
Intra-Host Diversity of SARS-Cov-2 Should Not Be Neglected: Case of the State of Victoria, Australia



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Identification of iSNVs
2.3. Temporal Dynamics of iSNV Diversity
2.4. Identification of the iSNV Haplotypes
2.5. Validation of iSNVs Haplotypes with Other Sequence Datasets
2.6. Phylogenetic Analysis
2.7. SNVs Identification
3. Results
3.1. Synonymous and Nonsynonymous iSNVs
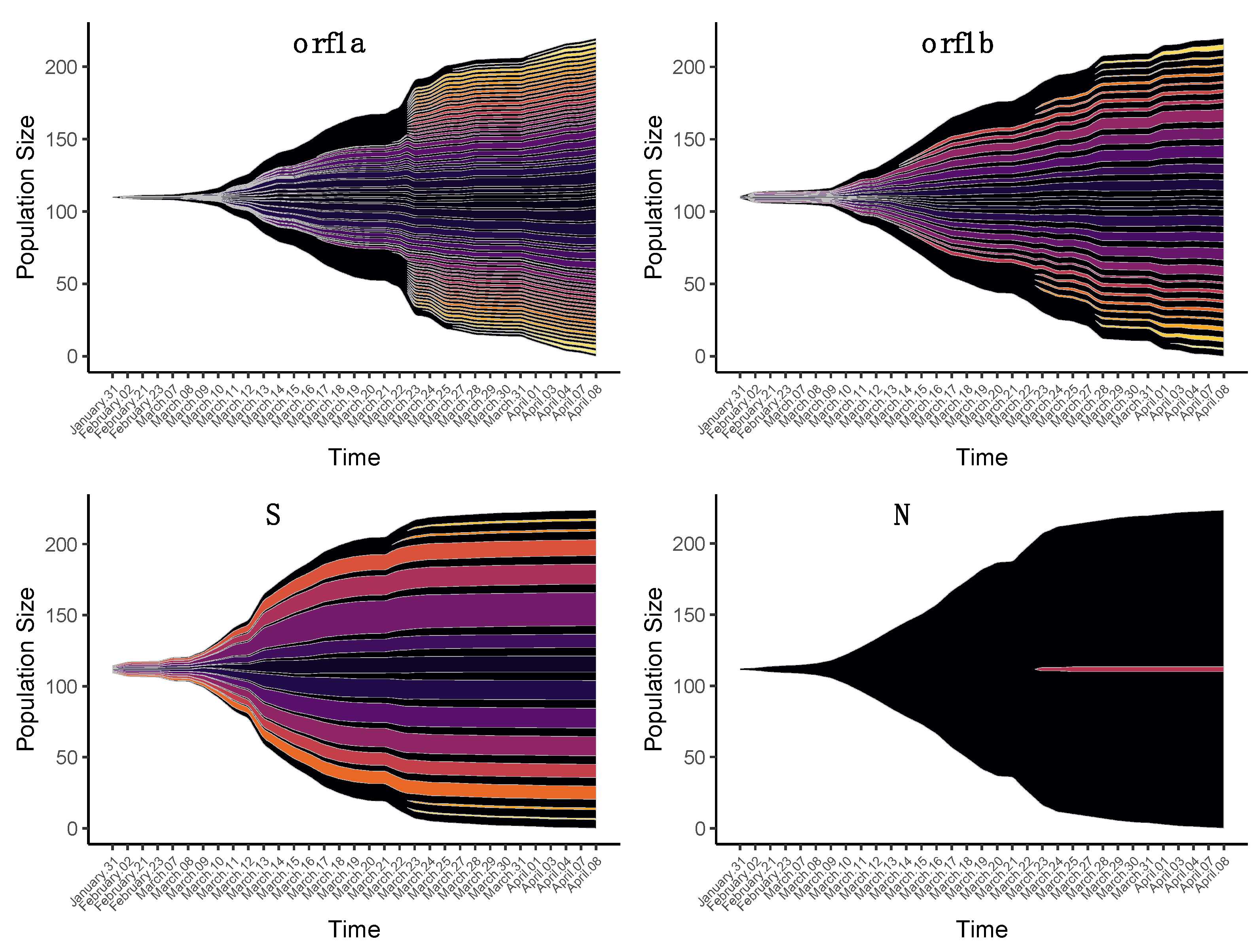
3.2. Changes in iSNVs Diversity over Time
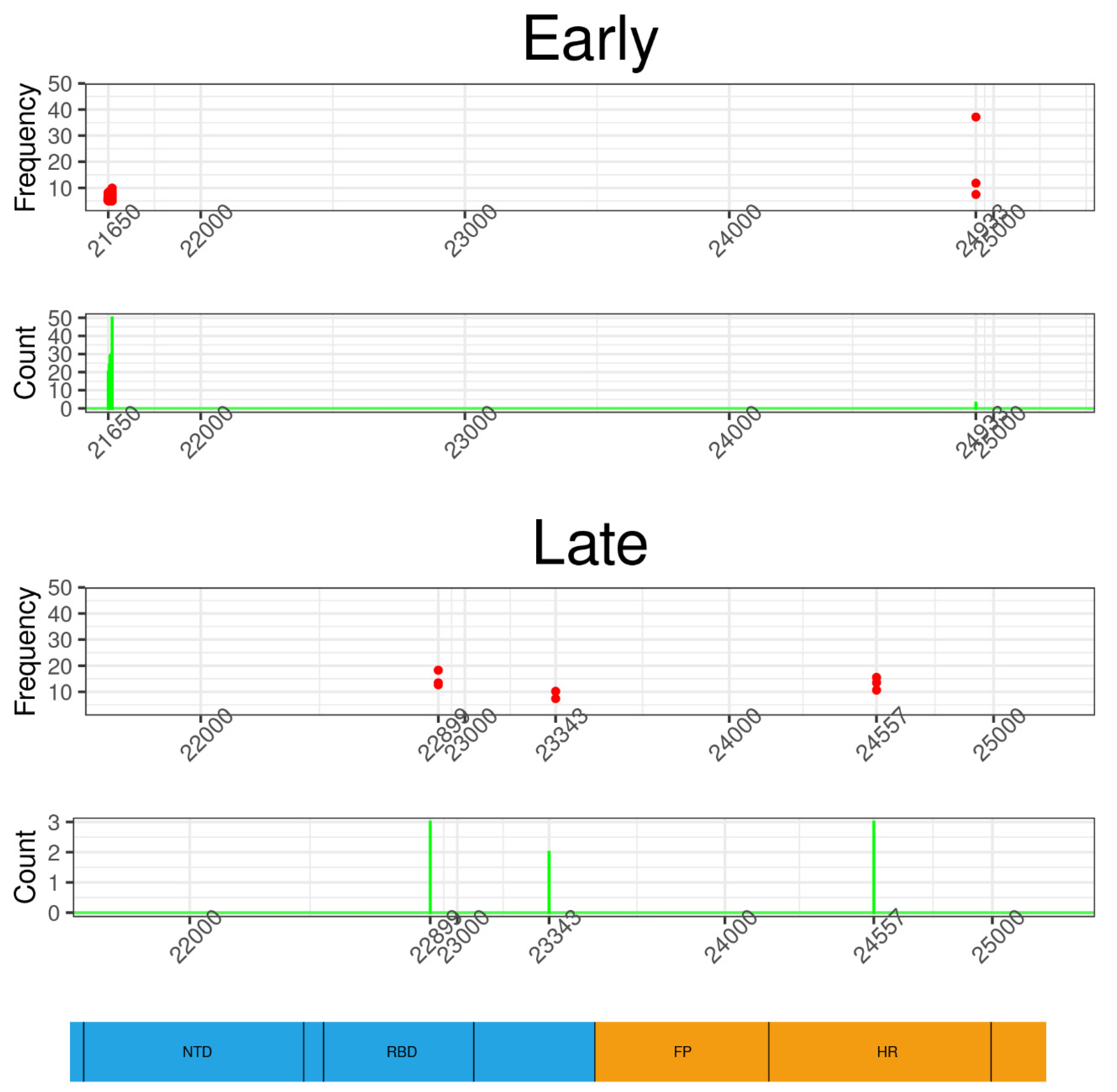
3.2.1. Nonsynonymous Substitutions
3.2.2. Synonymous Substitutions
3.2.3. Origin of iSNVs in the Late Group
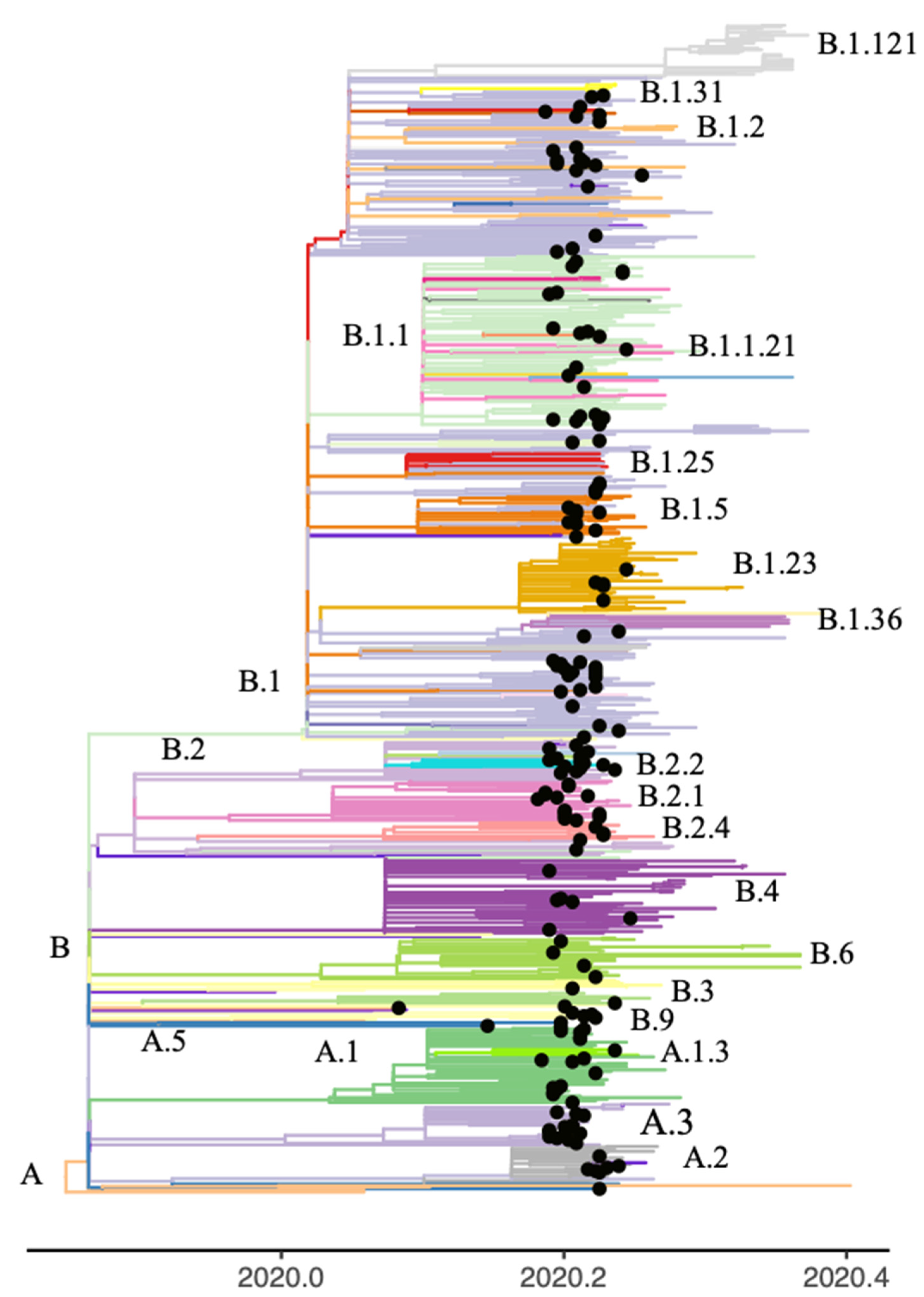
3.3. Phylogenetic Analysis of Consensus Sequences and iSNVs
4. Discussion
4.1. Diversity of SARS-CoV-2 iSNVs
4.2. Viral Haplotypes and Quasispecies
4.3. Changes in SARS-CoV-2 iSNVs Diversity over Time
4.4. Transmission and Bottleneck of SARS-CoV-2 iSNVs
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Jiang, S.; Shi, Z.-L. The First Disease X is Caused by a Highly Transmissible Acute Respiratory Syndrome Coronavirus. Virol. Sin. 2020, 35, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-C.; Shih, T.-P.; Ko, W.-C.; Tang, H.-J.; Hsueh, P.-R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zai, J.; Zhao, Q.; Nie, Q.; Li, Y.; Foley, B.T.; Chaillon, A. Evolutionary history, potential intermediate animal host, and cross-species analyses of SARS-CoV-2. J. Med. Virol. 2020, 92, 602–611. [Google Scholar] [CrossRef]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and Sources of Endemic Human Coronaviruses. Adv. Virus Res. 2018, 100, 163–188. [Google Scholar] [CrossRef] [PubMed]
- Blackstone, N.W.; Blackstone, S.R.; Berg, A.T. Variation and multilevel selection of SARS-CoV-2. Evol. Int. J. Org. Evol. 2020. [Google Scholar] [CrossRef]
- Koyama, T.; Platt, D.; Parida, L. Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ. 2020, 98, 495–504. [Google Scholar] [CrossRef]
- Zhang, C.-Y.; Wei, J.-F.; He, S.-H. Adaptive evolution of the spike gene of SARS coronavirus: Changes in positively selected sites in different epidemic groups. BMC Microbiol. 2006, 6, 88. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Rangarajan, E.S.; Izard, T.; Farzan, M.; Choe, H. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hu, J.; He, C.-L.; Gao, Q.-Z.; Zhang, G.-J.; Cao, X.-X.; Long, Q.-X.; Deng, H.-J.; Huang, L.-Y.; Chen, J.; Wang, K.; et al. D614G mutation of SARS-CoV-2 spike protein enhances viral infectivity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Volz, E.M.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, A.; Southgate, J.A.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the effects of SARS-CoV-2 Spike mutation D614G on transmissibility and pathogenicity. medRxiv 2020. [Google Scholar] [CrossRef]
- Van Dorp, L.; Richard, D.; Tan, C.S.; Shaw, L.P.; Acman, M.; Balloux, F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Forni, D.; Cagliani, R.; Mozzi, A.; Pozzoli, U.; Al-Daghri, N.; Clerici, M.; Sironi, M. Extensive Positive Selection Drives the Evolution of Nonstructural Proteins in Lineage C Betacoronaviruses. J. Virol. 2016, 90, 3627. [Google Scholar] [CrossRef]
- Hatta, M.; Gao, P.; Halfmann, P.; Kawaoka, Y. Molecular Basis for High Virulence of Hong Kong H5N1 Influenza A Viruses. Science 2001, 293, 1840. [Google Scholar] [CrossRef]
- Gabriel, G.; Dauber, B.; Wolff, T.; Planz, O.; Klenk, H.-D.; Stech, J. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proc. Natl. Acad. Sci. USA 2005, 102, 18590. [Google Scholar] [CrossRef]
- Dutta, N.K.; Mazumdar, K.; Gordy, J.T. The Nucleocapsid Protein of SARS–CoV-2: A Target for Vaccine Development. J. Virol. 2020, 94, e00647-20. [Google Scholar] [CrossRef]
- To, K.K.-W.; Tsang, O.T.-Y.; Leung, W.-S.; Tam, A.R.; Wu, T.-C.; Lung, D.C.; Yip, C.C.-Y.; Cai, J.-P.; Chan, J.M.-C.; Chik, T.S.-H.; et al. Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: An observational cohort study. Lancet Infect. Dis. 2020, 20, 565–574. [Google Scholar] [CrossRef]
- Cortey, M.; Li, Y.; Díaz, I.; Clilverd, H.; Darwich, L.; Mateu, E. SARS-CoV-2 amino acid substitutions widely spread in the human population are mainly located in highly conserved segments of the structural proteins. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lauring, A.S.; Andino, R. Quasispecies Theory and the Behavior of RNA Viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhang, Z.; Wang, F.-S. SARS-Associated Coronavirus Quasispecies in Individual Patients. N. Engl. J. Med. 2004, 350, 1366–1367. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, Z.; Chu, F.; Li, Y.; Jin, L.; Zhang, L.; Gao, G.F.; Wang, F.-S. Genetic variation of SARS coronavirus in Beijing Hospital. Emerg. Infect. Dis. 2004, 10, 789–794. [Google Scholar] [CrossRef]
- Tang, J.W.; Cheung, J.L.K.; Chu, I.M.T.; Sung, J.J.Y.; Peiris, M.; Chan, P.K.S. The large 386-nt deletion in SARS-associated coronavirus: Evidence for quasispecies? J. Infect. Dis. 2006, 194, 808–813. [Google Scholar] [CrossRef]
- Liu, J.; Lim, S.L.; Ruan, Y.; Ling, A.E.; Ng, L.F.P.; Drosten, C.; Liu, E.T.; Stanton, L.W.; Hibberd, M.L. SARS Transmission Pattern in Singapore Reassessed by Viral Sequence Variation Analysis. PLoS Med. 2005, 2, e43. [Google Scholar] [CrossRef][Green Version]
- Park, D.; Huh, H.J.; Kim, Y.J.; Son, D.-S.; Jeon, H.-J.; Im, E.-H.; Kim, J.-W.; Lee, N.Y.; Kang, E.-S.; Kang, C.I.; et al. Analysis of intrapatient heterogeneity uncovers the microevolution of Middle East respiratory syndrome coronavirus. Cold Spring Harb. Mol. Case Stud. 2016, 2, a001214. [Google Scholar] [CrossRef]
- Kleine-Weber, H.; Elzayat, M.T.; Wang, L.; Graham, B.S.; Müller, M.A.; Drosten, C.; Pöhlmann, S.; Hoffmann, M. Mutations in the Spike Protein of Middle East Respiratory Syndrome Coronavirus Transmitted in Korea Increase Resistance to Antibody-Mediated Neutralization. J. Virol. 2019, 93, e01381-18. [Google Scholar] [CrossRef] [PubMed]
- Briese, T.; Mishra, N.; Jain, K.; Zalmout, I.S.; Jabado, O.J.; Karesh, W.B.; Daszak, P.; Mohammed, O.B.; Alagaili, A.N.; Lipkin, W.I. Middle East respiratory syndrome coronavirus quasispecies that include homologues of human isolates revealed through whole-genome analysis and virus cultured from dromedary camels in Saudi Arabia. mBio 2014, 5, e01146. [Google Scholar] [CrossRef] [PubMed]
- Borucki, M.K.; Lao, V.; Hwang, M.; Gardner, S.; Adney, D.; Munster, V.; Bowen, R.; Allen, J.E. Middle East Respiratory Syndrome Coronavirus Intra-Host Populations Are Characterized by Numerous High Frequency Variants. PLoS ONE 2016, 11, e0146251. [Google Scholar] [CrossRef]
- Lu, X.; Rowe, L.A.; Frace, M.; Stevens, J.; Abedi, G.R.; Elnile, O.; Banassir, T.; Al-Masri, M.; Watson, J.T.; Assiri, A.; et al. Spike gene deletion quasispecies in serum of patient with acute MERS-CoV infection. J. Med. Virol. 2017, 89, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T.; Lane, C.; Sherry, N.; Duchene, S.; Goncalves da Silva, A.; Caly, L.; Sait, M.; Ballard, S.A.; Horan, K.; Schultz, M.B.; et al. Tracking the COVID-19 pandemic in Australia using genomics. medRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinform. Oxf. Engl. 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Verbist, B.M.P.; Thys, K.; Reumers, J.; Wetzels, Y.; Van der Borght, K.; Talloen, W.; Aerssens, J.; Clement, L.; Thas, O. VirVarSeq: A low-frequency virus variant detection pipeline for Illumina sequencing using adaptive base-calling accuracy filtering. Bioinformatics 2014, 31, 94–101. [Google Scholar] [CrossRef]
- Köster, J.; Rahmann, S. Snakemake—A scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef]
- Yang, X.; Charlebois, P.; Macalalad, A.; Henn, M.R.; Zody, M.C. V-Phaser 2: Variant inference for viral populations. BMC Genom. 2013, 14, 674. [Google Scholar] [CrossRef]
- Wickham, H. Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Gatenbee, C.D.; Schenck, R.O.; Bravo, R.R.; Anderson, A.R.A. EvoFreq: Visualization of the Evolutionary Frequencies of sequence and model data. BMC Bioinform. 2019, 20, 710. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M. The UGENE team Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Mavian, C.; Marini, S.; Prosperi, M.; Salemi, M. A Snapshot of SARS-CoV-2 Genome Availability up to April 2020 and its Implications: Data Analysis. JMIR Public Health Surveill. 2020, 6, e19170. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2. [Google Scholar] [CrossRef]
- Volz, E.M.; Frost, S.D.W. Scalable relaxed clock phylogenetic dating. Virus Evol. 2017, 3. [Google Scholar] [CrossRef]
- Guerrero-Murillo, M.; Font, J.G. QSutils: Quasispecies Diversity; Bioconductor Version: Release (3.11). 2020. Available online: http://bioconductor.org/packages/release/bioc/html/QSutils.html (accessed on 25 November 2020).
- Lythgoe, K.A.; Hall, M.; Ferretti, L.; de Cesare, M.; MacIntyre-Cockett, G.; Trebes, A.; Andersson, M.; Otecko, N.; Wise, E.L.; Moore, N.; et al. Shared SARS-CoV-2 diversity suggests localised transmission of minority variants. bioRxiv 2020. [Google Scholar] [CrossRef]
- Rose, R.; Nolan, D.J.; Moot, S.; Feehan, A.; Cross, S.; Garcia-Diaz, J.; Lamers, S.L. Intra-host site-specific polymorphisms of SARS-CoV-2 is consistent across multiple samples and methodologies. medRxiv 2020. [Google Scholar] [CrossRef]
- Sapoval, N.; Mahmoud, M.; Jochum, M.D.; Liu, Y.; Leo Elworth, R.A.; Wang, Q.; Albin, D.; Ogilvie, H.; Lee, M.D.; Villapol, S.; et al. Hidden genomic diversity of SARS-CoV-2: Implications for qRT-PCR diagnostics and transmission. bioRxiv 2020. [Google Scholar] [CrossRef]
- Shen, Z.; Xiao, Y.; Kang, L.; Ma, W.; Shi, L.; Zhang, L.; Zhou, Z.; Yang, J.; Zhong, J.; Yang, D.; et al. Genomic diversity of SARS-CoV-2 in Coronavirus Disease 2019 patients. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2020, ciaa203. [Google Scholar] [CrossRef]
- Spencer, D.H.; Tyagi, M.; Vallania, F.; Bredemeyer, A.J.; Pfeifer, J.D.; Mitra, R.D.; Duncavage, E.J. Performance of common analysis methods for detecting low-frequency single nucleotide variants in targeted next-generation sequence data. J. Mol. Diagn. JMD 2014, 16, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, S.; Martignano, F.; Torcia, M.G.; Mattiuz, G.; Conticello, S.G. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci. Adv. 2020, 6, eabb5813. [Google Scholar] [CrossRef]
- Simmonds, P. Rampant C→U hypermutation in the genomes of SARS-CoV-2 and other coronaviruses—Causes and consequences for their short and long evolutionary trajectories. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef]
- Lyons, D.M.; Lauring, A.S. Evidence for the Selective Basis of Transition-to-Transversion Substitution Bias in Two RNA Viruses. Mol. Biol. Evol. 2017, 34, 3205–3215. [Google Scholar] [CrossRef]
- Li, Z.; Wu, J.; DeLeo, C.J. RNA damage and surveillance under oxidative stress. IUBMB Life 2006, 58, 581–588. [Google Scholar] [CrossRef]
- Ramazzotti, D.; Angaroni, F.; Maspero, D.; Gambacorti-Passerini, C.; Antoniotti, M.; Graudenzi, A.; Piazza, R. Characterization of intra-host SARS-CoV-2 variants improves phylogenomic reconstruction and may reveal functionally convergent mutations. bioRxiv 2020. [Google Scholar] [CrossRef]
- Du, P.; Song, C.; Li, R.; Song, Y.; Li, J.; Ding, N.; Zhang, J.; Song, R.; Han, J.; Gao, G.; et al. Specific Redistribution of Severe Acute Respiratory Syndrome Coronavirus 2 Variants in the Respiratory System and Intestinal Tract. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, D.; Zhang, L.; Sun, W.; Zhang, Z.; Chen, W.; Zhu, A.; Huang, Y.; Xiao, F.; Yao, J.; et al. Intra-Host Variation and Evolutionary Dynamics of SARS-CoV-2 Population in COVID-19 Patients. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wong, Y.C.; Lau, S.Y.; Wang To, K.K.; Mok, B.W.Y.; Li, X.; Wang, P.; Deng, S.; Woo, K.F.; Du, Z.; Li, C.; et al. Natural Transmission of Bat-like Severe Acute Respiratory Syndrome Coronavirus 2 without Proline-Arginine-Arginine-Alanine Variants in Coronavirus Disease 2019 Patients. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Quick, J. NCoV-2019 Sequencing Protocol v1 (Protocols.Io.Bbmuik6w). Available online: https://www.protocols.io/view/ncov-2019-sequencing-protocol-bbmuik6w?version_warning=no (accessed on 25 November 2020).
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An Amplicon-Based Sequencing Framework for Accurately Measuring Intrahost Virus Diversity Using PrimalSeq and Ivar. Genome Biol. 2019, 20. [Google Scholar] [CrossRef]
- Issues with SARS-CoV-2 Sequencing Data. Available online: https://virological.org/t/issues-with-sars-cov-2-sequencing-data/473 (accessed on 30 December 2020).
- Andrés, C.; Garcia-Cehic, D.; Gregori, J.; Piñana, M.; Rodriguez-Frias, F.; Guerrero-Murillo, M.; Esperalba, J.; Rando, A.; Goterris, L.; Codina, M.G.; et al. Naturally occurring SARS-CoV-2 gene deletions close to the spike S1/S2 cleavage site in the viral quasispecies of COVID19 patients. Emerg. Microbes Infect. 2020, 9, 1900–1911. [Google Scholar] [CrossRef]
- Al Khatib, H.A.; Benslimane, F.M.; Elbashir, I.E.; Coyle, P.V.; Al Maslamani, M.A.; Al-Khal, A.; Al Thani, A.A.; Yassine, H.M. Within-Host Diversity of SARS-CoV-2 in COVID-19 Patients with Variable Disease Severities. Front. Cell. Infect. Microbiol. 2020, 10, 534. [Google Scholar] [CrossRef]
- Kuipers, J.; Batavia, A.A.; Jablonski, K.P.; Bayer, F.; Borgsmüller, N.; Dondi, A.; Drăgan, M.-A.; Ferreira, P.; Jahn, K.; Lamberti, L.; et al. Within-Patient Genetic Diversity of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sun, F.; Wang, X.; Tan, S.; Dan, Y.; Lu, Y.; Zhang, J.; Xu, J.; Tan, Z.; Xiang, X.; Zhou, Y.; et al. SARS-CoV-2 Quasispecies Provides Insight into Its Genetic Dynamics during Infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wang, D.; Wang, Y.; Sun, W.; Zhang, L.; Ji, J.; Zhang, Z.; Cheng, X.; Li, Y.; Xiao, F.; Zhu, A.; et al. Population Bottlenecks and Intra-Host Evolution during Human-to-Human Transmission of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Redd, A.D.; Collinson-Streng, A.N.; Chatziandreou, N.; Mullis, C.E.; Laeyendecker, O.; Martens, C.; Ricklefs, S.; Kiwanuka, N.; Nyein, P.H.; Lutalo, T.; et al. Previously transmitted HIV-1 strains are preferentially selected during subsequent sexual transmissions. J. Infect. Dis. 2012, 206, 1433–1442. [Google Scholar] [CrossRef]
- Carlson, J.M.; Schaefer, M.; Monaco, D.C.; Batorsky, R.; Claiborne, D.T.; Prince, J.; Deymier, M.J.; Ende, Z.S.; Klatt, N.R.; DeZiel, C.E.; et al. HIV transmission. Selection bias at the heterosexual HIV-1 transmission bottleneck. Science 2014, 345, 1254031. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armero, A.; Berthet, N.; Avarre, J.-C. Intra-Host Diversity of SARS-Cov-2 Should Not Be Neglected: Case of the State of Victoria, Australia. Viruses 2021, 13, 133. https://doi.org/10.3390/v13010133
Armero A, Berthet N, Avarre J-C. Intra-Host Diversity of SARS-Cov-2 Should Not Be Neglected: Case of the State of Victoria, Australia. Viruses. 2021; 13(1):133. https://doi.org/10.3390/v13010133
Chicago/Turabian StyleArmero, Alix, Nicolas Berthet, and Jean-Christophe Avarre. 2021. "Intra-Host Diversity of SARS-Cov-2 Should Not Be Neglected: Case of the State of Victoria, Australia" Viruses 13, no. 1: 133. https://doi.org/10.3390/v13010133
APA StyleArmero, A., Berthet, N., & Avarre, J.-C. (2021). Intra-Host Diversity of SARS-Cov-2 Should Not Be Neglected: Case of the State of Victoria, Australia. Viruses, 13(1), 133. https://doi.org/10.3390/v13010133