
Biochemical and Biophysical Characterization of the dsDNA Packaging Motor from the Lactococcus lactis Bacteriophage Asccphi28


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cloning
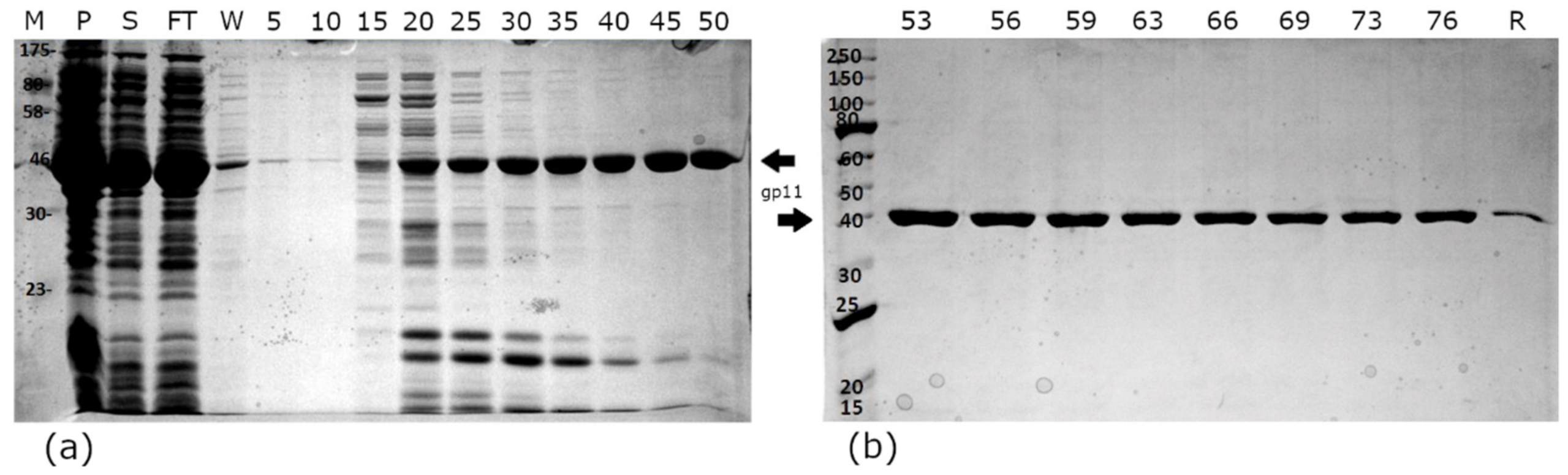
2.2. Protein Expression and Purification
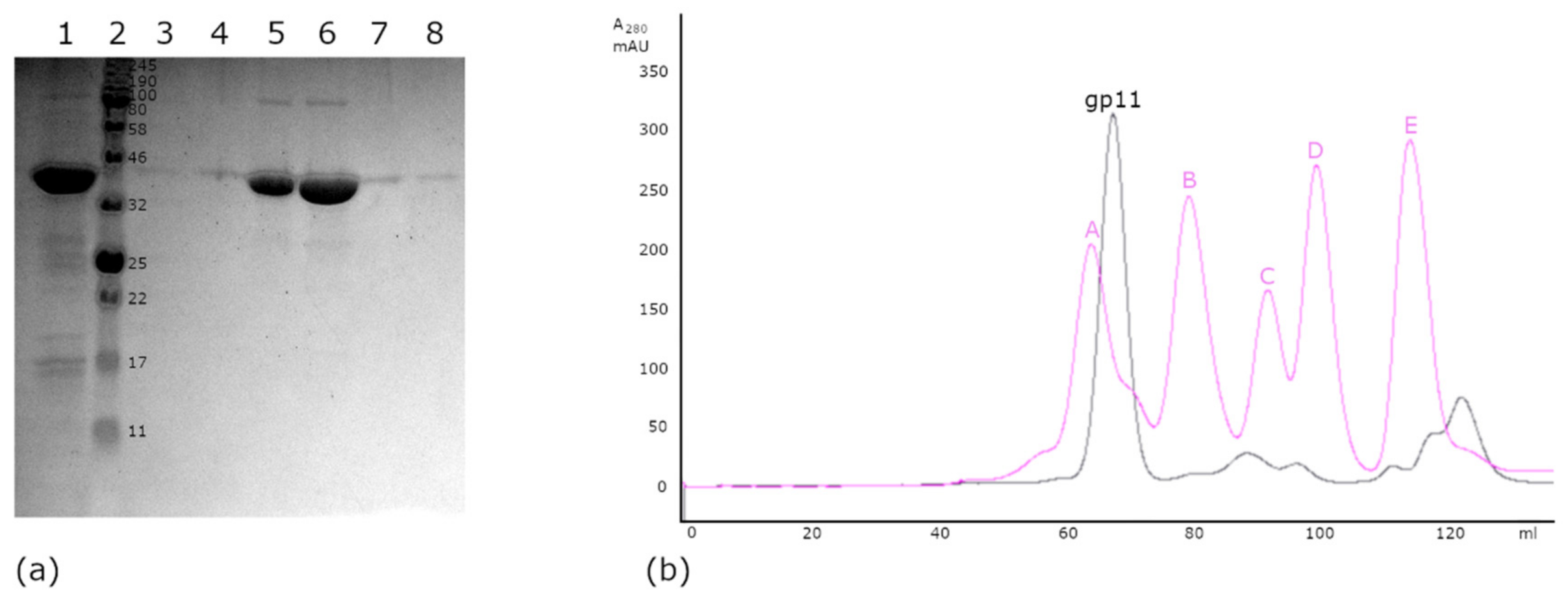
2.3. Size Estimation by Gel Filtration Chromatography
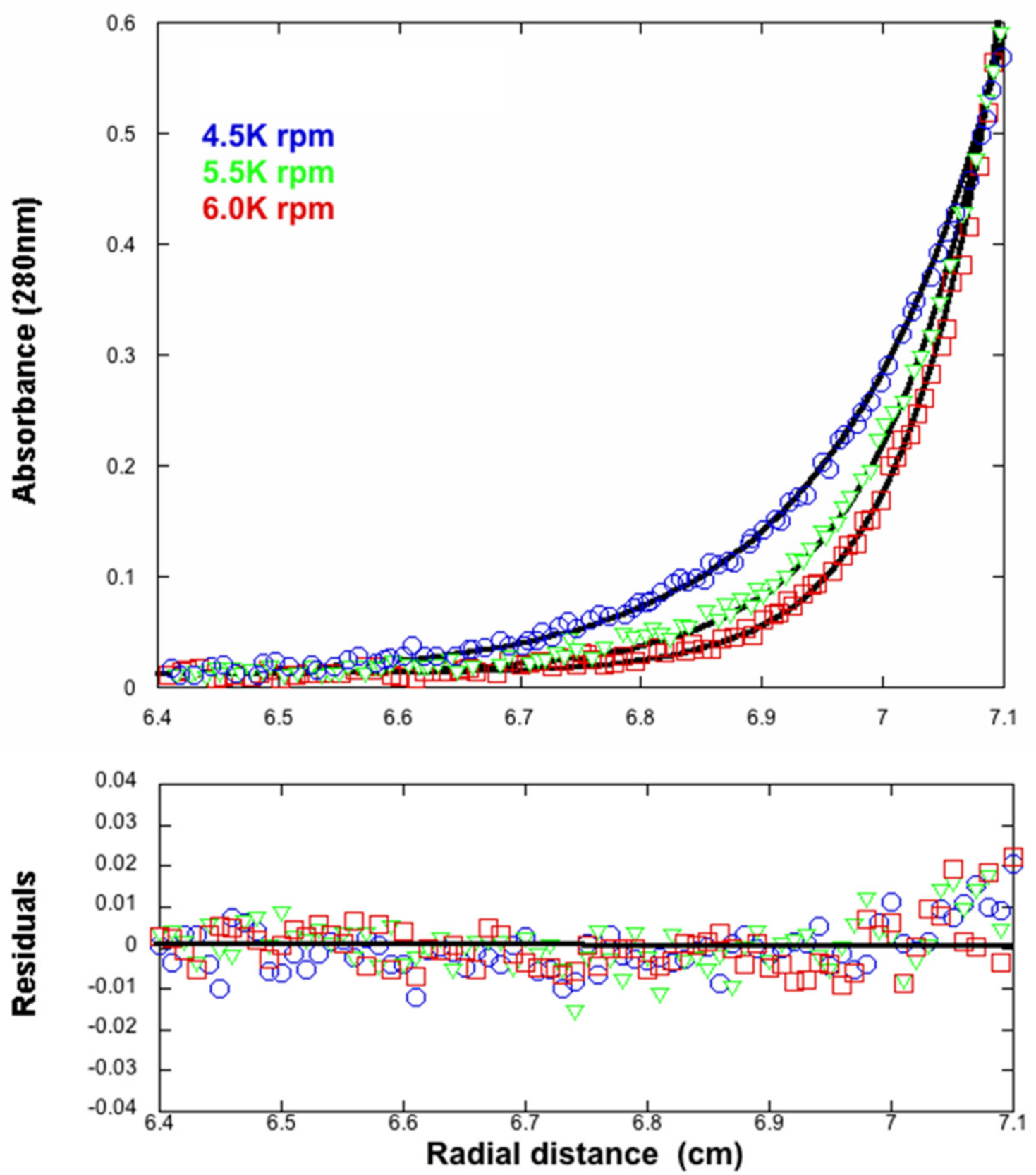
2.4. Analytical Ultracentrifugation
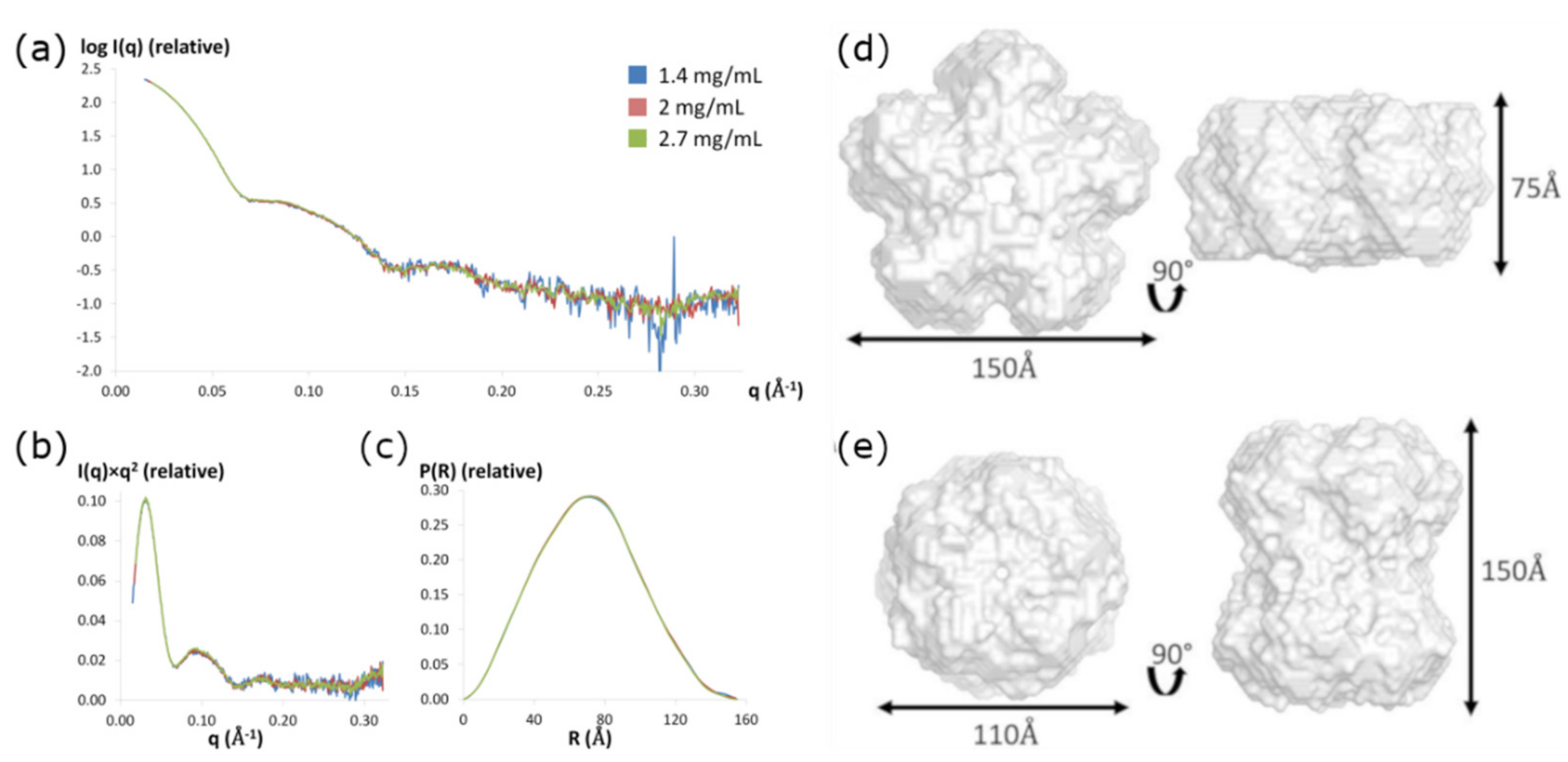
2.5. Solution Small-Angle X-ray Scattering
2.6. Electron Microscopy
2.7. Crystallization
2.8. X-ray Diffraction Data Collection and Processing
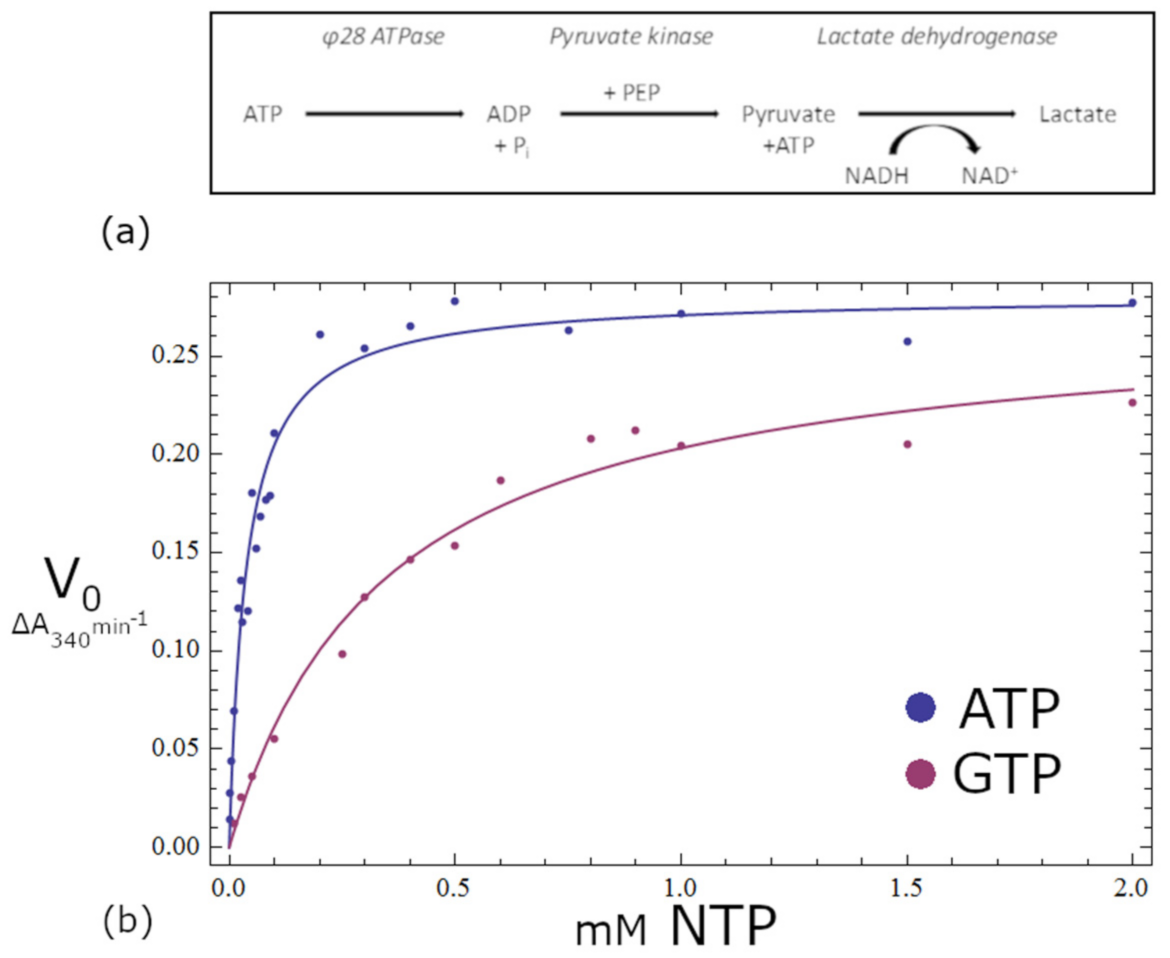
2.9. Activity Assays
3. Results
3.1. Identification of the Asccφ28 ATPase
3.2. Cloning, Expression, and Purification
3.3. Size Estimation by Gel Filtration Chromatography
3.4. Analytical Ultracentrifugation
3.5. Small-Angle X-ray Scattering:
3.6. SAXS Ab Initio Shape Calculations
3.7. Electron Microscopy
3.8. Crystallization
3.9. Crystal X-ray Diffraction
3.10. Activity Assays
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Catalano, C.E. The terminase enzyme from bacteriophage lambda: A DNA-packaging machine. Cell. Mol. Life Sci. 2000, 57, 128–148. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.B.; Feiss, M. The Bacteriophage DNA Packaging Motor. Annu. Rev. Genet. 2008, 42, 647–681. [Google Scholar] [CrossRef] [PubMed]
- Morais, M.C. The dsDNA Packaging Motor in Bacteriophage ø29. Adv. Exp. Med. Biol. 2012, 726, 511–547. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.B.; Feiss, M. Mechanisms of DNA Packaging by Large Double-Stranded DNA Viruses. Annu. Rev. Virol. 2015, 2, 351–378. [Google Scholar] [CrossRef] [PubMed]
- Myers, C.G.; Pettitt, B. Communication: Origin of the contributions to DNA structure in phages. J. Chem. Phys. 2013, 138, 071103. [Google Scholar] [CrossRef]
- Bores, C.; Pettitt, B.M. Structure and the role of filling rate on model dsDNA packed in a phage capsid. Phys. Rev. E 2020, 101, 012406. [Google Scholar] [CrossRef]
- Mitchell, M.S.; Matsuzaki, S.; Imai, S.; Rao, V.B. Sequence analysis of bacteriophage T4 DNA packaging/terminase genes 16 and 17 reveals a common ATPase center in the large subunit of viral terminases. Nucleic Acids Res. 2002, 30, 4009–4021. [Google Scholar] [CrossRef]
- Koti, J.S.; Morais, M.C.; Rajagopal, R.; Owen, B.A.L.; McMurray, C.T.; Anderson, D.L. DNA Packaging Motor Assembly Intermediate of Bacteriophage ϕ29. J. Mol. Biol. 2008, 381, 1114–1132. [Google Scholar] [CrossRef]
- Leipe, D.D.; Wolf, Y.I.; Koonin, E.V.; Aravind, L. Classification and evolution of P-loop GTPases and related ATPases. J. Mol. Biol. 2002, 317, 41–72. [Google Scholar] [CrossRef]
- Iyer, L.M.; Leipe, D.D.; Koonin, E.V.; Aravind, L. Evolutionary history and higher order classification of AAA + ATPases. J. Struct. Biol. 2004, 146, 11–31. [Google Scholar] [CrossRef]
- Iyer, L.M. Comparative genomics of the FtsK-HerA superfamily of pumping ATPases: Implications for the origins of chromosome segregation, cell division and viral capsid packaging. Nucleic Acids Res. 2004, 32, 5260–5279. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, A.; Iyer, L.; Aravind, L. Comparative Genomics and Evolutionary Trajectories of Viral ATP Dependent DNA-Packaging Systems. Genome Dyn. 2007, 3, 48–65. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.E.; Tans, S.J.; Smith, S.B.; Grimes, S.; Anderson, D.L.; Bustamante, C.D. The bacteriophage φ29 portal motor can package DNA against a large internal force. Nature 2001, 413, 748–752. [Google Scholar] [CrossRef]
- Liu, S.; Chistol, G.; Hetherington, C.L.; Tafoya, S.; Aathavan, K.; Schnitzbauer, J.; Grimes, S.; Jardine, P.J.; Bustamante, C. A Viral Packaging Motor Varies Its DNA Rotation and Step Size to Preserve Subunit Coordination as the Capsid Fills. Cell 2014, 157, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, Q.; Wang, M.; Jia, R.; Chen, S.; Zhu, D.; Liu, M.; Wu, Y.; Zhao, X.; Zhang, S.; et al. Terminase Large Subunit Provides a New Drug Target for Herpesvirus Treatment. Viruses 2019, 11, 219. [Google Scholar] [CrossRef] [PubMed]
- Mahler, B.P.; Bujalowski, P.J.; Mao, H.; Dill, E.A.; Jardine, P.J.; Choi, K.H.; Morais, M.C. NMR structure of a vestigial nuclease provides insight into the evolution of functional transitions in viral dsDNA packaging motors. bioRxiv 2020. [CrossRef] [PubMed]
- Simpson, A.A.; Tao, Y.; Leiman, P.G.; Badasso, M.O.; He, Y.; Jardine, P.J.; Olson, N.H.; Morais, M.C.; Grimes, S.; Anderson, D.L.; et al. Structure of the bacteriophage φ29 DNA packaging motor. Nature 2000, 408, 745–750. [Google Scholar] [CrossRef]
- Chistol, G.; Liu, S.; Hetherington, C.L.; Moffitt, J.R.; Grimes, S.; Jardine, P.J.; Bustamante, C. High Degree of Coordination and Division of Labor among Subunits in a Homomeric Ring ATPase. Cell 2012, 151, 1017–1028. [Google Scholar] [CrossRef]
- Liu, S.; Chistol, G.; Bustamante, C. Mechanical Operation and Intersubunit Coordination of Ring-Shaped Molecular Motors: Insights from Single-Molecule Studies. Biophys. J. 2014, 106, 1844–1858. [Google Scholar] [CrossRef]
- Pajak, J.; Dill, E.; White, M.A.; Kelch, B.A.; Jardine, P.; Arya, G.; Morais, M.C. Atomistic Mechanism of Force Generation, Translocation, and Coordination in a Viral Genome Packaging Motor. bioRxiv 2020. [CrossRef]
- Woodson, M.; Pajak, J.; Zhao, W.; Zhang, W.; Arya, G.; White, M.A.; Jardine, P.J.; Morais, M.C. A viral genome packaging motor transitions between cyclic and helical symmetry to translocate dsDNA. bioRxiv 2020. [CrossRef]
- Guo, P.X.; Bailey, S.; Bodley, J.W.; Anderson, D. Characterization of the small RNA of the bacteriophage phi 29 DNA packaging machine. Nucleic Acids Res. 1987, 15, 7081–7090. [Google Scholar] [CrossRef] [PubMed]
- Morais, M.C.; Tao, Y.; Olson, N.H.; Grimesb, S.; Jardinebc, P.J.; Anderson, D.L.; Baker, T.S.; Rossmann, M.G. Cryoelectron-Microscopy Image Reconstruction of Symmetry Mismatches in Bacteriophage φ29. J. Struct. Biol. 2001, 135, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Morais, M.C.; Choi, K.H.; Koti, J.S.; Chipman, P.R.; Anderson, D.L.; Rossmann, M.G. Conservation of the Capsid Structure in Tailed dsDNA Bacteriophages: The Pseudoatomic Structure of ϕ29. Mol. Cell 2005, 18, 149–159. [Google Scholar] [CrossRef]
- Morais, M.C.; Koti, J.S.; Bowman, V.D.; Reyes-Aldrete, E.; Anderson, D.L.; Rossmann, M.G. Defining Molecular and Domain Boundaries in the Bacteriophage ϕ29 DNA Packaging Motor. Structure 2008, 16, 1267–1274. [Google Scholar] [CrossRef]
- Sun, S.; Kondabagil, K.; Draper, B.; Alam, T.I.; Bowman, V.D.; Zhang, Z.; Hegde, S.; Fokine, A.; Rossmann, M.G.; Rao, V.B. The Structure of the Phage T4 DNA Packaging Motor Suggests a Mechanism Dependent on Electrostatic Forces. Cell 2008, 135, 1251–1262. [Google Scholar] [CrossRef]
- Andrews, B.T.; Catalano, C.E. The Enzymology of a Viral Genome Packaging Motor Is Influenced by the Assembly State of the Motor Subunits. Biochemistry 2012, 51, 9342–9353. [Google Scholar] [CrossRef]
- Mao, H.; Saha, M.; Reyes-Aldrete, E.; Sherman, M.B.; Woodson, M.; Atz, R.; Grimes, S.; Jardine, P.J.; Morais, M.C. Structural and Molecular Basis for Coordination in a Viral DNA Packaging Motor. Cell Rep. 2016, 14, 2017–2029. [Google Scholar] [CrossRef]
- Kotsonis, S.E.; Powell, I.B.; Pillidge, C.J.; Limsowtin, G.K.Y.; Hillier, A.J.; Davidson, B.E. Characterization and Genomic Analysis of Phage asccφ28, a Phage of the Family Podoviridae Infecting Lactococcus lactis. Appl. Environ. Microbiol. 2008, 74, 3453–3460. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Hochuli, E. Large-scale chromatography of recombinant proteins. J. Chromatogr. 1988, 444, 293–302. [Google Scholar] [CrossRef]
- Edelhoch, H. Spectroscopic Determination of Tryptophan and Tyrosine in Proteins. Biochemistry 1967, 6, 1948–1954. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.C.; Von Hippel, P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989, 182, 319–326. [Google Scholar] [CrossRef]
- Hura, G.L.; Menon, A.L.; Hammel, M.; Rambo, R.P.; Poole, F.L., 2nd; Tsutakawa, S.E.; Jenney, F.E., Jr.; Classen, S.; Frankel, K.A.; Hopkins, R.C.; et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nat. Methods 2009, 6, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Konarev, P.V.; Volkov, V.V.; Sokolova, A.V.; Koch, M.H.J.; Svergun, D.I. PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003, 36, 1277–1282. [Google Scholar] [CrossRef]
- Konarev, P.V.; Petoukhov, M.V.; Volkov, V.V.; Svergun, D. ATSAS2.1, a program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2006, 39, 277–286. [Google Scholar] [CrossRef]
- Svergun, D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Porod, G. Chapter 2: General Theory. In Small Angle X-ray Scattering; Glatter, O., Ed.; Academic Press: London, UK, 1982. [Google Scholar]
- Mertens, H.D.; Svergun, D. Structural characterization of proteins and complexes using small-angle X-ray solution scattering. J. Struct. Biol. 2010, 172, 128–141. [Google Scholar] [CrossRef]
- Svergun, D.; Petoukhov, M.V.; Koch, M.H.J. Determination of Domain Structure of Proteins from X-Ray Solution Scattering. Biophys. J. 2001, 80, 2946–2953. [Google Scholar] [CrossRef]
- Minor, W.; Cymborowski, M.; Otwinowski, Z.; Chruszcz, M. HKL-3000: The integration of data reduction and structure solution—From diffraction images to an initial model in minutes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 859–866. [Google Scholar] [CrossRef]
- Bücher, T.; Pfleiderer, G. Pyruvate kinase from muscle. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1955; Volume 1, pp. 435–440. [Google Scholar]
- Slabinski, L.; Jaroszewski, L.; Rychlewski, L.; Wilson, I.A.; Lesley, S.A.; Godzik, A. XtalPred: A web server for prediction of protein crystallizability. Bioinformatics 2007, 23, 3403–3405. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.; Waterman, M. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Timasheff, S.N. Partial specific volumes and interactions with solvent components of proteins in guanidine hydrochloride. Biochemistry 1974, 13, 257–265. [Google Scholar] [CrossRef]
- Volkov, V.V.; Svergun, D.I. Uniqueness ofab initioshape determination in small-angle scattering. J. Appl. Crystallogr. 2003, 36, 860–864. [Google Scholar] [CrossRef]
- Putnam, C.D.; Hammel, M.; Hura, G.L.; Tainer, J.A. X-ray solution scattering (SAXS) combined with crystallography and computation: Defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 2007, 40, 191–285. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Concentration | RG (Guinier) (Å) | Dmax (P(R)) (Å) | RG (Real Space, P(R)) (Å) | Hydrated Volume (Porod) (nm3) | Estimated Molecular Mass (kDa) | Number of Monomers |
|---|---|---|---|---|---|---|
| 1.4 mg/mL | 54.8 | 155 | 53.8 | 745.6 | 426.1 | 9.5 |
| 2.0 mg/mL | 55.1 | 155 | 53.7 | 778.6 | 444.9 | 9.9 |
| 2.7 mg/mL | 55.2 | 155 | 53.6 | 760.6 | 434.6 | 9.7 |
| Diffraction Source | APS 21 ID-F | APS 21 ID-F |
|---|---|---|
| Wavelength (Å) | 0.97872 | 0.97872 |
| Temperature (K) | 100 | 100 |
| Detector | MAR 225 CCD | MAR 225 CCD |
| Crystal-detector distance (mm) | 550 | 500 |
| Rotation range per image (°) | 1 | 0.5 |
| Total rotation range (°) | 720 | 720 |
| Exposure time per image (s) | 1 | 0.5 |
| Space group | P3x21 | P4x212 |
| a, b, c (Å) | 135.0, 135.0, 276.7 | 110.9, 110.9, 351.8 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 90 |
| Mosaicity (°) | 0.4 | 0.5 |
| Resolution range (Å) | 50—3.3 | 50—2.8 |
| Total No. of reflections | 3,916,456 | 555,523 |
| No. of unique reflections | 38,921 | 44,804 |
| Completeness (%) | 99.7 (95.9) | 99.6 (98.8) |
| Redundancy | 39.7 (20.5) | 12.4 (6.2) |
| 〈 I/σ(I)〉 | 30.2 (1.4) | 11.4 (0.9) |
| Rr.i.m. | 0.024 (0.50.4) | 0.060 (0.69) |
| Overall B factor from Wilson plot (Å2) | 121 | 51 |
| ATP | GTP | |
|---|---|---|
| Km | 28 μM | 344 μM |
| Vmax | 7.53 × 10−7 Ms−1 | 7.31 × 10−7 Ms−1 |
| kcat | 0.849 s−1 | 0.817s−1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes-Aldrete, E.; Dill, E.A.; Bussetta, C.; Szymanski, M.R.; Diemer, G.; Maindola, P.; White, M.A.; Bujalowski, W.M.; Choi, K.H.; Morais, M.C. Biochemical and Biophysical Characterization of the dsDNA Packaging Motor from the Lactococcus lactis Bacteriophage Asccphi28. Viruses 2021, 13, 15. https://doi.org/10.3390/v13010015
Reyes-Aldrete E, Dill EA, Bussetta C, Szymanski MR, Diemer G, Maindola P, White MA, Bujalowski WM, Choi KH, Morais MC. Biochemical and Biophysical Characterization of the dsDNA Packaging Motor from the Lactococcus lactis Bacteriophage Asccphi28. Viruses. 2021; 13(1):15. https://doi.org/10.3390/v13010015
Chicago/Turabian StyleReyes-Aldrete, Emilio, Erik A. Dill, Cecile Bussetta, Michal R. Szymanski, Geoffrey Diemer, Priyank Maindola, Mark A. White, Wlodzimierz M. Bujalowski, Kyung H. Choi, and Marc C. Morais. 2021. "Biochemical and Biophysical Characterization of the dsDNA Packaging Motor from the Lactococcus lactis Bacteriophage Asccphi28" Viruses 13, no. 1: 15. https://doi.org/10.3390/v13010015
APA StyleReyes-Aldrete, E., Dill, E. A., Bussetta, C., Szymanski, M. R., Diemer, G., Maindola, P., White, M. A., Bujalowski, W. M., Choi, K. H., & Morais, M. C. (2021). Biochemical and Biophysical Characterization of the dsDNA Packaging Motor from the Lactococcus lactis Bacteriophage Asccphi28. Viruses, 13(1), 15. https://doi.org/10.3390/v13010015