
Structure and Hierarchy of SARS-CoV-2 Infection Dynamics Models Revealed by Reaction Network Analysis




Abstract
1. Introduction
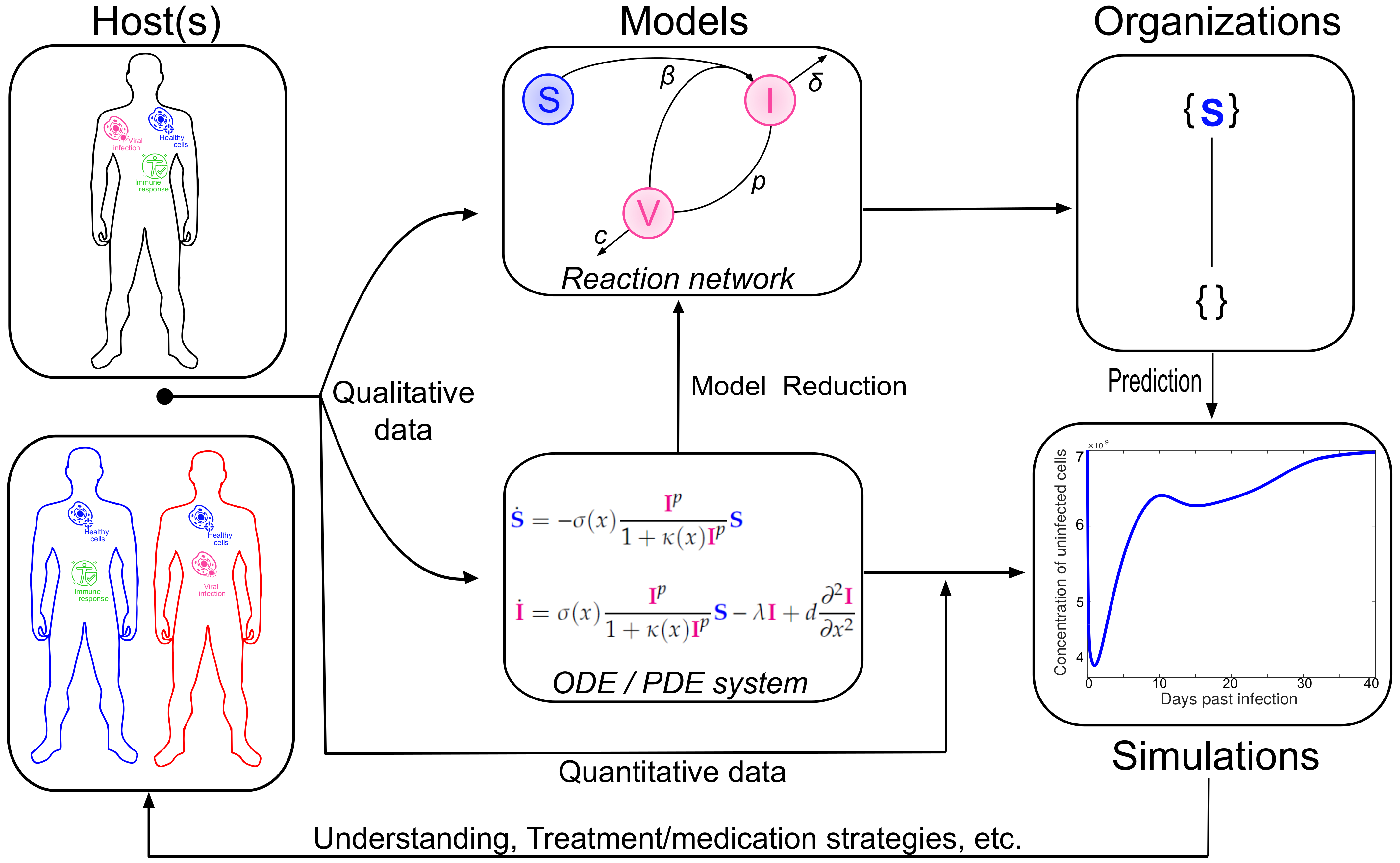
2. Materials and Methods: Procedure for the Organizational Analysis
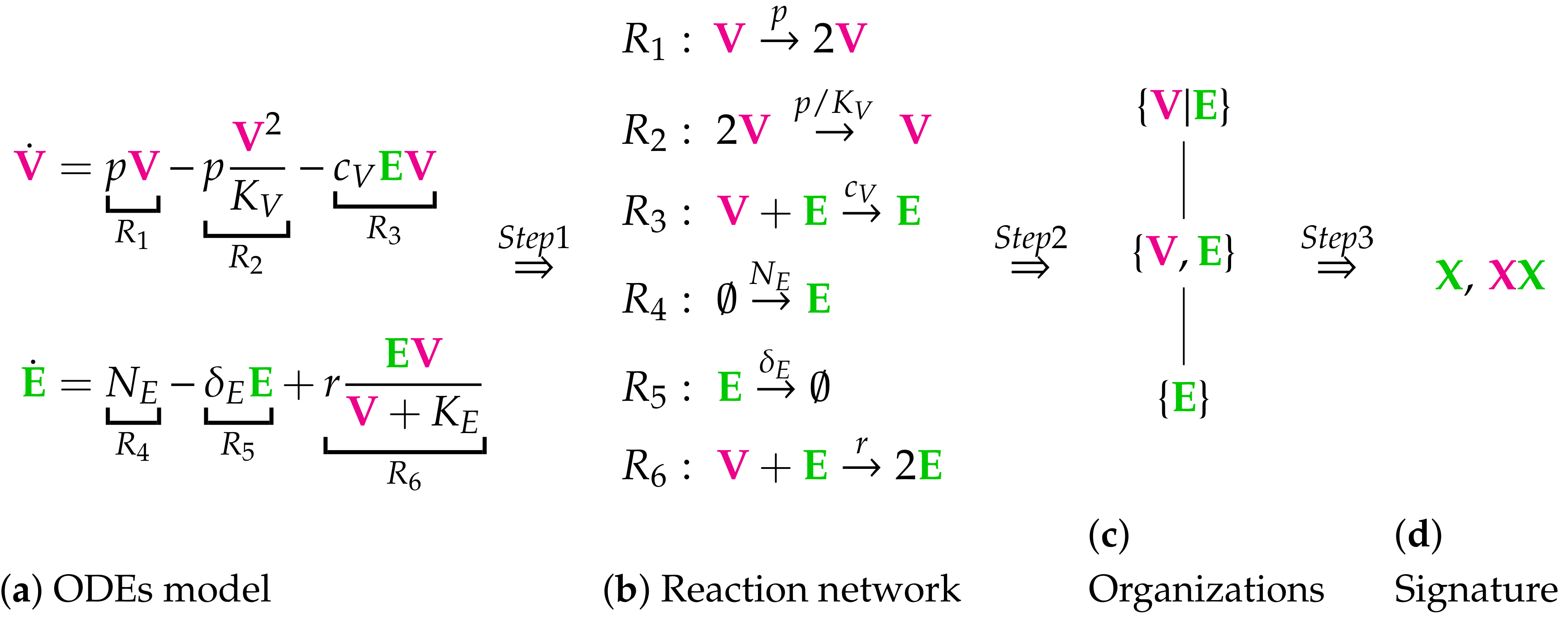
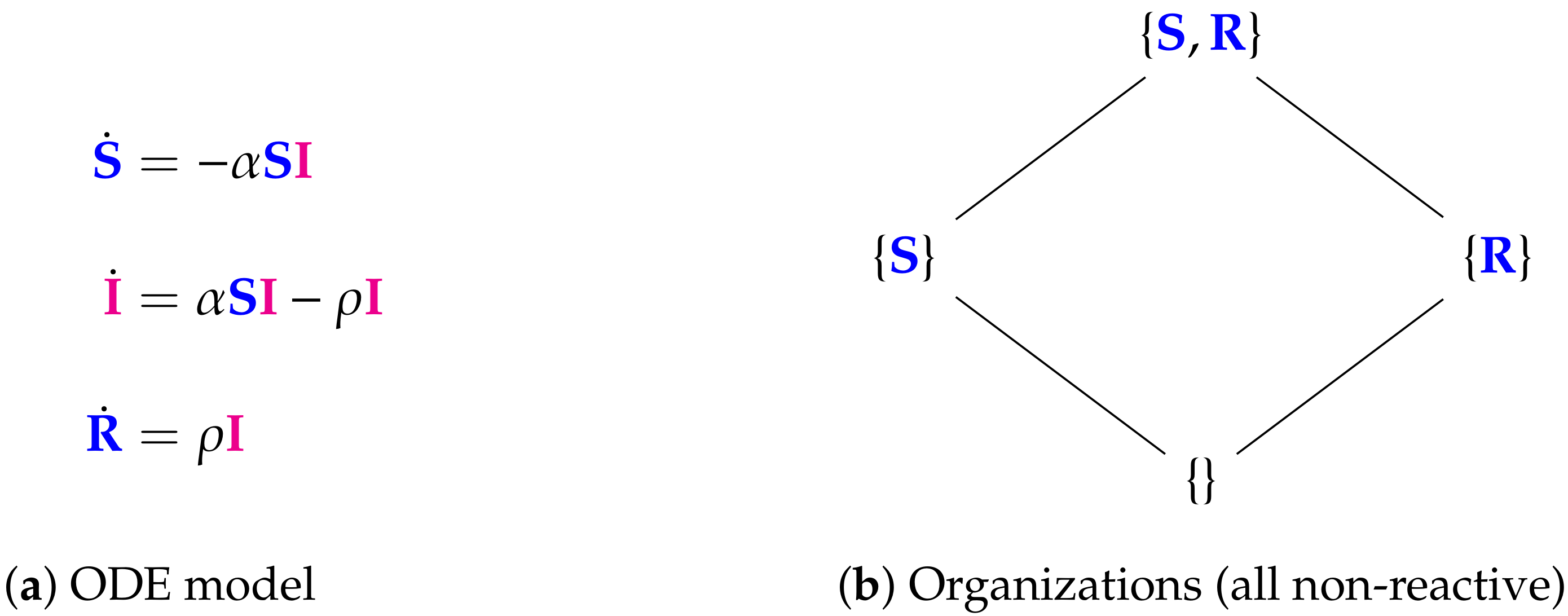
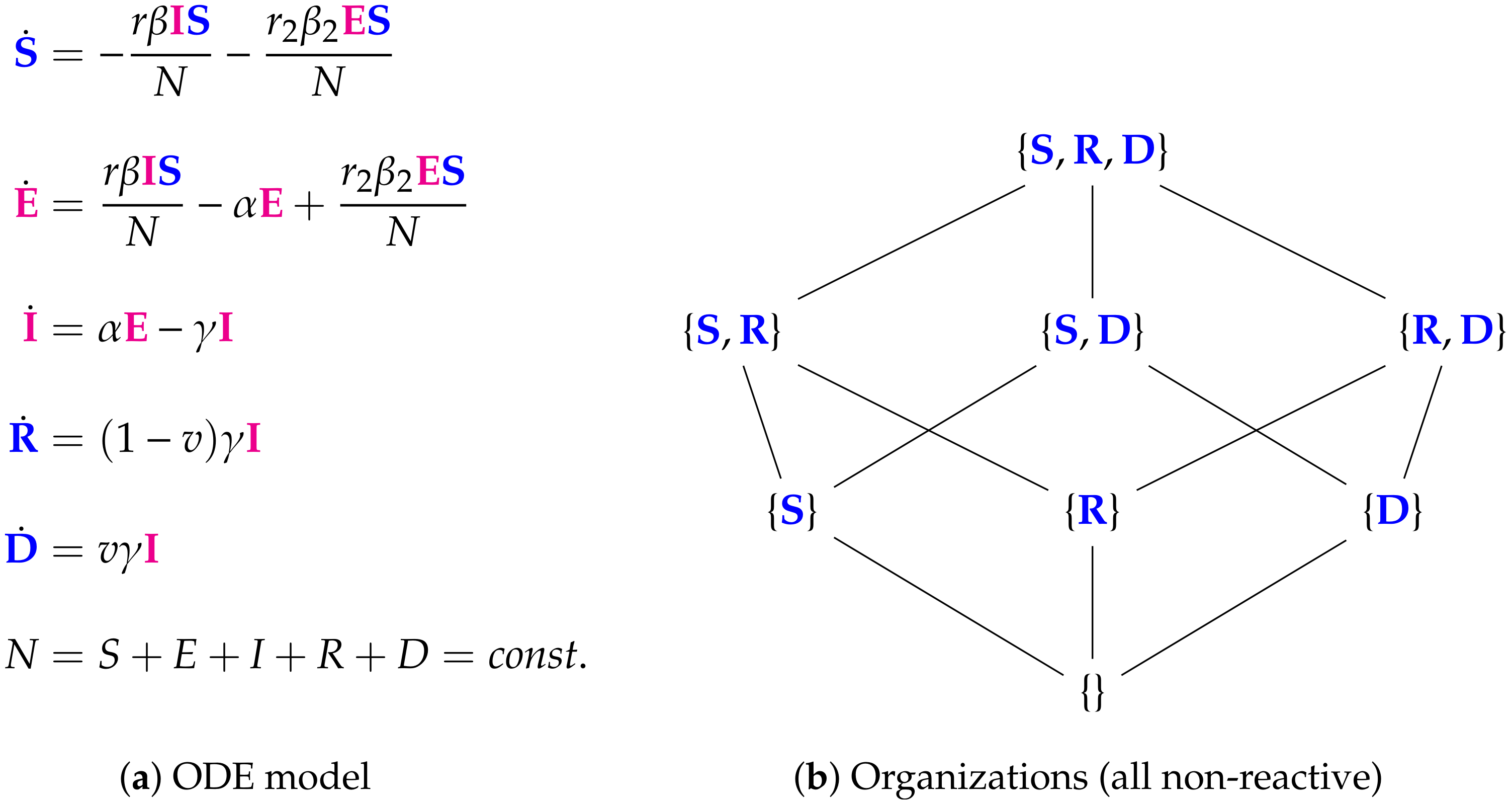
- Step 1—Deriving the set of reactions: Each summand of each ODE (or PDE) is translated into a reaction as illustrated by the transition from Subfigures (a) to (b) in Figure 1. On the left-hand side of each reaction formula, there is a set of species, the so-called support of a reaction. The support of a reaction is the unique set of species that are needed to run the reaction. If only one of the species of the support of the reaction is missing then that reaction is not active. The term (of the ODE (or PDE)) that belongs to that reaction must be zero if and only if the concentration of at least one of the species in the support of that reaction is zero. The number of the appearance of each species of a reaction on the right-hand side of a reaction is bigger or less than the number on the left-hand side depending on whether the regarding term has a positive or negative sign in the ODE (or PDE)) of the regarding species. As an example we consider reaction . The corresponding summand is . It is zero if and only if the concentration of at least one of or is zero. Thus the support of contains exactly the species E and V. On the left-hand side of the reaction equation of the species E resp. V appear only to the power of one because of the power of E resp. V is one in . Since the summand appears only in the ODE of V, namely with a negative sign, the right-hand side of the reaction equation of contains one less of V than the left-hand side. The number of E is equal on both sides of the reaction equation since the amount of E is not affected by the reaction .
- Step 2—Calculating the organizations from the set of reactions: The second step is to compute the organizations (as defined in [21]) from the derived reactions. Each organization consists of a subset of species that is
- closed and
- self-maintaining.
- A subset of species is closed if and only if for each reaction with its support contained in that subset, also all species appearing on the right-hand side of the reaction equation are contained in that subset. In other words, no reaction that is active on the subset S produces a species that is not contained in that subset.
- A subset of species is self-maintaining if and only if there is a feasible flux for S for S such that
- consisting of ODEs but also of PDEs,
- describing in-host dynamics but also host-to-host and mixed (in-host and host-to-host) models.
- Analyzed infection dynamics of SARS-CoV-2 but also compared to Influenza models
3. Analysis of the Models
3.1. In-Host Models
3.2. Host-To-Host Models
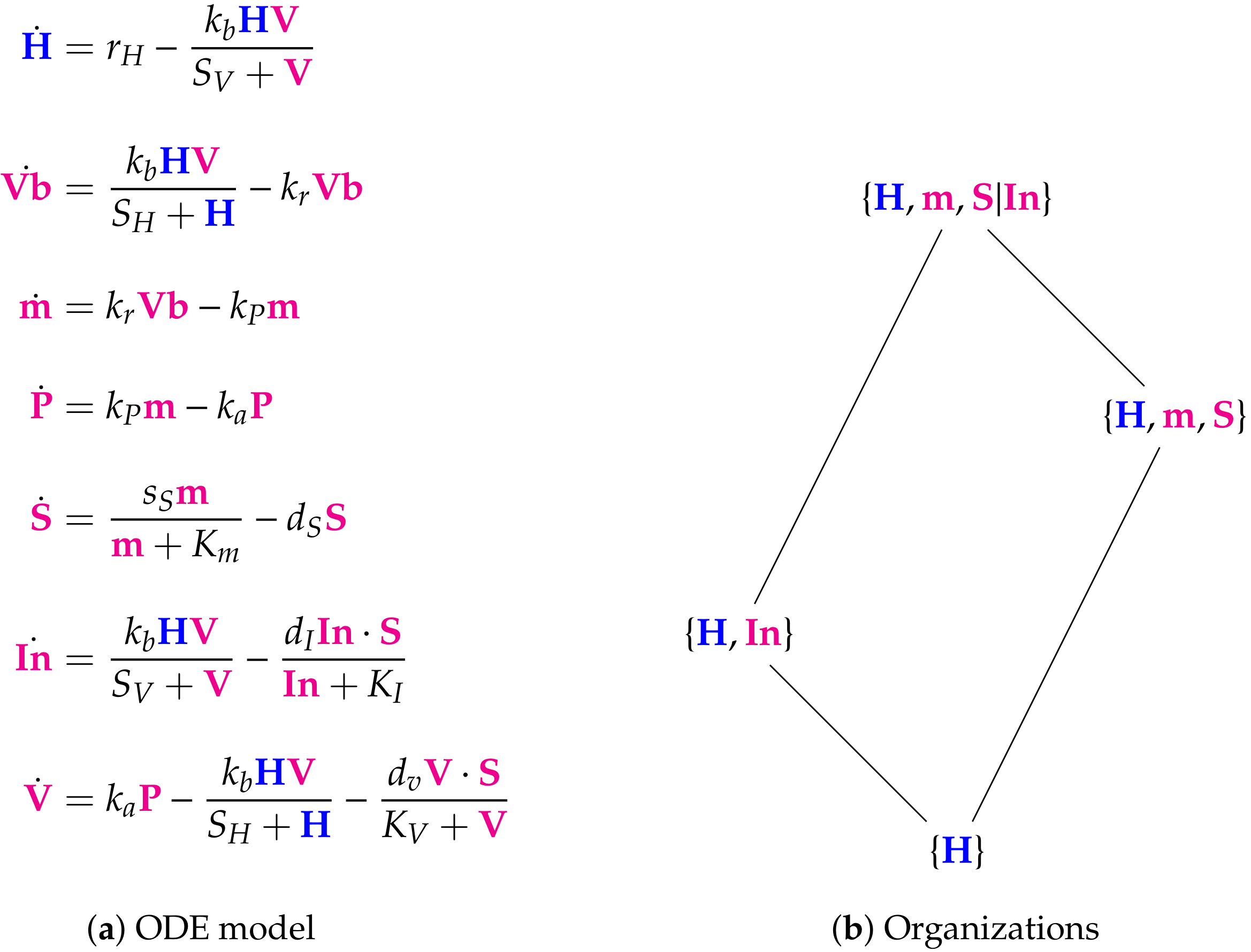
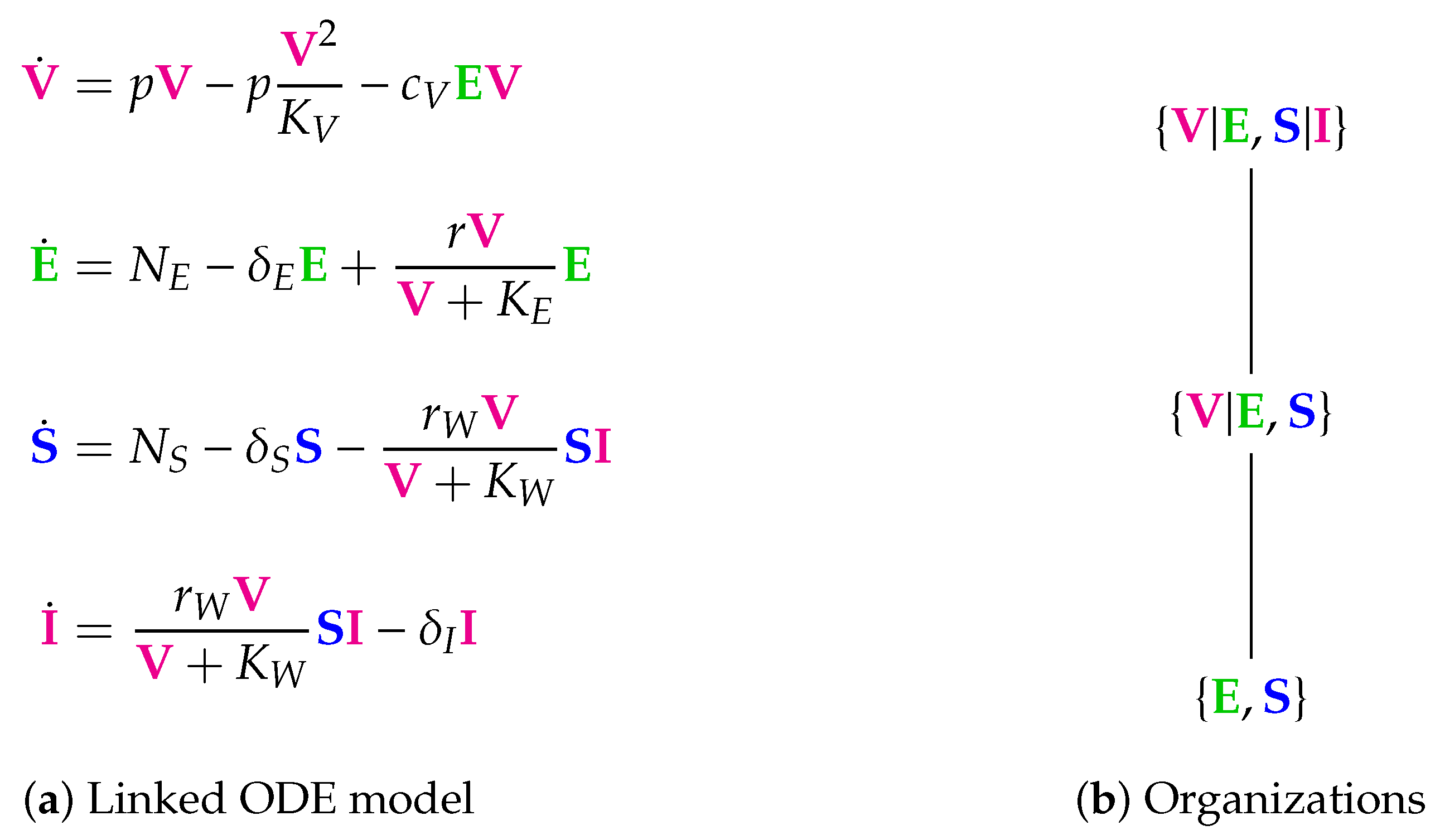
3.3. A Linked In-Host/Host-To-Host Model
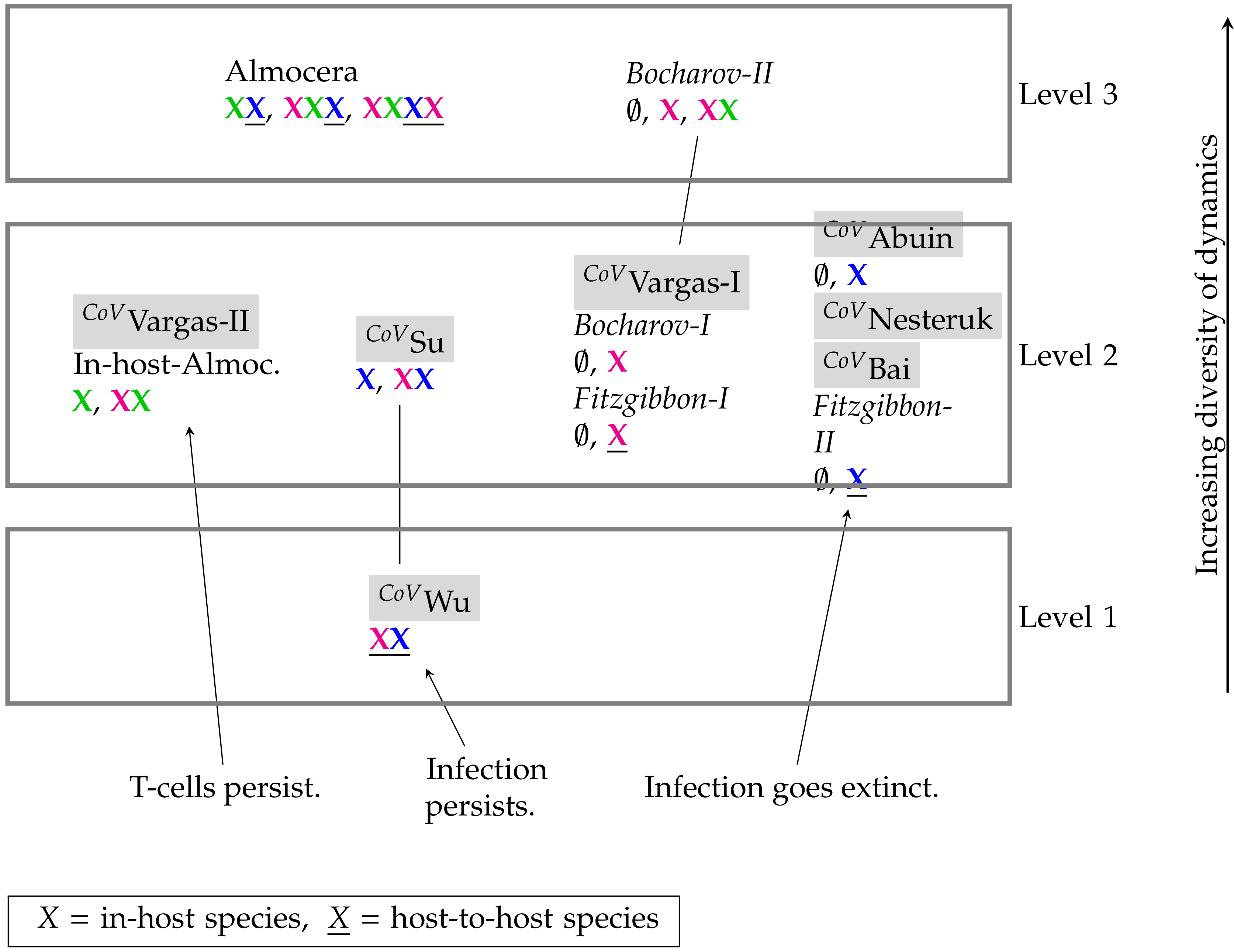
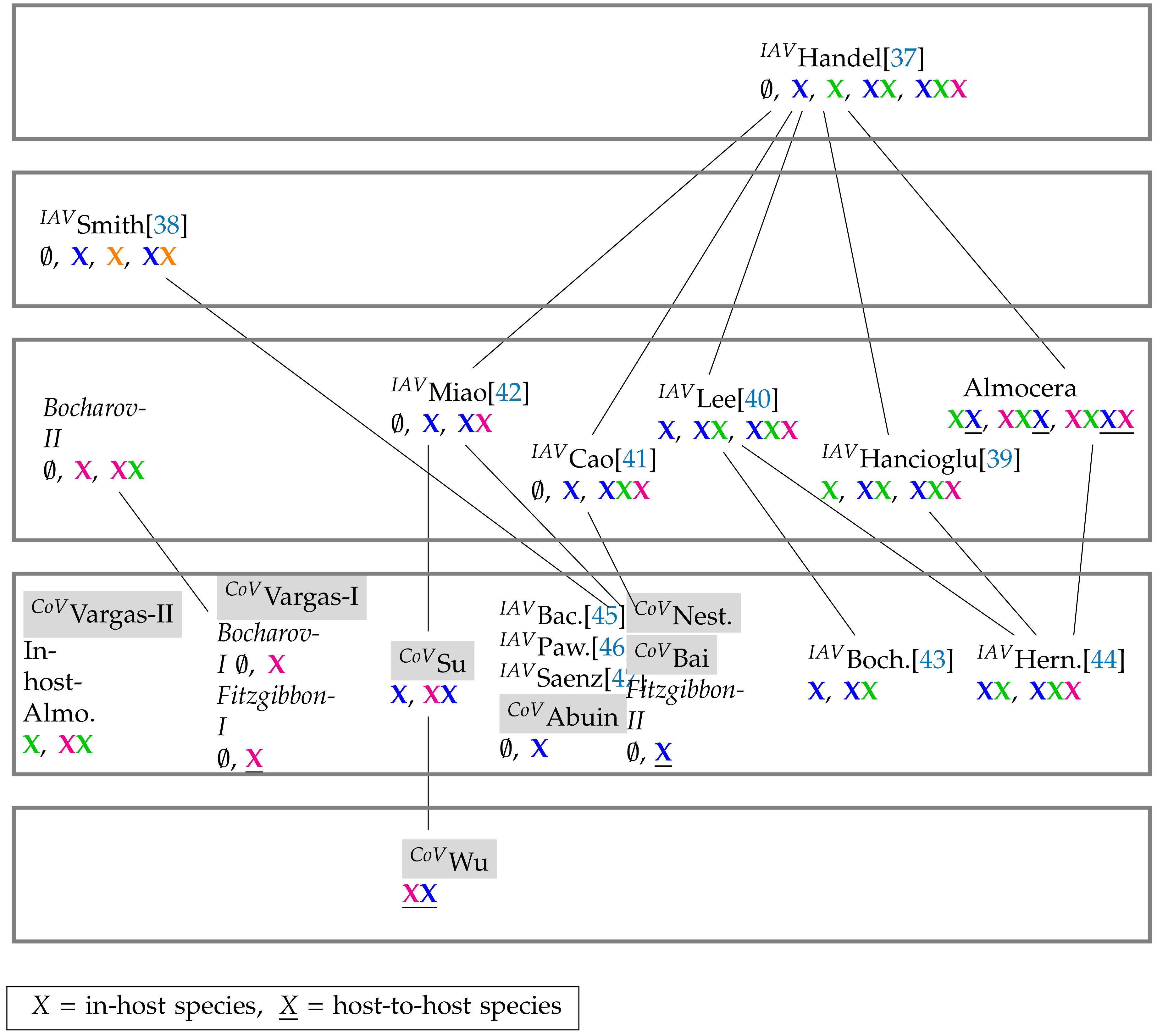
3.4. Hierarchy of Models
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. List of the Reactions of All Models with Reactions Constants in Brackets






References
- Almocera, A.E.S.; Hernandez-Vargas, E.A. Multiscale model within-host and between-host for viral infectious diseases. J. Math. Biol. 2018, 77, 1035–1057. [Google Scholar] [CrossRef] [PubMed]
- Boianelli, A.; Nguyen, V.K.; Ebensen, T.; Schulze, K.; Wilk, E.; Sharma, N.; Stegemann-Koniszewski, S.; Bruder, D.; Toapanta, F.R.; Guzman, C.A.; et al. Modeling Influenza Virus Infection: A Roadmap for Influenza Research. Viruses 2015, 7, 5274–5304. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Vargas, E.A.; Velasco-Hernandez, J.X. In-host Mathematical Modelling of COVID-19 in Humans. Annu. Rev. Control 2020, 50, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Du, S.Q.; Yuan, W. Mathematical modeling of interaction between innate and adaptive immune responses in COVID-19 and implications for viral pathogenesis. J. Med. Virol. 2020, 92, 1615–1628. [Google Scholar] [CrossRef]
- Vargas, E.A.H.; Velasco-Hernandez, J.X. In-host modelling of covid-19 kinetics in humans. medRxiv 2020, 44487. [Google Scholar] [CrossRef]
- Tasevich, A.L.; Bocharov, G.A.; Vol’pert, V.A. Reaction-diffusion equations in immunology. Zhurnal Vychislitel’noi Matematiki i Matematicheskoi Fiziki 2018, 58, 2048–2059. [Google Scholar] [CrossRef]
- Abuin, P.; Anderson, A.; Ferramosca, A.; Hernandez-Vargas, E.A.; Gonzalez, A.H. Characterization of SARS-CoV-2 Dynamics in the Host. arXiv 2020, arXiv2006.08447. [Google Scholar]
- Su, Z.; Wu, Y. A multiscale and comparative model for receptor binding of 2019 novel coronavirus and the implication of its life cycle in host cells. BioRxiv 2020. [Google Scholar] [CrossRef]
- Nesteruk, I. Statistics-Based Predictions of Coronavirus Epidemic Spreading in Mainland China. Innov. Biosyst. Bioeng. 2020. [Google Scholar] [CrossRef]
- Wu, J.T.; Leung, K.; Leung, G.M. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: A modelling study. Lancet 2020, 395, 689–697. [Google Scholar] [CrossRef]
- Fitzgibbon, W.; Morgan, J.; Webb, G.; Wu, Y. Analysis of a reaction–diffusion epidemic model with asymptomatic transmission. J. Biol. Syst. 2020, 28, 561–587. [Google Scholar] [CrossRef]
- Krishna, M.V.; Prakash, J. Mathematical modelling on phase based transmissibility of Coronavirus. Infect. Dis. Model. 2020, 5, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.; Sieber, P.; Garde, R.; Lang, S.N.; Schuster, S.; Ibrahim, B. Trends in mathematical modeling of host–pathogen interactions. Cell. Mol. Life Sci. 2020, 77, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Eker, S. Validity and usefulness of COVID-19 models. Humanit. Soc. Sci. Commun. 2020, 7, 1–5. [Google Scholar] [CrossRef]
- Ibrahim, B.; Henze, R.; Gruenert, G.; Egbert, M.; Huwald, J.; Dittrich, P. Spatial rule-based modeling: A method and its application to the human mitotic kinetochore. Cells 2013, 2, 506–544. [Google Scholar] [CrossRef]
- Wang, J. Mathematical models for COVID-19: Applications, limitations, and potentials. J. Public Health Emerg. 2020. [Google Scholar] [CrossRef]
- Hufsky, F.; Lamkiewicz, K.; Almeida, A.; Aouacheria, A.; Arighi, C.; Bateman, A.; Baumbach, J.; Beerenwinkel, N.; Brandt, C.; Cacciabue, M.; et al. Computational Strategies to Combat COVID-19: Useful Tools to Accelerate SARS-CoV-2 and Coronavirus Research. Briefings Bioinformat. 2020. [Google Scholar] [CrossRef]
- Ibrahim, B.; McMahon, D.P.; Hufsky, F.; Beer, M.; Deng, L.; Le Mercier, P.; Palmarini, M.; Thiel, V.; Marz, M. A new era of virus bioinformatics. Virus Res. 2018, 251, 86–90. [Google Scholar] [CrossRef]
- Peter, S.; Hölzer, M.; Lamkiewicz, K.; Di Fenizio, P.S.; Al Hwaeer, H.; Marz, M.; Schuster, S.; Dittrich, P.; Ibrahim, B. Structure and hierarchy of influenza virus models revealed by reaction network analysis. Viruses 2019, 11, 449. [Google Scholar] [CrossRef]
- Speroni di Fenizio, P.; Dittrich, P. Chemical Organizations at Different Spatial Scales. In Advances in Artificial Life; Springer: Berlin/Heidelberg, Germany, 2007; pp. 1–11. [Google Scholar]
- Di Fenizio, P.S.; Dittrich, P.; Banzhaf, W.; Ziegler, J. Towards a theory of organizations. In German Workshop on Artificial Life (GWAL 2000); DUV: Bayreuth, Germany, 2000. [Google Scholar]
- Peter, S.; Dittrich, P. On the Relation between Organizations and Limit Sets in Chemical Reaction Systems. Adv. Complex Syst. 2011, 14, 77–96. [Google Scholar] [CrossRef]
- Dittrich, P.; Speroni di Fenizio, P. Chemical Organization Theory. Bull. Math. Biol. 2007, 69, 1199–1231. [Google Scholar] [CrossRef] [PubMed]
- Kreyssig, P.; Wozar, C.; Peter, S.; Veloz, T.; Ibrahim, B.; Dittrich, P. Effects of small particle numbers on long-term behaviour in discrete biochemical systems. Bioinformatics 2014, 30, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Peter, S.; Ghanim, F.; Dittrich, P.; Ibrahim, B. Organizations in reaction-diffusion systems: Effects of diffusion and boundary conditions. Ecol. Complex. 2020, 43, 100855. [Google Scholar] [CrossRef]
- Bai, S. Simulations of COVID-19 spread by spatial agent-based model and ordinary differential equations. Int. J. Simul. Process. Model. 2020, 15, 268–277. [Google Scholar] [CrossRef]
- Wölfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Müller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef]
- Hernandez-Vargas, E.A. Modeling and Control of Infectious Diseases in the Host: With MATLAB and R; Academic Press: New York, NY, USA, 2019. [Google Scholar]
- Perelson, A.S. Modelling viral and immune system dynamics. Nat. Rev. Immunol. 2002, 2, 28–36. [Google Scholar] [CrossRef]
- Ciupe, S.M.; Heffernan, J.M. In-host modeling. Infect. Dis. Model. 2017, 2, 188–202. [Google Scholar] [CrossRef]
- Kreyssig, P.; Escuela, G.; Reynaert, B.; Veloz, T.; Ibrahim, B.; Dittrich, P. Cycles and the qualitative evolution of chemical systems. PLoS ONE 2012, 7, e45772. [Google Scholar] [CrossRef]
- Murray, J.D. Mathematical biology: I. An introduction. Interdisciplinary applied mathematics. In Mathematical Biology; Springer: New York, NY, USA, 2002. [Google Scholar]
- Bailey, N.T. The Mathematical Theory of Epidemics; Technical Report; Griffin: London, UK, 1957. [Google Scholar]
- Weitz, J.S.; Dushoff, J. Modeling post-death transmission of Ebola: Challenges for inference and opportunities for control. Sci. Rep. 2015, 5, 8751. [Google Scholar] [CrossRef]
- Shao, P.; Shan, Y. Beware of asymptomatic transmission: Study on 2019-nCoV prevention and control measures based on extended SEIR model. BioRxiv 2020. [Google Scholar] [CrossRef]
- Anderson, R.M.; Anderson, B.; May, R.M. Infectious Diseases of Humans: Dynamics and Control; Oxford University Press: New York, NY, USA, 1992. [Google Scholar]
- Handel, A.; Longini, I.M., Jr.; Antia, R. Towards a quantitative understanding of the within-host dynamics of influenza A infections. J. R. Soc. Interface 2009, 7, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Smith, A.P. A Critical, Nonlinear Threshold Dictates Bacterial Invasion and Initial Kinetics During Influenza. Sci. Rep. 2016, 6, 38703. [Google Scholar] [CrossRef] [PubMed]
- Hancioglu, B.; Swigon, D.; Clermont, G. A dynamical model of human immune response to influenza A virus infection. J. Theor. Biol. 2007, 246, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Topham, D.J.; Park, S.Y.; Hollenbaugh, J.; Treanor, J.; Mosmann, T.R.; Jin, X.; Ward, B.M.; Miao, H.; Holden-Wiltse, J.; et al. Simulation and prediction of the adaptive immune response to influenza A virus infection. J. Virol. 2009, 83, 7151–7165. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Yan, A.W.; Heffernan, J.M.; Petrie, S.; Moss, R.G.; Carolan, L.A.; Guarnaccia, T.A.; Kelso, A.; Barr, I.G.; McVernon, J.; et al. Innate Immunity and the Inter-exposure Interval Determine the Dynamics of Secondary Influenza Virus Infection and Explain Observed Viral Hierarchies. PLoS Comput. Biol. 2015, 11, e1004334. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Hollenbaugh, J.A.; Zand, M.S.; Holden-Wiltse, J.; Mosmann, T.R.; Perelson, A.S.; Wu, H.; Topham, D.J. Quantifying the early immune response and adaptive immune response kinetics in mice infected with influenza A virus. J. Virol. 2010, 84, 6687–6698. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, G.A.; Romanyukha, A.A. Mathematical model of antiviral immune response. III. Influenza A virus infection. J. Theor. Biol. 1994, 167, 323–360. [Google Scholar] [CrossRef]
- Hernandez-Vargas, A.E.; Meyer-Hermann, M. Innate immune system dynamics to influenza virus. Ifac Proc. Vol. 2012, 45, 260–265. [Google Scholar] [CrossRef]
- Baccam, P.; Beauchemin, C.; Macken, C.A.; Hayden, F.G.; Perelson, A.S. Kinetics of influenza A virus infection in humans. J. Virol. 2006, 80, 7590–7599. [Google Scholar] [CrossRef]
- Pawelek, K.A.; Huynh, G.T.; Quinlivan, M.; Cullinane, A.; Rong, L.; Perelson, A.S. Modeling within-host dynamics of influenza virus infection including immune responses. PLoS Comput. Biol. 2012, 8, e1002588. [Google Scholar] [CrossRef]
- Saenz, R.A.; Quinlivan, M.; Elton, D.; Macrae, S.; Blunden, A.S.; Mumford, J.A.; Daly, J.M.; Digard, P.; Cullinane, A.; Grenfell, B.T.; et al. Dynamics of influenza virus infection and pathology. J. Virol. 2010, 84, 3974–3983. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
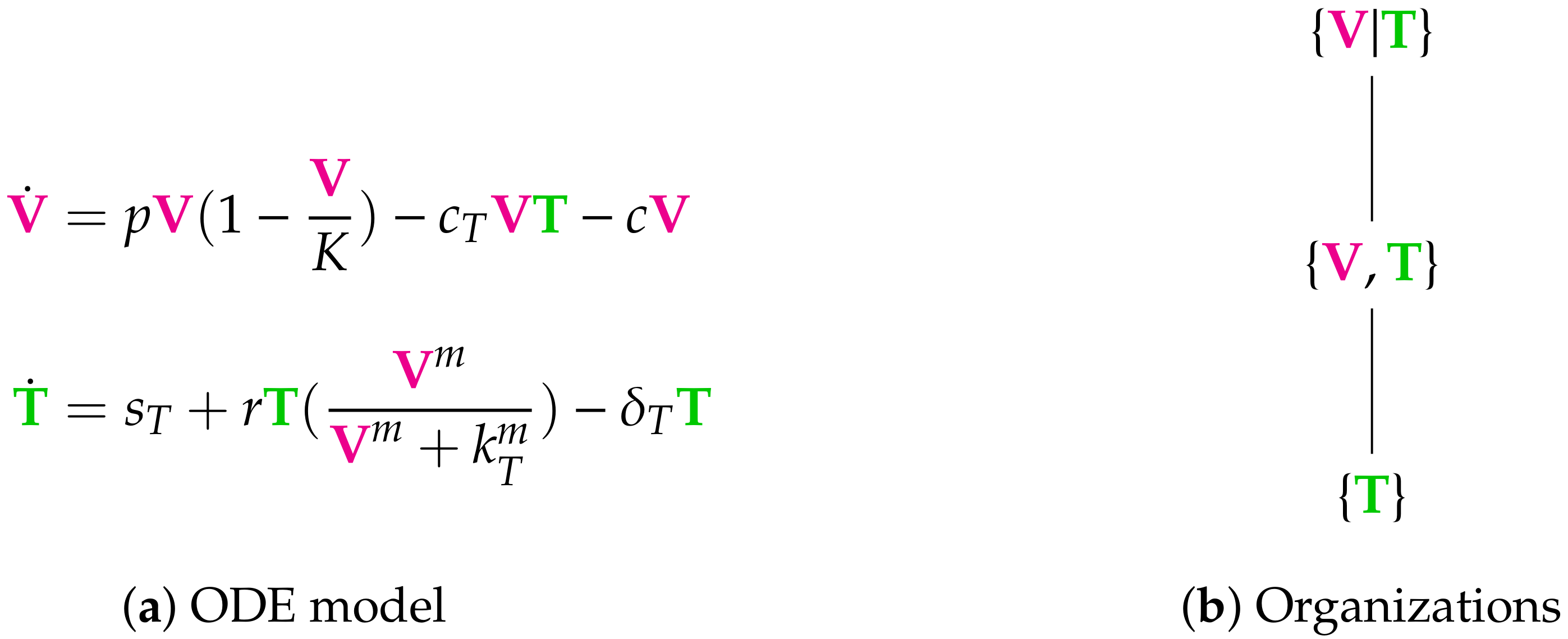{kind=link}
{kind=link}
{kind=link}
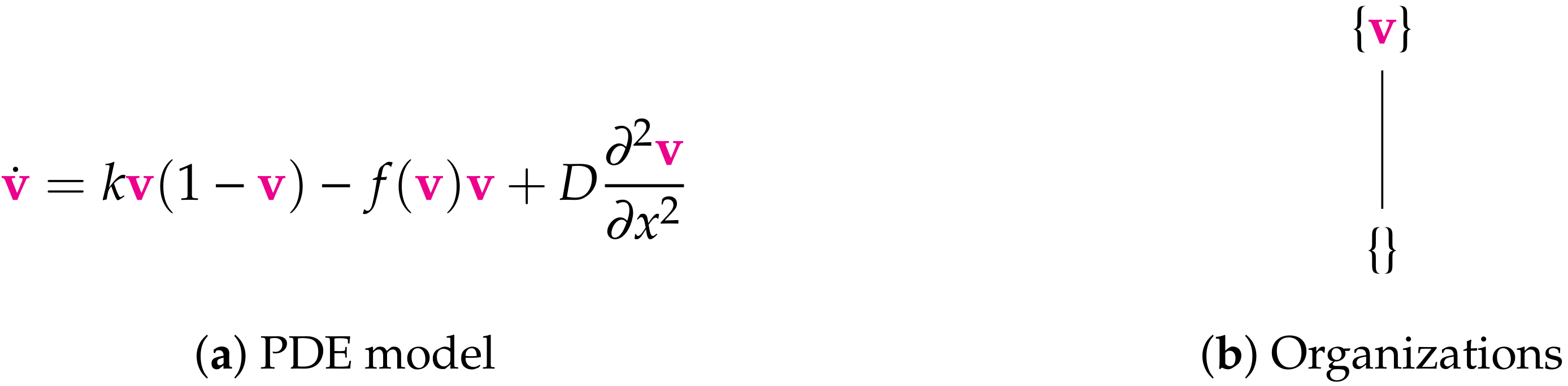{kind=link}
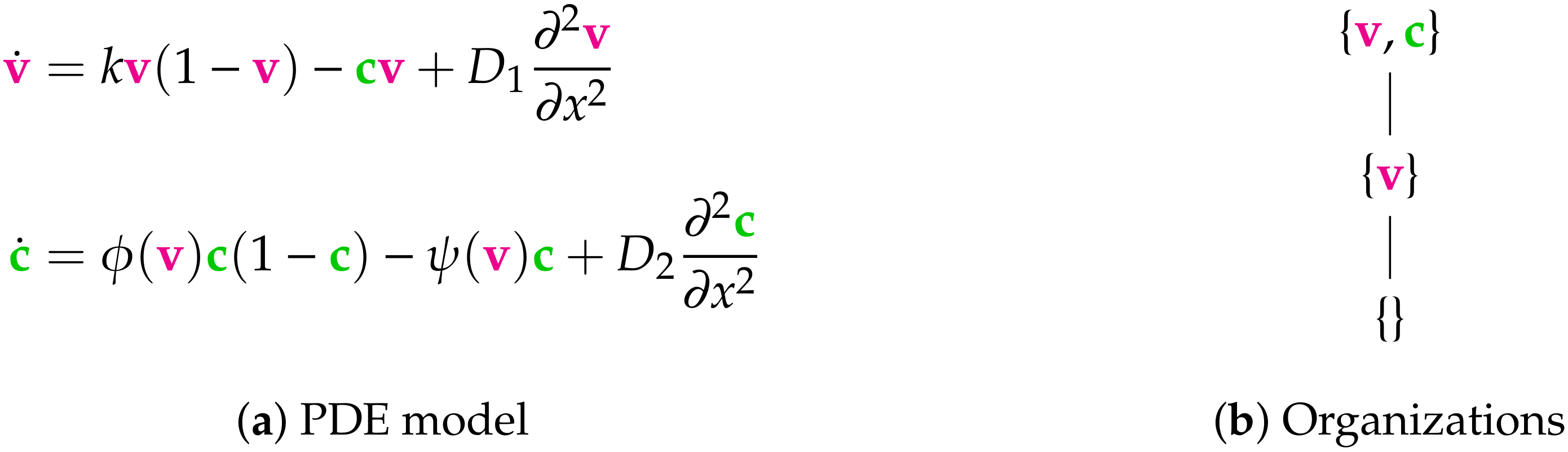{kind=link}
{kind=link}
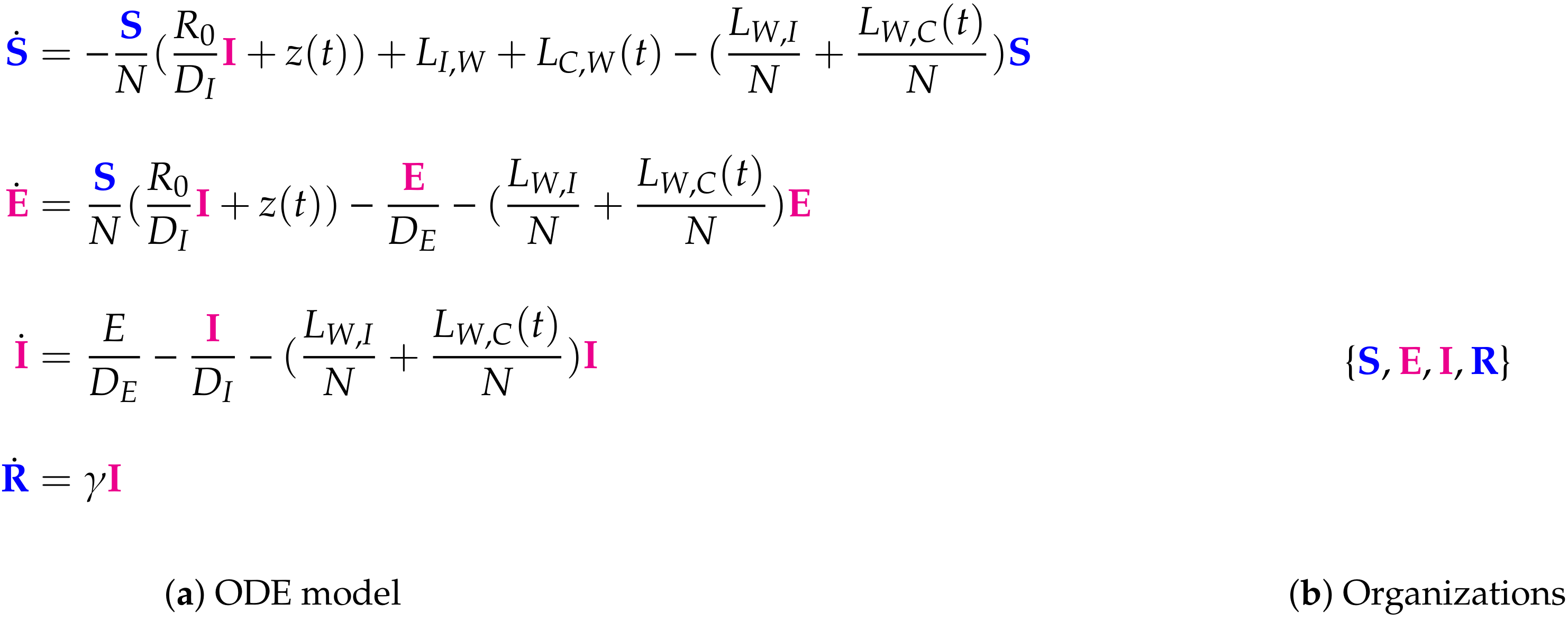{kind=link}
{kind=link}
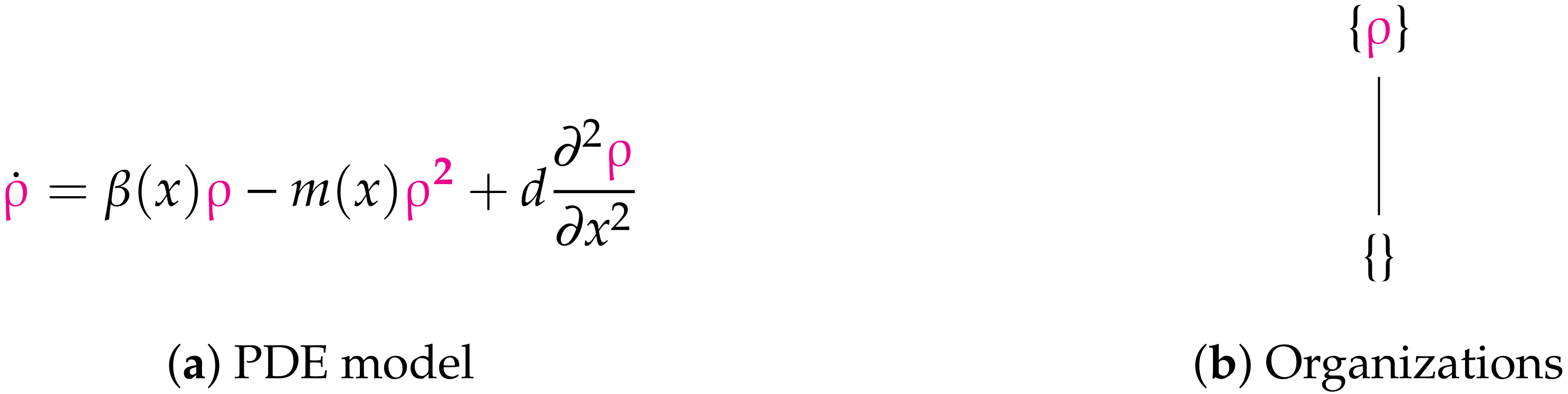{kind=link}
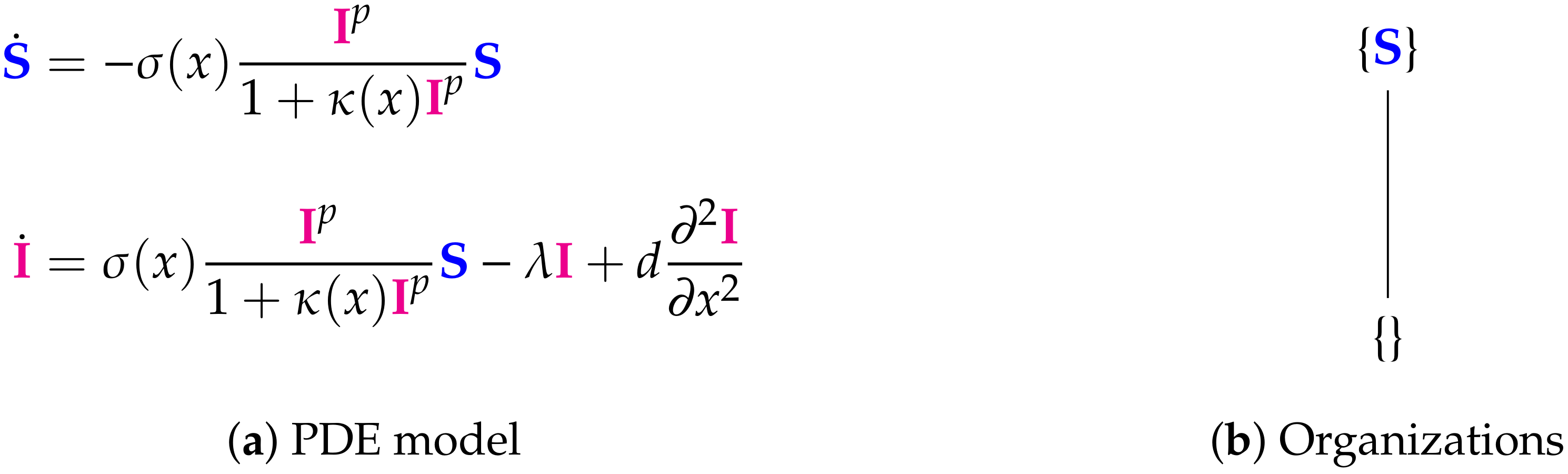{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
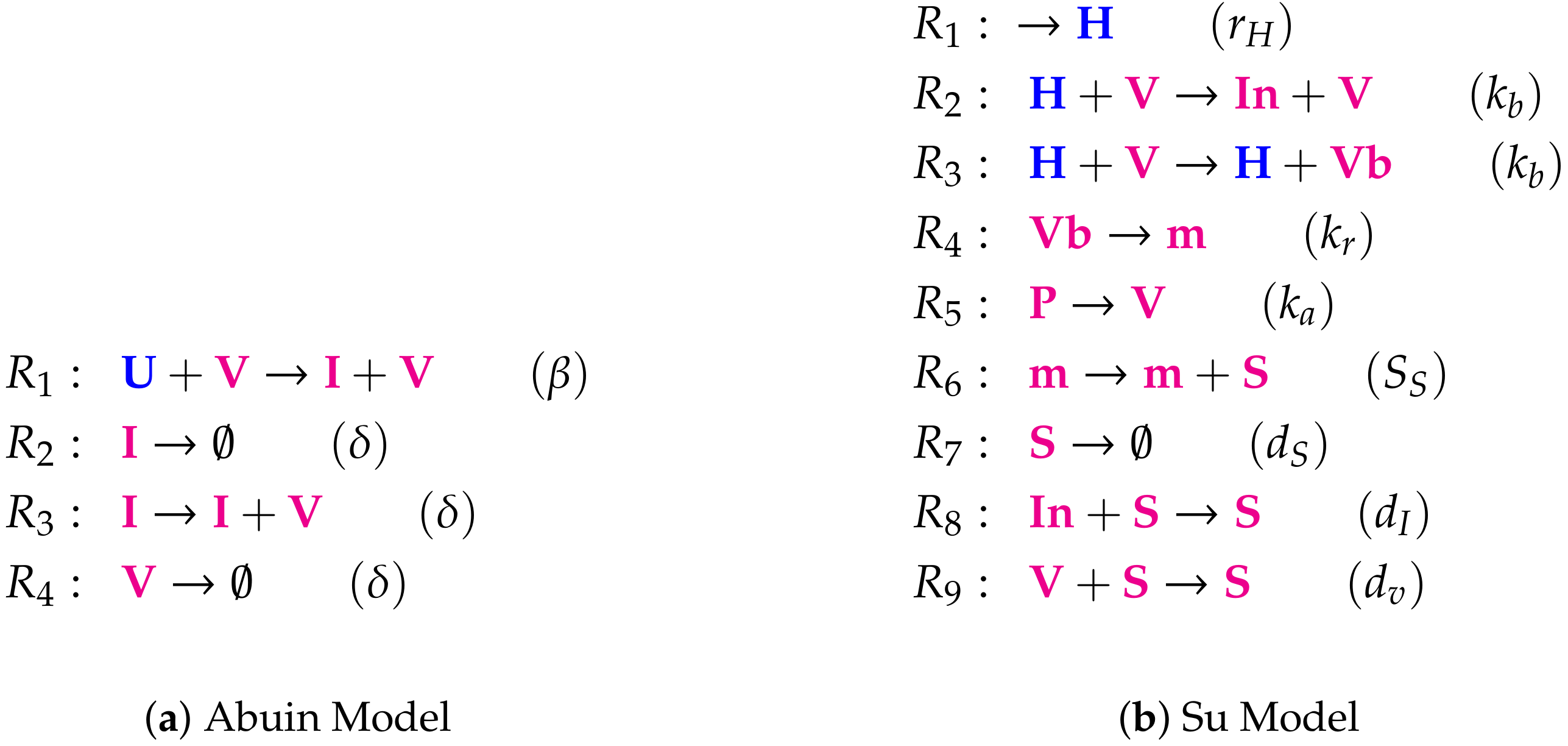{kind=link}
{kind=link}
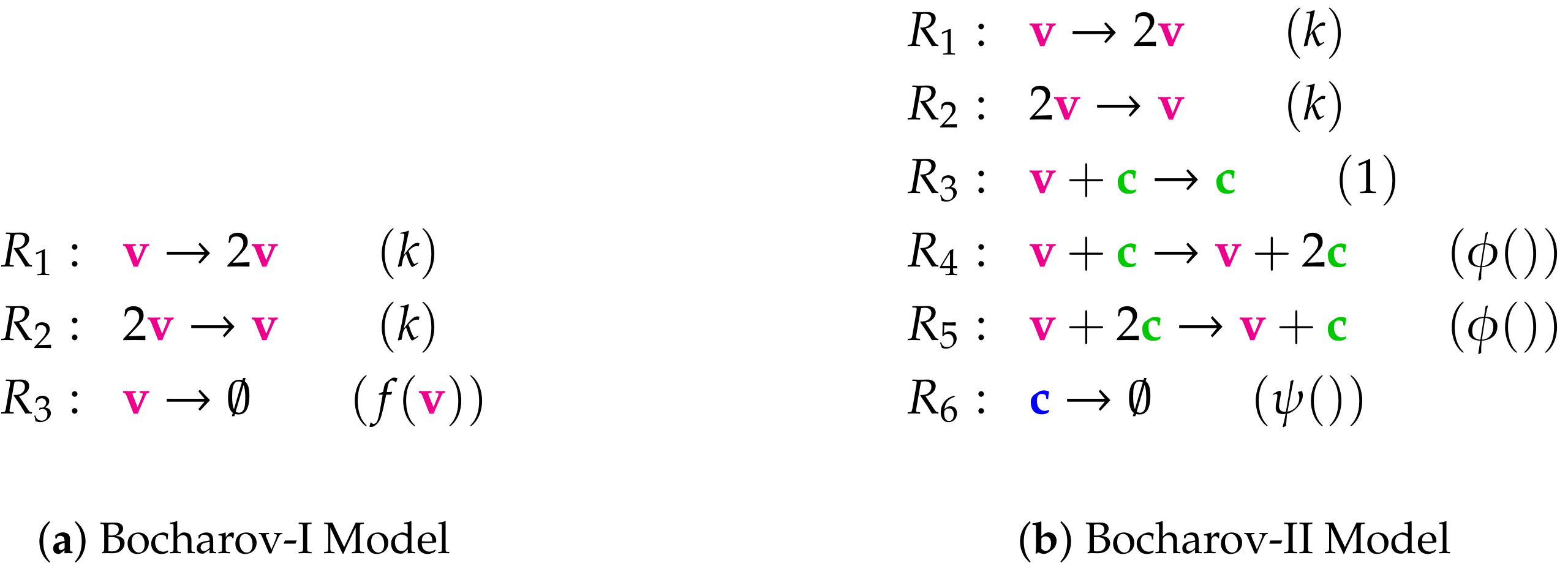{kind=link}
{kind=link}
{kind=link}
| Model Type | ODE | PDE |
|---|---|---|
| In-host | [5] CoVVargas-I: | [6] Bocharov-I: |
| [5] CoVVargas-II: | [6] Bocharov-II: | |
| [7]CoVAbuin: | ||
| [8] CoVSu: | ||
| Host-to-host | [9] CoVNesteruk: | [11] CoVFitzgibbon-I: |
| [10] CoVWu: | [11] CoVFitzgibbon-II: | |
| [26] CoVBai: | ||
| Linked | [1] Almocera: | |
| In-Host: ; Linked: |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peter, S.; Dittrich, P.; Ibrahim, B. Structure and Hierarchy of SARS-CoV-2 Infection Dynamics Models Revealed by Reaction Network Analysis. Viruses 2021, 13, 14. https://doi.org/10.3390/v13010014
Peter S, Dittrich P, Ibrahim B. Structure and Hierarchy of SARS-CoV-2 Infection Dynamics Models Revealed by Reaction Network Analysis. Viruses. 2021; 13(1):14. https://doi.org/10.3390/v13010014
Chicago/Turabian StylePeter, Stephan, Peter Dittrich, and Bashar Ibrahim. 2021. "Structure and Hierarchy of SARS-CoV-2 Infection Dynamics Models Revealed by Reaction Network Analysis" Viruses 13, no. 1: 14. https://doi.org/10.3390/v13010014
APA StylePeter, S., Dittrich, P., & Ibrahim, B. (2021). Structure and Hierarchy of SARS-CoV-2 Infection Dynamics Models Revealed by Reaction Network Analysis. Viruses, 13(1), 14. https://doi.org/10.3390/v13010014