] [CrossRef]
- Maelfait, J.; Roose, K.; Vereecke, L.; McGuire, C.; Sze, M.; Schulijs, M.J.; Willart, M.; Ibañez, L.I.; Hammad, H.; Lambrecht, B.N.; et al. A20 deficiency inlung epithelial cells protects against influenza A virus infection. PLoS Pathog. 2016, 12, e1005410. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, A.; Kuba, K.; Morita, M.; Chida, S.; Tezuka, H.; Hara, H.; Sasaki, T.; Ohteki, T.; Ranieri, V.M.; Dos Santos, C.C.; et al. CXCL10-CXCR3 Enhances the Development of Neutrophil-mediated Fulminant Lung Injury of Viral and Nonviral Origin. Am. J. Respir. Crit. Care Med. 2013, 187, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.; Wang, K.; Zhao, Z.; Zhang, L.; Gu, H.; Yang, P.; Wang, X. C-C Motif Chemokine Ligand 2 (CCL2) Mediates Acute Lung Injury Induced by Lethal Influenza H7N9 Virus. Front. Microbiol. 2017, 8, 587. [Google Scholar] [CrossRef] [PubMed]
- Tavares, L.P.; Garcia, C.C.; Machado, M.G.; Queiroz-Junior, C.M.; Barthelemy, A.; Trottein, F.; Siqueira, M.M.; Brandolini, L.; Allegretti, M. CXCR1/2 antagonism is protective during influenza and post-influenza pneumococcal infection. Front Immunol. 2017, 8, 1799. [Google Scholar] [CrossRef]
- Tomita, S.; Furihata, K.; Tanaka, N.; Satoh, E.; Nukada, T.; Okada, S. Determination of strain-specific wall teichoic acid structures in Lactobacillus plantarum reveals diverse α-d-glucosyl substitutions and high structural uniformity of the repeating units. Microbiology 2012, 158, 2712–2723. [Google Scholar] [CrossRef]
- Fernandez, E.M.; Valenti, V.; Rockel, C.; Hermann, C.; Pot, B.; Boneca, I.G.; Grangette, C. Anti-inflammatory capacity of selected lactobacilli in experimental colitis is driven by NOD2-mediated recognition of a specific peptidoglycan-derived muropeptide. Gut 2011, 60, 1050–1059. [Google Scholar] [CrossRef]
- Riehl, T.E.; Alvarado, D.; Ee, X.; Zuckerman, A.; Foster, L.; Kapoor, V.; Thotala, D.; Ciorba, M.A.; Stenson, W.F. Lactobacillus rhamnosus GG protects the intestinal epithelium from radiation injury through release of lipoteichoic acid, macrophage activation and the migration of mesenchymal stem cells. Gut 2019, 68, 1003–1013. [Google Scholar] [CrossRef]
- Trzpis, M.; McLaughlin, P.M.; de Leij, L.M.; Harmsen, M.C. Epithelial cell adhesion molecule: More than a carcinoma marker and adhesion molecule. Am. J. Pathol. 2007, 171, 386–395. [Google Scholar] [CrossRef]
- Liu, H.; Xie, Q.; Li, Y.; Wang, J. Stimulating Toll-like receptor 2 promotes the cell apoptosis through augmenting the expression of NIPK in lung epithelial cells. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2014, 30, 806–809. [Google Scholar]
- Neagos, J.; Standiford, T.J.; Newstead, M.W.; Zeng, X.; Huang, S.K.; Ballinger, M.N. Epigenetic Regulation of Tolerance to Toll-Like Receptor Ligands in Alveolar Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2015, 53, 872–881. [Google Scholar] [CrossRef]
- McClure, R.; Massari, P. TLR-Dependent Human Mucosal Epithelial Cell Responses to Microbial Pathogens. Front. Immunol. 2014, 5, 386. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Kawai, T.; Mühlradt, P.F.; Morr, M.; Radolf, J.D.; Zychlinsky, A.; Takeda, K.; Akira, S. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 2001, 13, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Brenner, T.A.; Rice, T.A.; Anderson, E.D.; Percopo, C.M.; Rosenberg, H.F. Immortalized MH-S cells lack defining features of primary alveolar macrophages and do not support mouse pneumovirus replication. Immunol. Lett. 2016, 172, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, S.; Ohto, U.; Shibata, T.; Miyake, T.S.K.; Shimizu, T. Crystal structure of NOD2 and its implications in human disease. Nat. Commun. 2016, 7, 11813. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kitani, A.; Murray, P.J.; Strober, W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat. Immunol. 2004, 8, 800–808. [Google Scholar] [CrossRef]
- Borm, M.E.A.; van Bodegraven, A.A.; Mulder, C.J.J.; Kraal, G.; Bouma, G. The effect of NOD2 activation on TLR2-medidated cytokine responses is dependent on activation dose and NOD2 genotype. Genes Immun. 2008, 9, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Percopo, C.M.; Dyer, K.D.; Karpe, K.A.; Domachowske, J.B.; Rosenberg, H.F. Eosinophils and respiratory virus infection: A dual-standard curve qRT-PCR-based method for determining virus recovery from mouse lung tissue. Methods Mol. Biol. 2014, 1178, 257–266. [Google Scholar]
- Dyer, K.D.; Drummond, R.A.; Rice, T.A.; Percopo, C.M.; Brenner, T.A.; Barisas, D.A.G.; Karpe, K.A.; Moore, M.L.; Rosenberg, H.F. Priming of the Respiratory Tract with Immunobiotic Lactobacillus plantarum Limits Infection of Alveolar Macrophages with Recombinant Pneumonia Virus of Mice (rK2-PVM). J. Virol. 2016, 90, 979–991. [Google Scholar] [CrossRef]
- Bonville, C.A.; Bennett, N.J.; Koehnlein, M.; Haines, D.M.; Ellis, J.A.; DelVecchio, A.M.; Rosenberg, H.F.; Domachowske, J.B. Respiratory dysfunction and proinflammatory chemokines in the pneumonia virus of mice (PVM) model of viral bronchiolitis. Virology 2006, 349, 87–95. [Google Scholar] [CrossRef]
- Dyer, K.D.; Schellens, I.M.; Bonville, C.A.; Martin, B.V.; Domachowske, J.B.; Rosenberg, H.F. Efficient replication of pneumonia virus of mice (PVM) in a mouse macrophage cell line. Virol. J. 2007, 4, 48. [Google Scholar] [CrossRef]
- Buttignol, M.; Pires-Neto, R.C.; Silva, R.C.R.E.; Albino, M.B.; Dolhnikoff, M.; Mauad, T. Airway and parenchyma immune cells in influenza A(H1N1)pdm09 viral and non-viral diffuse alveolar damage. Respir. Res. 2017, 18, 147. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, P.P.; Samarasinghe, A.E. The role of innate leukoyctes during influenza virus infection. J. Immunol. Res. 2019, 2019, 8028725. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.A. Respiratory Epithelial Cells as Master Communicators during Viral Infections. Curr. Clin. Microbiol. Rep. 2019, 6, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.; Tirouvanziam, R.; Laval, J.; Greene, C.M.; Habiel, D.; Sharma, L.; Yildirim, A.Ö.; Cruz, C.S.D.; Hogaboam, C.M. Innate Immunity of the Lung: From Basic Mechanisms to Translational Medicine. J. Innate Immun. 2018, 10, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.M.; Nakamura, S.; Weiser, J.N. Nod2 sensing of lysozyme-digested peptidoglycan promotes macrophage recruitment and clearance of S. pneumoniae colonization in mice. J. Clin. Investig. 2011, 121, 3666–3676. [Google Scholar] [CrossRef] [PubMed]
- Leissinger, M.; Kulkarni, R.; Zemans, R.L.; Downey, G.P.; Jeyaseelan, S. Investigating the Role of Nucleotide-Binding Oligomerization Domain–Like Receptors in Bacterial Lung Infection. Am. J. Respir. Crit. Care Med. 2014, 189, 1461–1468. [Google Scholar] [CrossRef]
- Yu, X.; Zeng, J.; Xie, J. Navigating through the maze of TLR2 mediated signaling network for better mycobacterium infection control. Biochimie 2014, 102, 1–8. [Google Scholar] [CrossRef]
- Royet, J.; Gupta, D.; Dziarski, R. Peptidoglycan recognition proteins: Modulators of the microbiome and inflammation. Nat. Rev. Immunol. 2011, 11, 837–851. [Google Scholar] [CrossRef]
- Wolf, A.J.; Underhill, D.M. Peptidoglycan recognition by the innate immune system. Nat. Rev. Immunol. 2018, 18, 243–254. [Google Scholar] [CrossRef]
- Dziarski, R.; Gupta, D. The peptidoglycan recognition proteins (PGRPs). Genome Biol. 2006, 7, 232. [Google Scholar] [CrossRef]
- Levy, O. Antimicrobial proteins and peptides: Anti-infective molecules of mammalian leukocytes. J. Leukoc. Biol. 2004, 76, 909–925. [Google Scholar] [CrossRef] [PubMed]
- Bonville, C.; Rosenberg, H.; Domachowske, J.B. Ribavirin and cysteinyl leukotriene-1 receptor blockade as treatment for severe bronchiolitis. Antivir. Res. 2006, 69, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H.; Frigault, M.J.; Serling-Boyd, N.J.; Fernandes, A.D.; Harvey, L.; Foulkes, A.S.; Horick, N.K.; Healy, B.C.; Shah, R.; Bensaci, A.M.; et al. Efficacy of Tocilizumab in Patients Hospitalized with Covid-19. N. Engl. J. Med. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Mora, M.G.; Cabello Úbeda, A.; Pérez, L.P.; Álvarez, F.V.; Álvarez, B.Á.; Nieto, M.J.R.; Acosta, I.C.; Ormaechea, I.F.; Al-Hayani, A.W.M.; Carballosa, P.; et al. Compassionate use of tocilizumab in severe SARS-CoV2 pneumonia. Int. J. Infect. Dis. 2020, S1201, 32249. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mai, E.; Percopo, C.M.; Limkar, A.R.; Sek, A.C.; Ma, M.; Rosenberg, H.F. Respiratory Epithelial Cells Respond to Lactobacillus plantarum but Provide No Cross-Protection against Virus-Induced Inflammation. Viruses 2021, 13, 2. https://doi.org/10.3390/v13010002
Mai E, Percopo CM, Limkar AR, Sek AC, Ma M, Rosenberg HF. Respiratory Epithelial Cells Respond to Lactobacillus plantarum but Provide No Cross-Protection against Virus-Induced Inflammation. Viruses. 2021; 13(1):2. https://doi.org/10.3390/v13010002
Chicago/Turabian StyleMai, Eric, Caroline M. Percopo, Ajinkya R. Limkar, Albert C. Sek, Michelle Ma, and Helene F. Rosenberg. 2021. "Respiratory Epithelial Cells Respond to Lactobacillus plantarum but Provide No Cross-Protection against Virus-Induced Inflammation" Viruses 13, no. 1: 2. https://doi.org/10.3390/v13010002
APA StyleMai, E., Percopo, C. M., Limkar, A. R., Sek, A. C., Ma, M., & Rosenberg, H. F. (2021). Respiratory Epithelial Cells Respond to Lactobacillus plantarum but Provide No Cross-Protection against Virus-Induced Inflammation. Viruses, 13(1), 2. https://doi.org/10.3390/v13010002