
Single-Molecule Super-Resolution Imaging of T-Cell Plasma Membrane CD4 Redistribution upon HIV-1 Binding

 ,
,  , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Antibody Conjugation
2.3. Phorbol Ester Stimulation
2.4. Virus Preparation
2.5. Virus Titration
2.6. HIV Binding
2.7. gp120 Binding
2.8. Imaging
2.9. Localisation Algorithm
2.10. Cluster Analysis
2.11. Cluster Manual Annotation
2.12. Channel Registration
2.13. Colocalisation Analysis
2.14. Molecular Counting
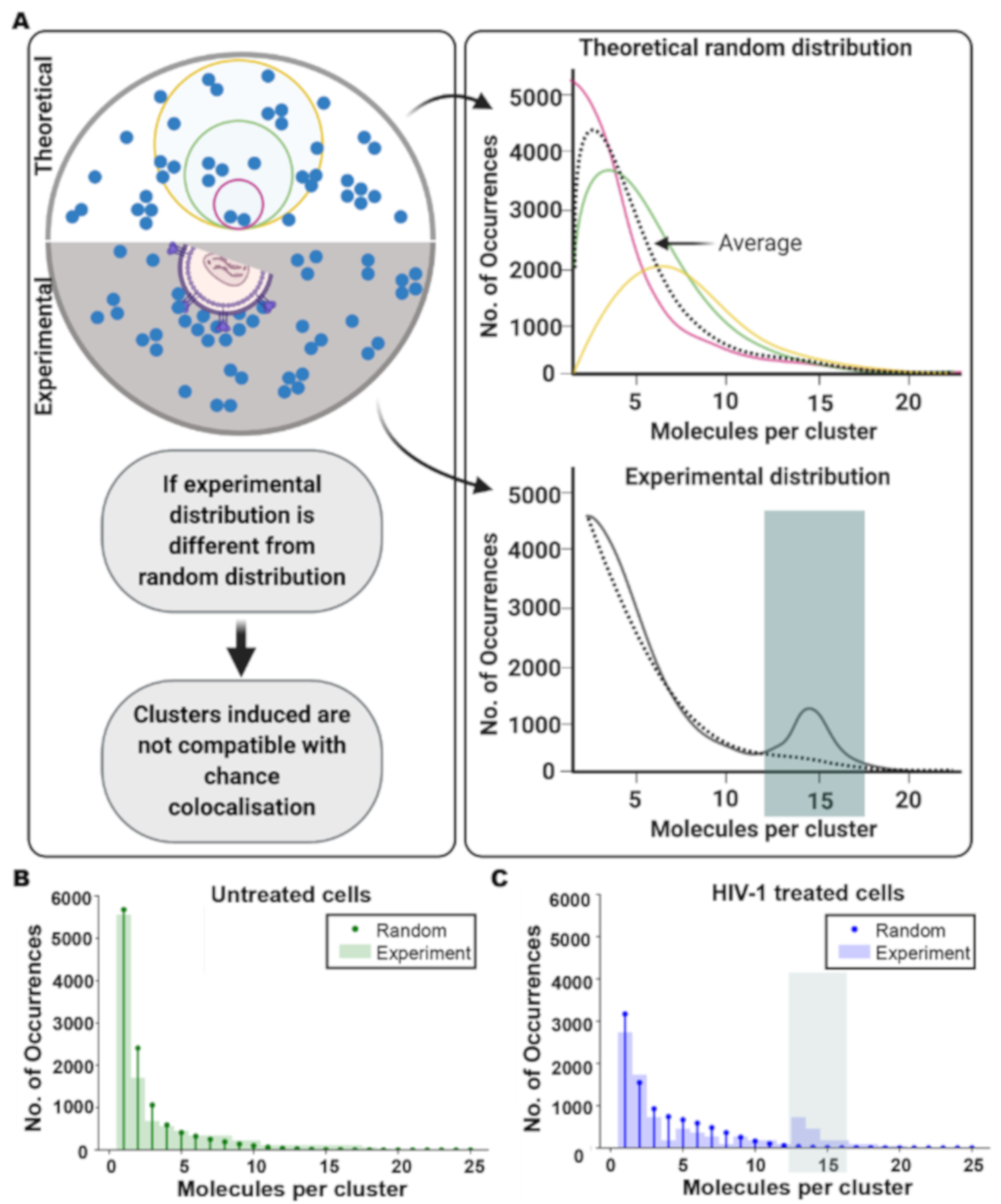
2.15. Theoretical Poisson Statistical Model
2.16. Statistical Analysis
3. Results
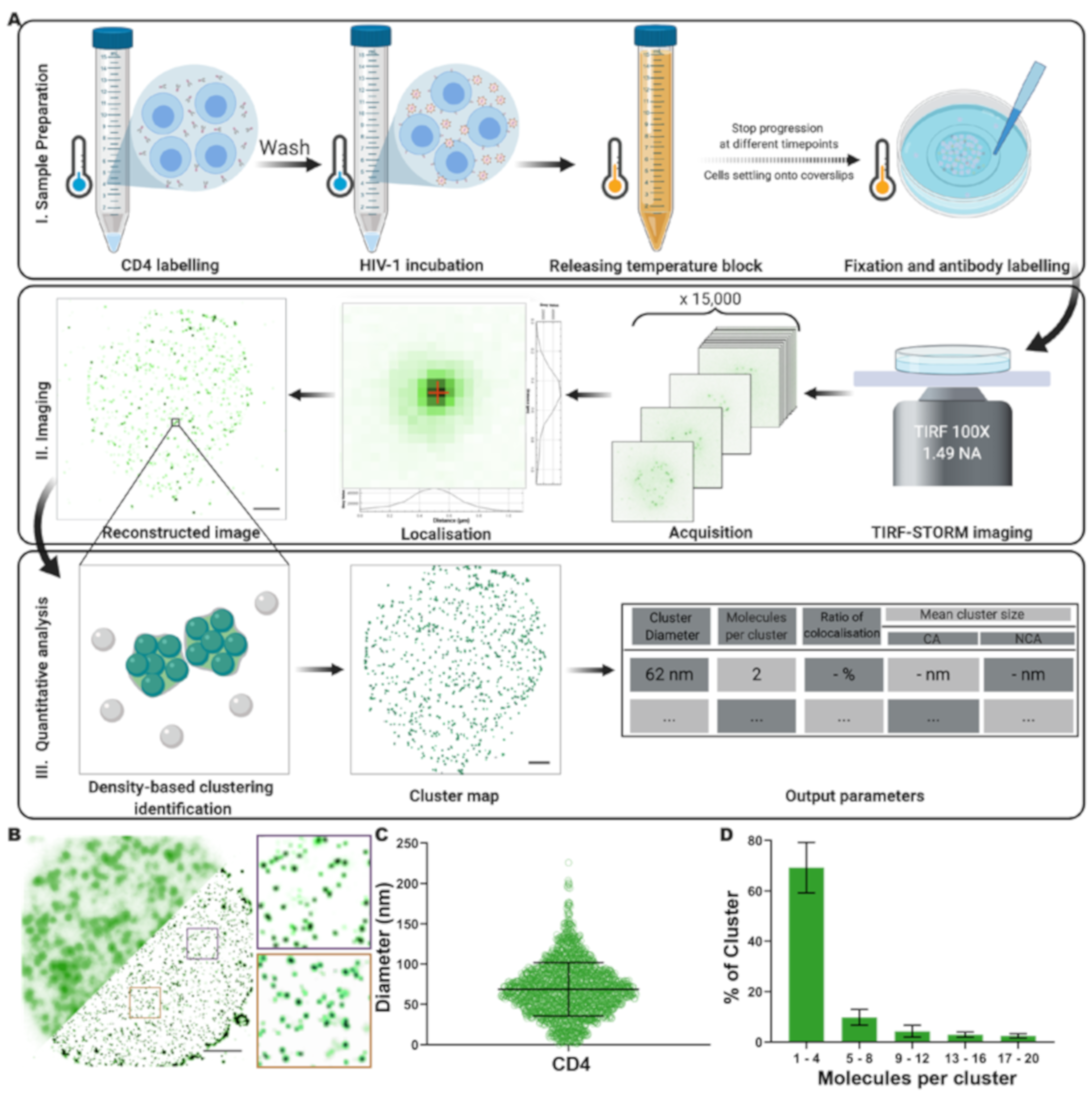
3.1. Analysis of CD4 Nanoclusters on CD4+ T Cells by Quantitative Super-Resolution Imaging
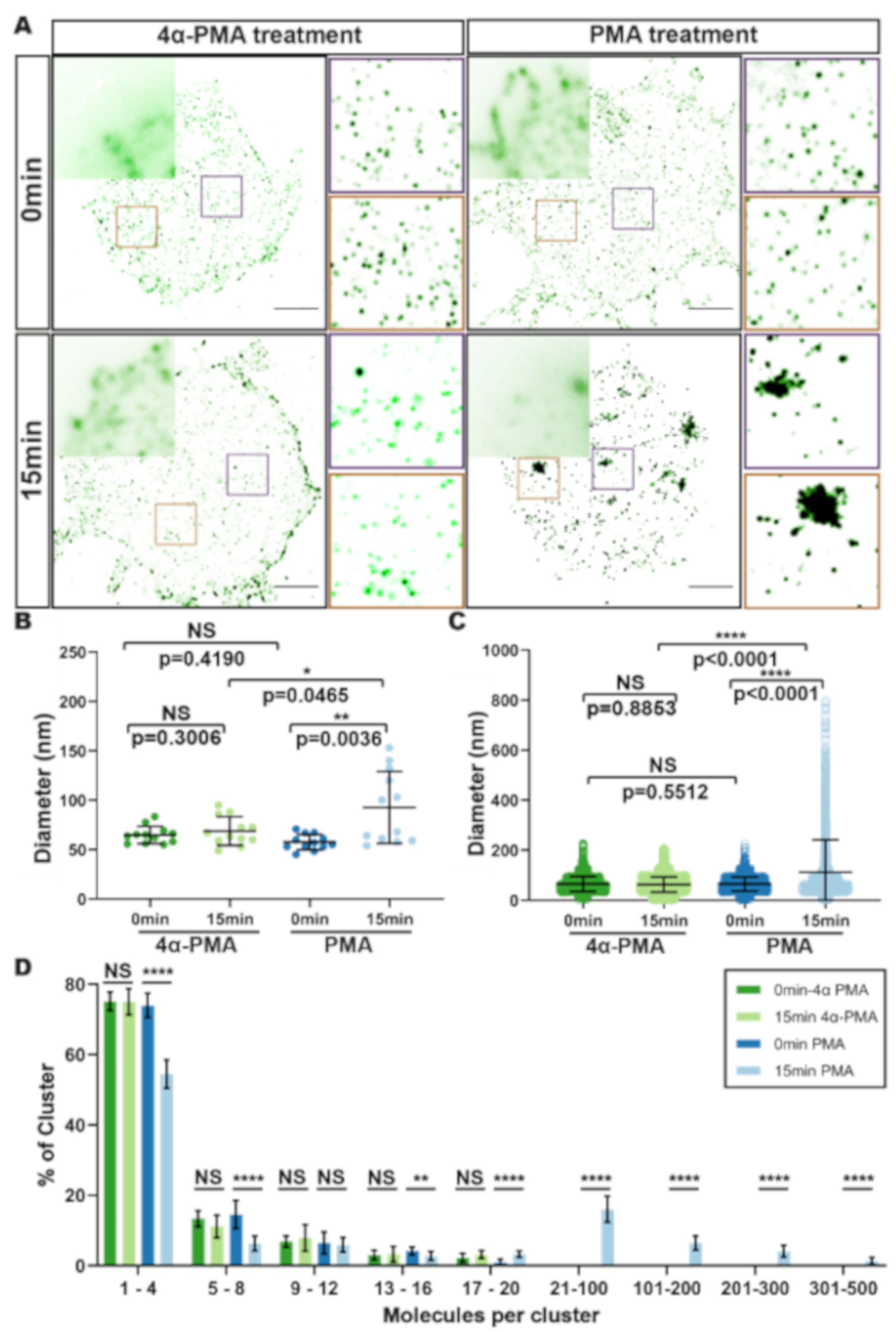
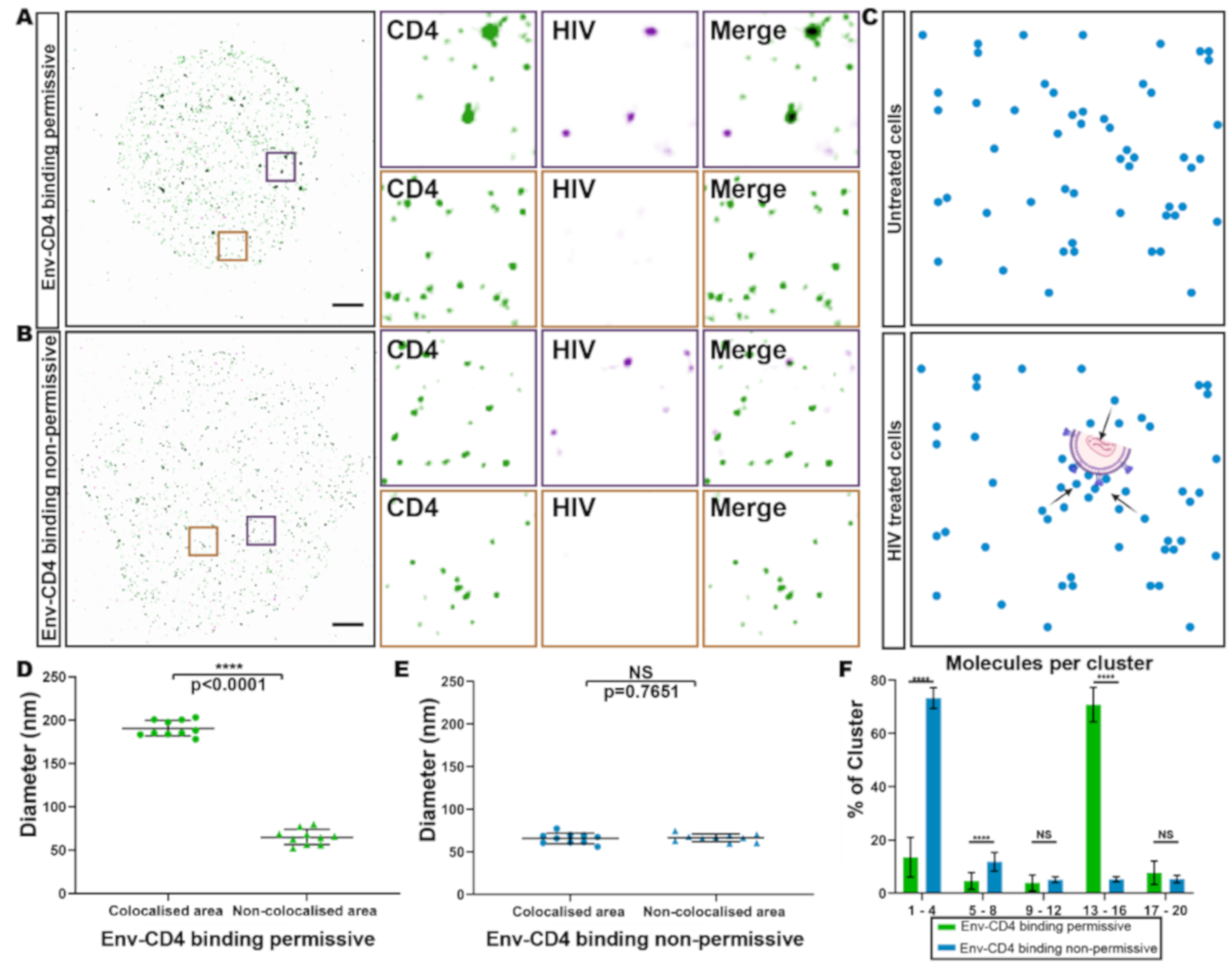
3.2. HIV Receptor Binding Induces Changes in CD4 Clustering
3.3. HIV gp120-Induced CD4 Clustering is Independent of CCR5
4. Discussions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grove, J.; Marsh, M. The cell biology of receptor-mediated virus entry. J. Cell Biol. 2011, 195, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Llorente García, I.; Marsh, M. A biophysical perspective on receptor-mediated virus entry with a focus on HIV. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183158. [Google Scholar] [CrossRef] [PubMed]
- Hartman, N.C.; Groves, J.T. Signaling clusters in the cell membrane. Curr. Opin. Cell Biol. 2011, 23, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Roh, K.H.; Lillemeier, B.F.; Wang, F.; Davis, M.M. The coreceptor CD4 is expressed in distinct nanoclusters and does not colocalize with T-cell receptor and active protein tyrosine kinase p56lck. Proc. Natl. Acad. Sci. USA 2015, 112, E1604–E1613. [Google Scholar] [CrossRef]
- Lillemeier, B.F.; Mörtelmaier, M.A.; Forstner, M.B.; Huppa, J.B.; Groves, J.T.; Davis, M.M. TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat. Immunol. 2010, 11, 90–96. [Google Scholar] [CrossRef]
- Razvag, Y.; Neve-Oz, Y.; Sajman, J.; Yakovian, O.; Reches, M.; Sherman, E. T Cell Activation through Isolated Tight Contacts. Cell Rep. 2019, 29, 3506–3521. [Google Scholar] [CrossRef]
- Layne, S.P.; Merges, M.J.; Dembo, M.; Spouge, J.L.; Nara, P.L. HIV requires multiple gp120 molecules for CD4-mediated infection. Nature 1990, 346, 277–279. [Google Scholar] [CrossRef]
- Kuhmann, S.E.; Platt, E.J.; Kozak, S.L.; Kabat, D. Cooperation of Multiple CCR5 Coreceptors Is Required for Infections by Human Immunodeficiency Virus Type 1. J. Virol. 2000, 74, 7005–7015. [Google Scholar] [CrossRef]
- Platt, E.J.; Durnin, J.P.; Shinde, U.; Kabat, D. An Allosteric Rheostat in HIV-1 gp120 Reduces CCR5 Stoichiometry Required for Membrane Fusion and Overcomes Diverse Entry Limitations. J. Mol. Biol. 2007, 374, 64–79. [Google Scholar] [CrossRef]
- Chang, M.I.; Panorchan, P.; Dobrowsky, T.M.; Tseng, Y.; Wirtz, D. Single-Molecule Analysis of Human Immunodeficiency Virus Type 1 gp120-Receptor Interactions in Living Cells. J. Virol. 2005, 79, 14748–14755. [Google Scholar] [CrossRef]
- Dobrowsky, T.M.; Zhou, Y.; Sun, S.X.; Siliciano, R.F.; Wirtz, D. Monitoring Early Fusion Dynamics of Human Immunodeficiency Virus Type 1 at Single-Molecule Resolution. J. Virol. 2008, 82, 7022–7033. [Google Scholar] [CrossRef] [PubMed]
- Chojnacki, J.; Waithe, D.; Carravilla, P.; Huarte, N.; Galiani, S.; Enderlein, J.; Eggeling, C. Envelope glycoprotein mobility on HIV-1 particles depends on the virus maturation state. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Chertova, E.; Bess, J.; Lifson, J.D.; Arthur, L.O.; Liu, J.; Taylor, K.A.; Roux, K.H. Electron tomography analysis of envelope glycoprotein trimers on HIV and simian immunodeficiency virus virions. Proc. Natl. Acad. Sci. USA 2003, 100, 15812–15817. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Liu, J.; Bess, J.; Chertova, E.; Lifson, J.D.; Grisé, H.; Ofek, G.A.; Taylor, K.A.; Roux, K.H. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 2006, 441, 847–852. [Google Scholar] [CrossRef]
- Chertova, E.; Bess, J.W.; Crise, B.J.; Sowder, R.C.; Schaden, T.M.; Hilburn, J.M.; Hoxie, J.A.; Benveniste, R.E.; Lifson, J.D.; Henderson, L.E.; et al. Envelope Glycoprotein Incorporation, Not Shedding of Surface Envelope Glycoprotein (gp120/SU), Is the Primary Determinant of SU Content of Purified Human Immunodeficiency Virus Type 1 and Simian Immunodeficiency Virus. J. Virol. 2002, 76, 5315–5325. [Google Scholar] [CrossRef]
- Barrero-Villar, M.; Cabrero, J.R.; Gordón-Alonso, M.; Barroso-González, J.; Álvarez-Losada, S.; Muñoz-Fernández, M.Á.; Sánchez-Madrid, F.; Valenzuela-Fernández, A. Moesin is required for HIV-1-induced CD4-CXCR4 interaction, F-actin redistribution, membrane fusion and viral infection in lymphocytes. J. Cell Sci. 2009, 122, 103–113. [Google Scholar] [CrossRef]
- Jiménez-Baranda, S.; Gómez-Moutón, C.; Rojas, A.; Martínez-Prats, L.; Mira, E.; Ana Lacalle, R.; Valencia, A.; Dimitrov, D.S.; Viola, A.; Delgado, R.; et al. Filamin-A regulates actin-dependent clustering of HIV receptors. Nat. Cell Biol. 2007, 9, 838–846. [Google Scholar] [CrossRef]
- Ugolini, S.; Moulard, M.; Mondor, I.; Barois, N.; Demandolx, D.; Hoxie, J.; Brelot, A.; Alizon, M.; Davoust, J.; Sattentau, Q.J. HIV-1 gp120 induces an association between CD4 and the chemokine receptor CXCR4. J. Immunol. 1997, 159, 159. [Google Scholar]
- Mañes, S.; del Real, G.; Lacalle, R.A.; Lucas, P.; Gómez-Moutón, C.; Sánchez-Palomino, S.; Delgado, R.; Alcamí, J.; Mira, E.; Martínez-A, C. Membrane raft microdomains mediate lateral assemblies required for HIV-1 infection. EMBO Rep. 2000, 1, 190–196. [Google Scholar] [CrossRef]
- Iyengar, S.; Hildreth, J.E.K.; Schwartz, D.H. Actin-Dependent Receptor Colocalization Required for Human Immunodeficiency Virus Entry into Host Cells. J. Virol. 1998, 72, 5251–5255. [Google Scholar] [CrossRef]
- Yi, L.; Fang, J.; Isik, N.; Chim, J.; Jin, T. HIV gp120-induced interaction between CD4 and CCR5 requires cholesterol-rich microenvironments revealed by live cell fluorescence resonance energy transfer imaging. J. Biol. Chem. 2006, 281, 35446–35453. [Google Scholar] [CrossRef]
- Brandenberg, O.F.; Magnus, C.; Rusert, P.; Regoes, R.R.; Trkola, A. Different Infectivity of HIV-1 Strains Is Linked to Number of Envelope Trimers Required for Entry. PLoS Pathog. 2015, 11, e1004595. [Google Scholar] [CrossRef]
- Chojnacki, J.; Eggeling, C. Super-resolution fluorescence microscopy studies of human immunodeficiency virus. Retrovirology 2018, 15, 41. [Google Scholar] [CrossRef]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Schamel, W.W.A.; Alarcón, B. Organization of the resting TCR in nanoscale oligomers. Immunol. Rev. 2013, 251, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Sherman, E.; Barr, V.; Samelson, L.E. Super-resolution characterization of TCR-dependent signaling clusters. Immunol. Rev. 2013, 251, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Rossboth, B.; Arnold, A.M.; Ta, H.; Platzer, R.; Kellner, F.; Huppa, J.B.; Brameshuber, M.; Baumgart, F.; Schütz, G.J. TCRs are randomly distributed on the plasma membrane of resting antigen-experienced T cells. Nat. Immunol. 2018, 19, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Ester, M.; Kriegel, H.-P.; Sander, J.; Xu, X. A Density-Based Algorithm for Discovering Clusters in Large Spatial Databases with Noise. In Proceedings of the Second International Conference on Knowledge Discovery and Data Mining (KDD’96), Portland, OR, USA, 2–4 August 1996; AAAI Press: Palo Alto, CA, USA, 1996; pp. 226–231. [Google Scholar]
- Malkusch, S.; Endesfelder, U.; Mondry, J.; Gelléri, M.; Verveer, P.J.; Heilemann, M. Coordinate-based colocalization analysis of single-molecule localization microscopy data. Histochem. Cell Biol. 2012, 137, 1–10. [Google Scholar] [CrossRef]
- Pageon, S.V.; Tabarin, T.; Yamamoto, Y.; Ma, Y.; Bridgeman, J.S.; Cohnen, A.; Benzing, C.; Gao, Y.; Crowther, M.D.; Tungatt, K.; et al. Functional role of T-cell receptor nanoclusters in signal initiation and antigen discrimination. Proc. Natl. Acad. Sci. USA 2016, 113, E5454–E5463. [Google Scholar] [CrossRef]
- Siddig, S.; Aufmkolk, S.; Doose, S.; Jobin, M.L.; Werner, C.; Sauer, M.; Calebiro, D. Super-resolution imaging reveals the nanoscale organization of metabotropic glutamate receptors at presynaptic active zones. Sci. Adv. 2020, 6, eaay7193. [Google Scholar] [CrossRef]
- Sieben, C.; Sezgin, E.; Eggeling, C.; Manley, S. Influenza A viruses use multivalent sialic acid clusters for cell binding and receptor activation. PLoS Pathog. 2020, 16, e1008656. [Google Scholar] [CrossRef] [PubMed]
- Lukeš, T.; Glatzová, D.; Kvíčalová, Z.; Levet, F.; Benda, A.; Letschert, S.; Sauer, M.; Brdička, T.; Lasser, T.; Cebecauer, M. Quantifying protein densities on cell membranes using super-resolution optical fluctuation imaging. Nat. Commun. 2017, 8, 1731. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; di Bartolo, V.; Tubul, L.; Shimoni, E.; Kartvelishvily, E.; Dadosh, T.; Feigelson, S.W.; Alon, R.; Alcover, A.; Haran, G. ERM-Dependent Assembly of T Cell Receptor Signaling and Co-stimulatory Molecules on Microvilli prior to Activation. Cell Rep. 2020, 30, 3434–3447.e6. [Google Scholar] [CrossRef] [PubMed]
- Reinherz, E.L.; Kung, P.C.; Goldstein, G.; Schlossman, S.F. Separation of functional subsets of human T cells by a monoclonal antibody. Proc. Natl. Acad. Sci. USA 1979, 76, 4061–4065. [Google Scholar] [CrossRef]
- Pereira, P.M.; Albrecht, D.; Culley, S.; Jacobs, C.; Marsh, M.; Mercer, J.; Henriques, R. Fix Your Membrane Receptor Imaging: Actin Cytoskeleton and CD4 Membrane Organization Disruption by Chemical Fixation. Front. Immunol. 2019, 10, 675. [Google Scholar] [CrossRef]
- Ovesný, M.; Křížek, P.; Borkovec, J.; Švindrych, Z.; Hagen, G.M. ThunderSTORM: A comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 2014, 30, 2389–2390. [Google Scholar] [CrossRef]
- Pageon, S.V.; Nicovich, P.R.; Mollazade, M.; Tabarin, T.; Gaus, K. Clus-DoC: A combined cluster detection and colocalization analysis for single-molecule localization microscopy data. Mol. Biol. Cell 2016, 27, 3627–3636. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Pelchen-Matthews, A.; Armes, J.E.; Griffiths, G.; Marsh, M. Differential endocytosis of CD4 in lymphocytic and nonlymphocytic cells. J. Exp. Med. 1991, 173, 575–587. [Google Scholar] [CrossRef]
- Lee, B.; Sharron, M.; Montaner, L.J.; Weissman, D.; Doms, R.W. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc. Natl. Acad. Sci. USA 1999, 96, 5215–5220. [Google Scholar] [CrossRef]
- Frey, S.; Marsh, M.; Günther, S.; Pelchen-Matthews, A.; Stephens, P.; Ortlepp, S.; Stegmann, T. Temperature dependence of cell-cell fusion induced by the envelope glycoprotein of human immunodeficiency virus type 1. J. Virol. 1995, 69, 1462–1472. [Google Scholar] [CrossRef] [PubMed]
- Henderson, H.I.; Hope, T.J. The temperature arrested intermediate of virus-cell fusion is a functional step in HIV infection. Virol. J. 2006, 3, 1–7. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Markosyan, R.M.; Cohen, F.S.; Melikyan, G.B. Time-resolved imaging of HIV-1 Env-mediated lipid and content mixing between a single virion and cell membrane. Mol. Biol. Cell 2005, 16, 5502–5513. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, C.; Höning, S.; Fingerhut, A.; Bowers, K.; Marsh, M. Cluster of differentiation antigen 4 (CD4) endocytosis and adaptor complex binding require activation of the CD4 endocytosis signal by serine phosphorylation. Mol. Biol. Cell 1999, 10, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Pelchen-Matthews, A.; Parsons, I.J.; Marsh, M. Phorbol Ester-induced Downregulation of CD4 is a Multistep Process Involving Dissociation from p56 lck, Increased Association with Clathrin-coated Pits, and Altered Endosomal Sorting. J. Exp. Med. 1993, 178, 1209–1222. [Google Scholar] [CrossRef]
- Klasse, P.J.; Rosenkilde, M.M.; Signoret, N.; Pelchen-Matthews, A.; Schwartz, T.W.; Marsh, M. CD4-Chemokine Receptor Hybrids in Human Immunodeficiency Virus Type 1 Infection. J. Virol. 1999, 73, 7453–7466. [Google Scholar] [CrossRef]
- Lu, M.; Ma, X.; Castillo-Menendez, L.R.; Gorman, J.; Alsahafi, N.; Ermel, U.; Terry, D.S.; Chambers, M.; Peng, D.; Zhang, B.; et al. Associating HIV-1 envelope glycoprotein structures with states on the virus observed by smFRET. Nature 2019, 568, 415–419. [Google Scholar] [CrossRef]
- Arthos, J.; Cicala, C.; Martinelli, E.; Macleod, K.; van Ryk, D.; Wei, D.; Xiao, Z.; Veenstra, T.D.; Conrad, T.P.; Lempicki, R.A.; et al. HIV-1 envelope protein binds to and signals through integrin α4β7, the gut mucosal homing receptor for peripheral T cells. Nat. Immunol. 2008, 9, 301–309. [Google Scholar] [CrossRef]
- Cicala, C.; Martinelli, E.; McNally, J.P.; Goode, D.J.; Gopaul, R.; Hiatt, J.; Jelicic, K.; Kottilil, S.; Macleod, K.; O’Shea, A.; et al. The integrin α4β7 forms a complex with cell-surface CD4 and defines a T-cell subset that is highly susceptible to infection by HIV-1. Proc. Natl. Acad. Sci. USA 2009, 106, 20877–20882. [Google Scholar] [CrossRef]
- Saphire, A.C.S.; Bobardt, M.D.; Zhang, Z.; David, G.; Gallay, P.A. Syndecans Serve as Attachment Receptors for Human Immunodeficiency Virus Type 1 on Macrophages. J. Virol. 2001, 75, 9187–9200. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.H.; Kwon, D.S.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.F.; Middel, J.; Cornelissen, I.L.M.H.A.; Nottet, H.S.L.M.; KewalRamani, V.N.; Littman, D.R.; et al. DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 2000, 100, 587–597. [Google Scholar] [CrossRef]
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell binding and entry. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Gallaher, W.R.; Ball, J.M.; Garry, R.F.; Griffin, M.C.; Montelaro, R.C. A General Model for the Transmembrane Proteins of HIV and Other Retroviruses. Aids Res. Hum. Retrovir. 1989, 5, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Chojnacki, J.; Staudt, T.; Glass, B.; Bingen, P.; Engelhardt, J.; Anders, M.; Schneider, J.; Müller, B.; Hell, S.W.; Kräusslich, H.G. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science 2012, 338, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Giri, B.; Collins, G.; Taub, D.D. Dynamic reorganization of chemokine receptors, cholesterol, lipid rafts, and adhesion molecules to sites of CD4 engagement. Exp. Cell Res. 2005, 304, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulou, M.; Nolan, R.; Alvarez, L.; Watanabe, Y.; Coomer, C.A.; Jakobsdottir, G.M.; Bowden, T.A.; Padilla-Parra, S. A dynamic three-step mechanism drives the HIV-1 pre-fusion reaction. Nat. Struct. Mol. Biol. 2018, 25, 814–822. [Google Scholar] [CrossRef]
- Mazia, D.; Schatten, G.; Sale, W. Adhesion of cells to surfaces coated with polylysine: Applications to electron microscopy. J. Cell Biol. 1975, 66, 198–200. [Google Scholar] [CrossRef]
- Groves, J.T.; Dustin, M.L. Supported planar bilayers in studies on immune cell adhesion and communication. J. Immunol. Methods 2003, 278, 19–32. [Google Scholar] [CrossRef]
- Santos, A.M.; Ponjavic, A.; Fritzsche, M.; Fernandes, R.A.; de La Serna, J.B.; Wilcock, M.J.; Schneider, F.; Urbančič, I.; McColl, J.; Anzilotti, C.; et al. Capturing resting T cells: The perils of PLL correspondence. Nat. Immunol. 2018, 19, 203–205. [Google Scholar] [CrossRef]
- Oszmiana, A.; Williamson, D.J.; Cordoba, S.P.; Morgan, D.J.; Kennedy, P.R.; Stacey, K.; Davis, D.M. The Size of Activating and Inhibitory Killer Ig-like Receptor Nanoclusters Is Controlled by the Transmembrane Sequence and Affects Signaling. Cell Rep. 2016, 15, 1957–1972. [Google Scholar] [CrossRef]
- Lillemeier, B.F.; Pfeiffer, J.R.; Surviladze, Z.; Wilson, B.S.; Davis, M.M. Plasma membrane-associated proteins are clustered into islands attached to the cytoskeleton. Proc. Natl. Acad. Sci. USA 2006, 103, 18992–18997. [Google Scholar] [CrossRef] [PubMed]
- Saka, S.K.; Honigmann, A.; Eggeling, C.; Hell, S.W.; Lang, T.; Rizzoli, S.O. Multi-protein assemblies underlie the mesoscale organization of the plasma membrane. Nat. Commun. 2014, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.K.; Iwasawa, K.; Kalay, Z.; Tsunoyama, T.A.; Watanabe, Y.; Umemura, Y.M.; Murakoshi, H.; Suzuki, K.G.N.; Nemoto, Y.L.; Morone, N.; et al. Confined diffusion of transmembrane proteins and lipids induced by the same actin meshwork lining the plasma membrane. Mol. Biol. Cell 2016, 27, 1101–1119. [Google Scholar] [CrossRef] [PubMed]
- Feher, K.; Halstead, J.M.; Goyette, J.; Gaus, K. Can single molecule localization microscopy detect nanoclusters in T cells? Curr. Opin. Chem. Biol. 2019, 51, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.; Owen, D.M.; Cohen, E.A.K. Blinking Statistics and Molecular Counting in direct Stochastic Reconstruction Microscopy (dSTORM). bioRxiv 2019, 834572. [Google Scholar] [CrossRef]
- Jong, Y.-J.I.; Harmon, S.K.; O’Malley, K.L. GPCR signalling from within the cell. Br. J. Pharmacol. 2018, 175, 4026–4035. [Google Scholar] [CrossRef] [PubMed]
- Yoder, A.; Yu, D.; Dong, L.; Iyer, S.R.; Xu, X.; Kelly, J.; Liu, J.; Wang, W.; Vorster, P.J.; Agulto, L.; et al. HIV Envelope-CXCR4Signaling Activates Cofilin to Overcome Cortical Actin Restriction in Resting CD4 T Cells. Cell 2008, 134, 782–792. [Google Scholar] [CrossRef]
- Turoňová, B.; Sikora, M.; Schürmann, C.; Hagen, W.J.H.; Welsch, S.; Blanc, F.E.C.; von Bülow, S.; Gecht, M.; Bagola, K.; Hörner, C.; et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208. [Google Scholar] [CrossRef]
- Lucera, M.B.; Fleissner, Z.; Tabler, C.O.; Schlatzer, D.M.; Troyer, Z.; Tilton, J.C. HIV signaling through CD4 and CCR5 activates Rho family GTPases that are required for optimal infection of primary CD4+ T cells. Retrovirology 2017, 14, 4. [Google Scholar] [CrossRef]
- Amara, A.; Vidy, A.; Boulla, G.; Mollier, K.; Garcia-Perez, J.; Alcamí, J.; Blanpain, C.; Parmentier, M.; Virelizier, J.-L.; Charneau, P.; et al. G Protein-Dependent CCR5 Signaling Is Not Required for Efficient Infection of Primary T Lymphocytes and Macrophages by R5 Human Immunodeficiency Virus Type 1 Isolates. J. Virol. 2003, 77, 2550–2558. [Google Scholar] [CrossRef]
- Gibbons, M.M.; Chou, T.; D’Orsogna, M.R. Diffusion-Dependent Mechanisms of Receptor Engagement and Viral Entry. J. Phys. Chem. B 2010, 114, 15403–15412. [Google Scholar] [CrossRef] [PubMed]
- Dobrowsky, T.M.; Daniels, B.R.; Siliciano, R.F.; Sun, S.X.; Wirtz, D. Organization of Cellular Receptors into a Nanoscale Junction during HIV-1 Adhesion. PLoS Comput. Biol. 2010, 6, e1000855. [Google Scholar] [CrossRef] [PubMed]
- Trister, A.D.; Hammer, D.A. Role of Gp120 Trimerization on HIV Binding Elucidated with Brownian Adhesive Dynamics. Biophys. J. 2008, 95, 40–53. [Google Scholar] [CrossRef] [PubMed][Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Y.; Jacobs, C.A.; Llorente Garcia, I.; Pereira, P.M.; Lawrence, S.P.; Laine, R.F.; Marsh, M.; Henriques, R. Single-Molecule Super-Resolution Imaging of T-Cell Plasma Membrane CD4 Redistribution upon HIV-1 Binding. Viruses 2021, 13, 142. https://doi.org/10.3390/v13010142
Yuan Y, Jacobs CA, Llorente Garcia I, Pereira PM, Lawrence SP, Laine RF, Marsh M, Henriques R. Single-Molecule Super-Resolution Imaging of T-Cell Plasma Membrane CD4 Redistribution upon HIV-1 Binding. Viruses. 2021; 13(1):142. https://doi.org/10.3390/v13010142
Chicago/Turabian StyleYuan, Yue, Caron A. Jacobs, Isabel Llorente Garcia, Pedro M. Pereira, Scott P. Lawrence, Romain F. Laine, Mark Marsh, and Ricardo Henriques. 2021. "Single-Molecule Super-Resolution Imaging of T-Cell Plasma Membrane CD4 Redistribution upon HIV-1 Binding" Viruses 13, no. 1: 142. https://doi.org/10.3390/v13010142
APA StyleYuan, Y., Jacobs, C. A., Llorente Garcia, I., Pereira, P. M., Lawrence, S. P., Laine, R. F., Marsh, M., & Henriques, R. (2021). Single-Molecule Super-Resolution Imaging of T-Cell Plasma Membrane CD4 Redistribution upon HIV-1 Binding. Viruses, 13(1), 142. https://doi.org/10.3390/v13010142