
Domains and Functions of Spike Protein in SARS-Cov-2 in the Context of Vaccine Design


Abstract
1. Introduction
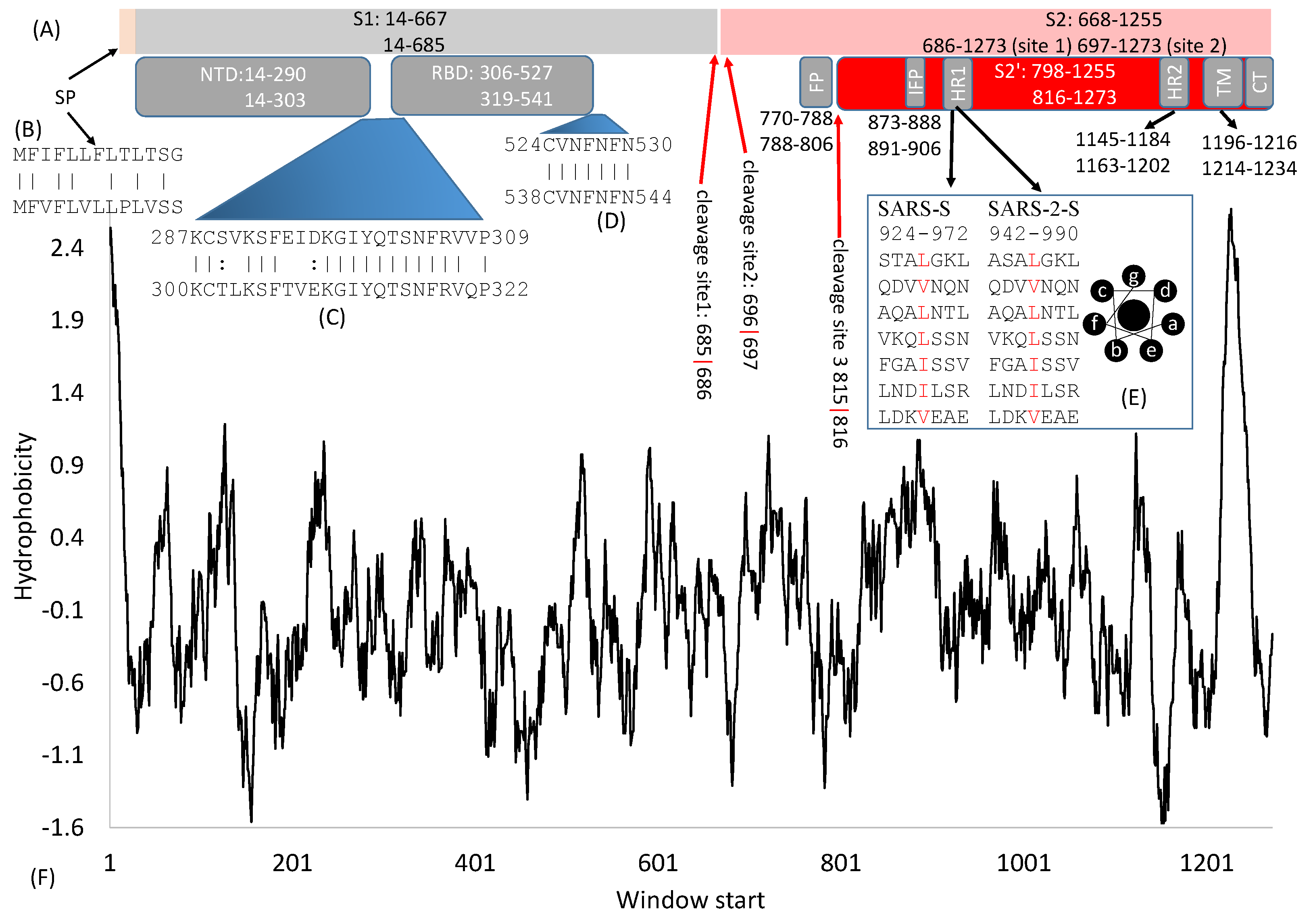
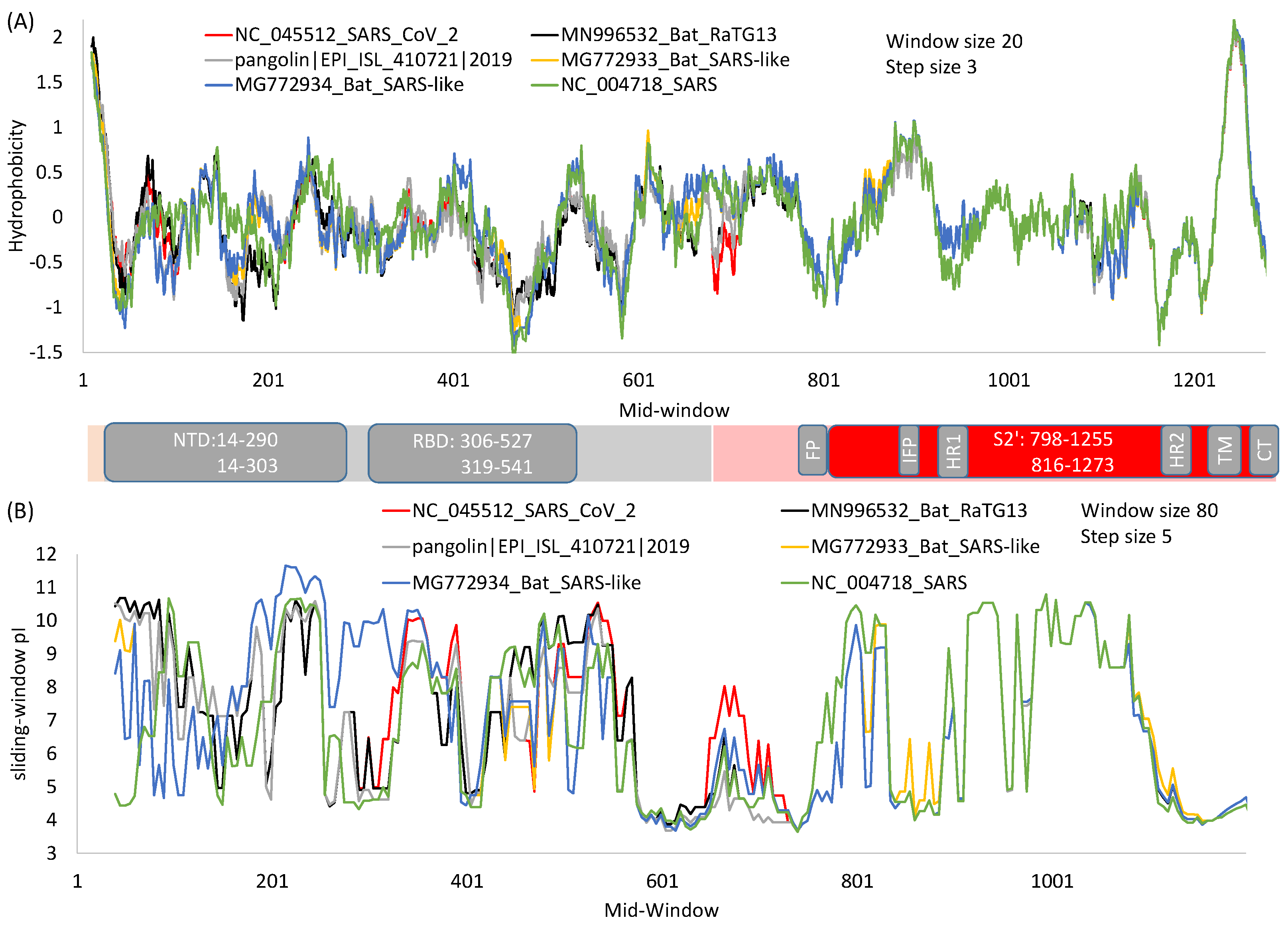
2. General Features of SARS-S and SARS-2S
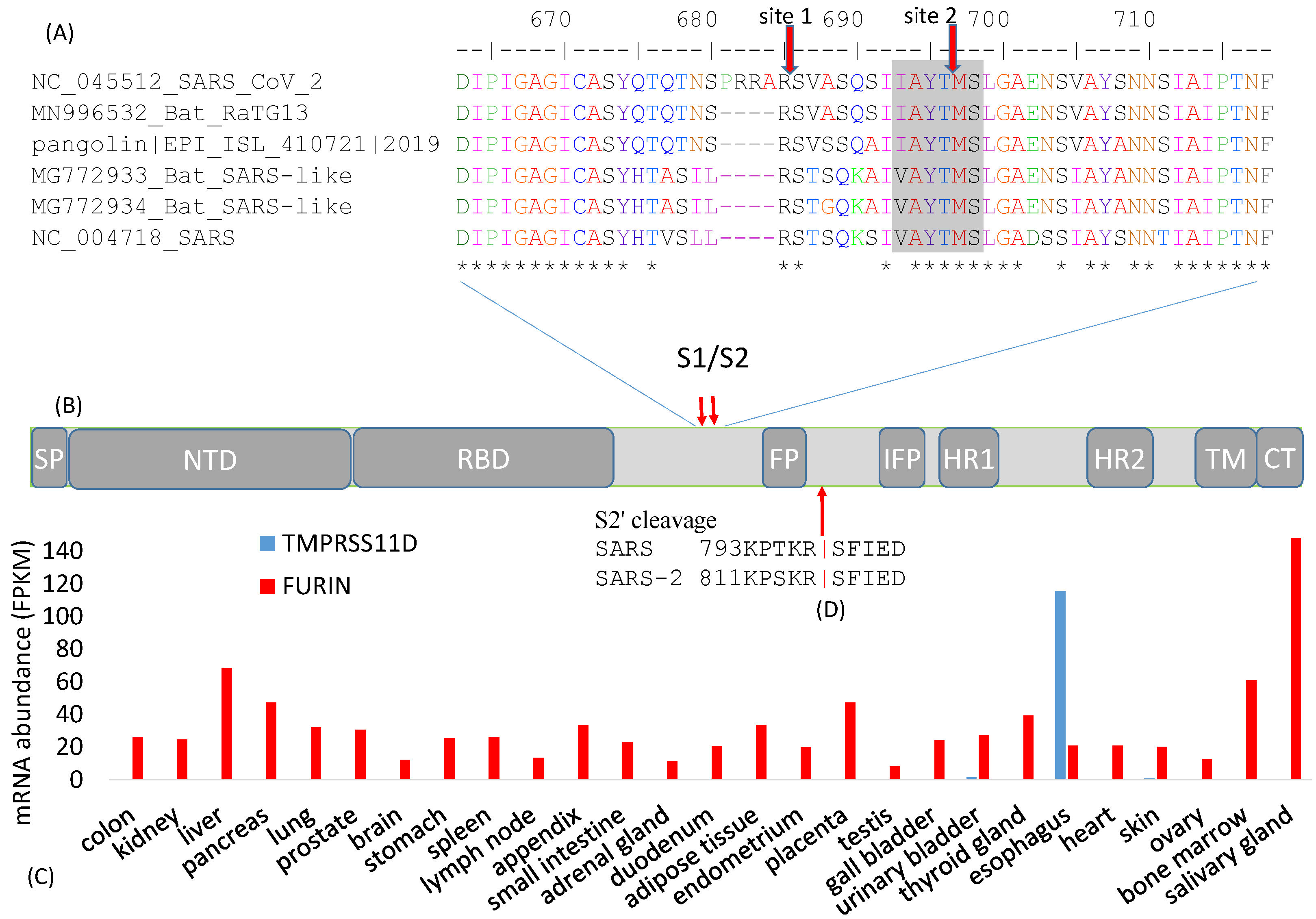
3. Cleavage Sites
4. The S1 Domain
4.1. The Signal Peptide
4.2. The N-Terminal Domain (NTD)
4.3. The Receptor-Binding Domain (RBD)
5. The S2 Domain
5.1. Fusion Peptides
5.2. The Heptad-Repeat Domains: HR-1 and HR-2
5.3. The Transmembrane Domain and Cytoplasmic Tail Domain
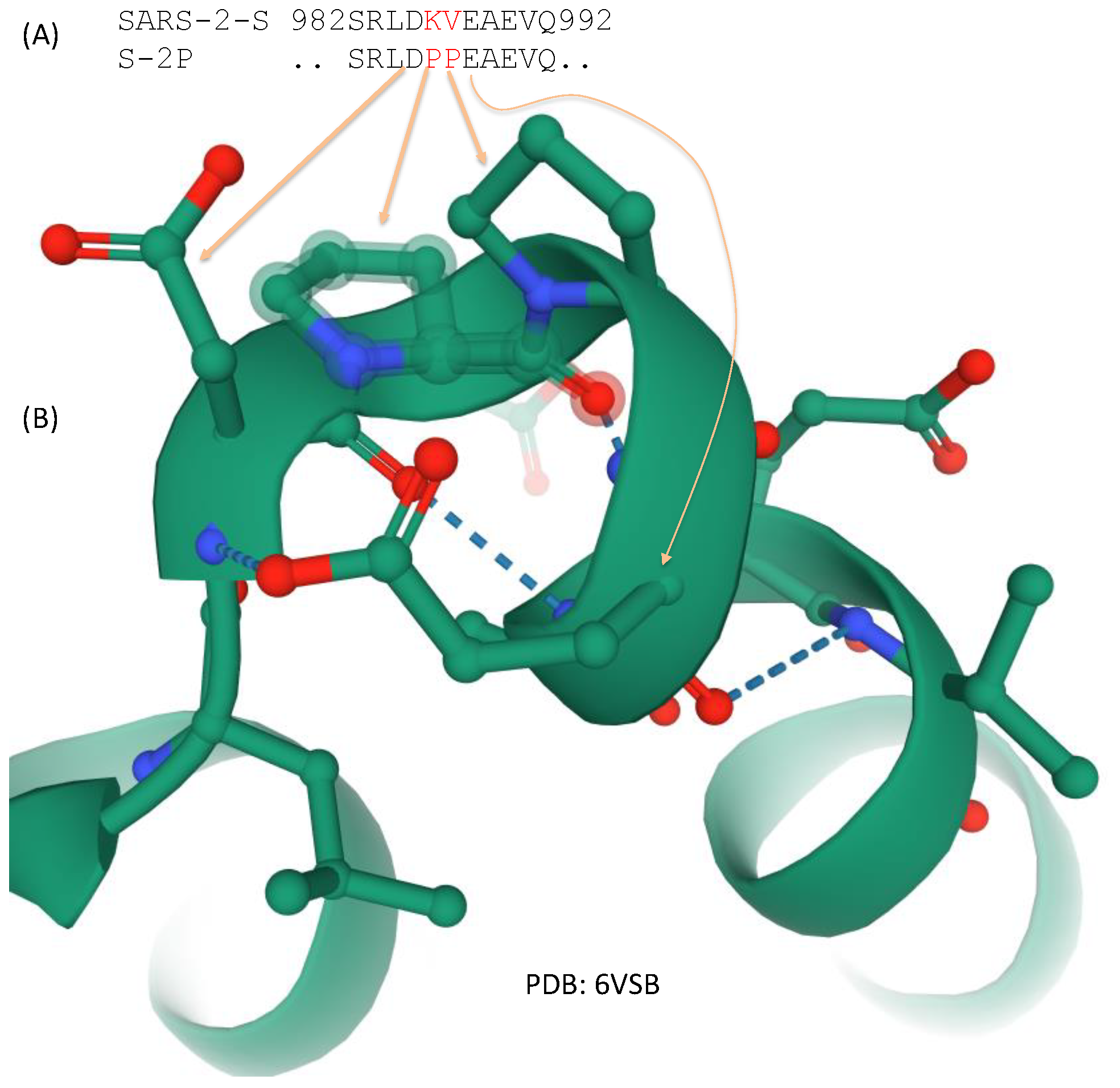
6. The Spike Protein in Vaccine Development
7. Structural Insights into the Emergence of New Viral Variants
8. The Spike Protein and the Conspiracy Theory
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e775. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [PubMed]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef]
- Lu, G.; Wang, Q.; Gao, G.F. Bat-to-human: Spike features determining ‘host jump’ of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 2015, 23, 468–478. [Google Scholar] [CrossRef]
- Hulswit, R.J.G.; de Haan, C.A.M.; Bosch, B.J. Coronavirus Spike Protein and Tropism Changes. In Advances in Virus Research; Ziebuhr, J., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 96, pp. 29–57. [Google Scholar]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar]
- Walls, A.C.; Tortorici, M.A.; Bosch, B.-J.; Frenz, B.; Rottier, P.J.M.; DiMaio, F.; Rey, F.A.; Veesler, D. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 2016, 531, 114–117. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Cottrell, C.A.; Wang, N.; Pallesen, J.; Yassine, H.M.; Turner, H.L.; Corbett, K.S.; Graham, B.S.; McLellan, J.S.; Ward, A.B. Pre-fusion structure of a human coronavirus spike protein. Nature 2016, 531, 118–121. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e286. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Li, C.; Huang, A.; Xia, S.; Lu, S.; Shi, Z.; Lu, L.; Jiang, S.; Yang, Z.; Wu, Y.; et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg. Microb. Infect. 2020, 9, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. DAMBE5: A comprehensive software package for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2013, 30, 1720–1728. [Google Scholar] [CrossRef]
- Lai, M.M.; Cavanagh, D. The molecular biology of coronaviruses. Adv. Virus Res. 1997, 48, 1–100. [Google Scholar]
- Chakraborti, S.; Prabakaran, P.; Xiao, X.; Dimitrov, D.S. The SARS coronavirus S glycoprotein receptor binding domain: Fine mapping and functional characterization. Virol. J. 2005, 2, 73. [Google Scholar] [CrossRef]
- Xiao, X.; Feng, Y.; Chakraborti, S.; Dimitrov, D.S. Oligomerization of the SARS-CoV S glycoprotein: Dimerization of the N-terminus and trimerization of the ectodomain. Biochem. Biophys. Res. Commun. 2004, 322, 93–99. [Google Scholar] [CrossRef]
- Beniac, D.R.; deVarennes, S.L.; Andonov, A.; He, R.; Booth, T.F. Conformational reorganization of the SARS coronavirus spike following receptor binding: Implications for membrane fusion. PLoS ONE 2007, 2, e1082. [Google Scholar] [CrossRef]
- Madu, I.G.; Belouzard, S.; Whittaker, G.R. SARS-coronavirus spike S2 domain flanked by cysteine residues C822 and C833 is important for activation of membrane fusion. Virology 2009, 393, 265–271. [Google Scholar] [CrossRef]
- Mitsuki, Y.Y.; Ohnishi, K.; Takagi, H.; Oshima, M.; Yamamoto, T.; Mizukoshi, F.; Terahara, K.; Kobayashi, K.; Yamamoto, N.; Yamaoka, S.; et al. A single amino acid substitution in the S1 and S2 Spike protein domains determines the neutralization escape phenotype of SARS-CoV. Microbes Infect. 2008, 10, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Ng, O.W.; Keng, C.T.; Leung, C.S.; Peiris, J.S.; Poon, L.L.; Tan, Y.J. Substitution at aspartic acid 1128 in the SARS coronavirus spike glycoprotein mediates escape from a S2 domain-targeting neutralizing monoclonal antibody. PLoS ONE 2014, 9, e102415. [Google Scholar] [CrossRef] [PubMed]
- Song, H.C.; Seo, M.Y.; Stadler, K.; Yoo, B.J.; Choo, Q.L.; Coates, S.R.; Uematsu, Y.; Harada, T.; Greer, C.E.; Polo, J.M.; et al. Synthesis and characterization of a native, oligomeric form of recombinant severe acute respiratory syndrome coronavirus spike glycoprotein. J. Virol. 2004, 78, 10328–10335. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.Y.; Zheng, B.J. Roles of spike protein in the pathogenesis of SARS coronavirus. Hong Kong Med. J. 2009, 15, 37–40. [Google Scholar] [PubMed]
- Xia, X. Translation Control of HAC1 by Regulation of Splicing in Saccharomyces cerevisiae. Int. J. Mol. Sci. 2019, 20, 2860. [Google Scholar] [CrossRef]
- Chow, K.Y.; Yeung, Y.S.; Hon, C.C.; Zeng, F.; Law, K.M.; Leung, F.C. Adenovirus-mediated expression of the C-terminal domain of SARS-CoV spike protein is sufficient to induce apoptosis in Vero E6 cells. FEBS Lett. 2005, 579, 6699–6704. [Google Scholar] [CrossRef]
- Shajahan, A.; Supekar, N.T.; Gleinich, A.S.; Azadi, P. Deducing the N- and O- glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 2020, 30, 981–988. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Sun, X.; Tse, L.V.; Ferguson, A.D.; Whittaker, G.R. Modifications to the Hemagglutinin Cleavage Site Control the Virulence of a Neurotropic H1N1 Influenza Virus. J. Virol. 2010, 84, 8683. [Google Scholar] [CrossRef]
- Kido, H.; Okumura, Y.; Takahashi, E.; Pan, H.-Y.; Wang, S.; Yao, D.; Yao, M.; Chida, J.; Yano, M. Role of host cellular proteases in the pathogenesis of influenza and influenza-induced multiple organ failure. Biochim. Biophys. Acta Proteins Proteom. 2012, 1824, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Silke, J.R.; Aris, P.; Xia, X. Coronavirus genomes carry the signatures of their habitats. PLoS ONE 2020, 15, e0244025. [Google Scholar]
- Belouzard, S.; Chu, V.C.; Whittaker, G.R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl. Acad. Sci. USA 2009, 106, 5871–5876. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.-C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.J.; Bartelink, W.; Rottier, P.J.M. Cathepsin L Functionally Cleaves the Severe Acute Respiratory Syndrome Coronavirus Class I Fusion Protein Upstream of Rather than Adjacent to the Fusion Peptide. J. Virol. 2008, 82, 8887. [Google Scholar] [CrossRef]
- Burkard, C.; Verheije, M.H.; Wicht, O.; van Kasteren, S.I.; van Kuppeveld, F.J.; Haagmans, B.L.; Pelkmans, L.; Rottier, P.J.; Bosch, B.J.; de Haan, C.A. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog. 2014, 10, e1004502. [Google Scholar] [CrossRef]
- Kirschke, H. Chapter 410—Cathepsin L. In Handbook of Proteolytic Enzymes, 3rd ed.; Rawlings, N.D., Salvesen, G., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 1808–1817. [Google Scholar]
- Jaimes, J.A.; André, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient Activation of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein by the Transmembrane Protease TMPRSS2. J. Virol. 2010, 84, 12658. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hofmann-Winkler, H.; Pöhlmann, S. Priming Time: How Cellular Proteases Arm Coronavirus Spike Proteins. In Activation of Viruses by Host Proteases; Böttcher-Friebertshäuser, E., Garten, W., Klenk, H.D., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 71–98. [Google Scholar]
- Glowacka, I.; Bertram, S.; Müller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef]
- Kleine-Weber, H.; Elzayat, M.T.; Hoffmann, M.; Pöhlmann, S. Functional analysis of potential cleavage sites in the MERS-coronavirus spike protein. Sci. Rep. 2018, 8, 16597. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.; Reeves, J.D.; Rennekamp, A.J.; Amberg, S.M.; Piefer, A.J.; Bates, P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. USA 2004, 101, 4240–4245. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Ujike, M.; Morikawa, S.; Tashiro, M.; Taguchi, F. Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc. Natl. Acad. Sci. USA 2005, 102, 12543. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Sun, D.; Rajashankar, K.R.; Qian, Z.; Holmes, K.V.; Li, F. Crystal structure of mouse coronavirus receptor-binding domain complexed with its murine receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 10696. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.K.; Jiang, G.S.; Holmes, K.V. Receptor for mouse hepatitis virus is a member of the carcinoembryonic antigen family of glycoproteins. Proc. Natl. Acad. Sci. USA 1991, 88, 5533. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. DAMBE7: New and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef]
- Gui, M.; Song, W.; Zhou, H.; Xu, J.; Chen, S.; Xiang, Y.; Wang, X. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 2017, 27, 119–129. [Google Scholar] [CrossRef]
- Zakhartchouk, A.N.; Sharon, C.; Satkunarajah, M.; Auperin, T.; Viswanathan, S.; Mutwiri, G.; Petric, M.; See, R.H.; Brunham, R.C.; Finlay, B.B.; et al. Immunogenicity of a receptor-binding domain of SARS coronavirus spike protein in mice: Implications for a subunit vaccine. Vaccine 2007, 25, 136–143. [Google Scholar] [CrossRef]
- He, Y.; Zhou, Y.; Liu, S.; Kou, Z.; Li, W.; Farzan, M.; Jiang, S. Receptor-binding domain of SARS-CoV spike protein induces highly potent neutralizing antibodies: Implication for developing subunit vaccine. Biochem. Biophys. Res. Commun. 2004, 324, 773–781. [Google Scholar] [CrossRef]
- He, Y.; Zhou, Y.; Siddiqui, P.; Jiang, S. Inactivated SARS-CoV vaccine elicits high titers of spike protein-specific antibodies that block receptor binding and virus entry. Biochem. Biophys. Res. Commun. 2004, 325, 445–452. [Google Scholar] [CrossRef]
- Du, L.; Zhao, G.; Chan, C.C.; Sun, S.; Chen, M.; Liu, Z.; Guo, H.; He, Y.; Zhou, Y.; Zheng, B.J.; et al. Recombinant receptor-binding domain of SARS-CoV spike protein expressed in mammalian, insect and E. coli cells elicits potent neutralizing antibody and protective immunity. Virology 2009, 393, 144–150. [Google Scholar] [CrossRef]
- Du, L.; Zhao, G.; He, Y.; Guo, Y.; Zheng, B.J.; Jiang, S.; Zhou, Y. Receptor-binding domain of SARS-CoV spike protein induces long-term protective immunity in an animal model. Vaccine 2007, 25, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhao, G.; Lin, Y.; Sui, H.; Chan, C.; Ma, S.; He, Y.; Jiang, S.; Wu, C.; Yuen, K.Y.; et al. Intranasal vaccination of recombinant adeno-associated virus encoding receptor-binding domain of severe acute respiratory syndrome coronavirus (SARS-CoV) spike protein induces strong mucosal immune responses and provides long-term protection against SARS-CoV infection. J. Immunol. 2008, 180, 948–956. [Google Scholar] [PubMed]
- Zhang, X.; Wang, J.; Wen, K.; Mou, Z.; Zou, L.; Che, X.; Ni, B.; Wu, Y. Antibody binding site mapping of SARS-CoV spike protein receptor-binding domain by a combination of yeast surface display and phage peptide library screening. Viral Immunol. 2009, 22, 407–415. [Google Scholar] [PubMed]
- Cao, Z.; Liu, L.; Du, L.; Zhang, C.; Jiang, S.; Li, T.; He, Y. Potent and persistent antibody responses against the receptor-binding domain of SARS-CoV spike protein in recovered patients. Virol. J. 2010, 7, 299. [Google Scholar]
- Prabakaran, P.; Gan, J.; Feng, Y.; Zhu, Z.; Choudhry, V.; Xiao, X.; Ji, X.; Dimitrov, D.S. Structure of severe acute respiratory syndrome coronavirus receptor-binding domain complexed with neutralizing antibody. J. Biol. Chem. 2006, 281, 15829–15836. [Google Scholar] [CrossRef]
- Chen, W.H.; Du, L.; Chag, S.M.; Ma, C.; Tricoche, N.; Tao, X.; Seid, C.A.; Hudspeth, E.M.; Lustigman, S.; Tseng, C.T.; et al. Yeast-expressed recombinant protein of the receptor-binding domain in SARS-CoV spike protein with deglycosylated forms as a SARS vaccine candidate. Hum. Vaccines Immunother. 2014, 10, 648–658. [Google Scholar]
- Du, L.; Ma, C.; Jiang, S. Antibodies induced by receptor-binding domain in spike protein of SARS-CoV do not cross-neutralize the novel human coronavirus hCoV-EMC. J. Infect. 2013, 67, 348–350. [Google Scholar]
- Zhu, Z.; Chakraborti, S.; He, Y.; Roberts, A.; Sheahan, T.; Xiao, X.; Hensley, L.E.; Prabakaran, P.; Rockx, B.; Sidorov, I.A.; et al. Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc. Natl. Acad. Sci. USA 2007, 104, 12123–12128. [Google Scholar]
- Elshabrawy, H.A.; Coughlin, M.M.; Baker, S.C.; Prabhakar, B.S. Human monoclonal antibodies against highly conserved HR1 and HR2 domains of the SARS-CoV spike protein are more broadly neutralizing. PLoS ONE 2012, 7, e50366. [Google Scholar]
- He, Y.; Li, J.; Jiang, S. A single amino acid substitution (R441A) in the receptor-binding domain of SARS coronavirus spike protein disrupts the antigenic structure and binding activity. Biochem. Biophys. Res. Commun. 2006, 344, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Poh, W.P.; Narasaraju, T.; Pereira, N.A.; Zhong, F.; Phoon, M.C.; Macary, P.A.; Wong, S.H.; Lu, J.; Koh, D.R.; Chow, V.T. Characterization of cytotoxic T-lymphocyte epitopes and immune responses to SARS coronavirus spike DNA vaccine expressing the RGD-integrin-binding motif. J. Med. Virol. 2009, 81, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Lin, C.F.; Fang, Y.T.; Kuo, Y.M.; Liao, P.C.; Yeh, T.M.; Hwa, K.Y.; Shieh, C.C.; Yen, J.H.; Wang, H.J.; et al. Antibody to severe acute respiratory syndrome (SARS)-associated coronavirus spike protein domain 2 cross-reacts with lung epithelial cells and causes cytotoxicity. Clin. Exp. Immunol. 2005, 141, 500–508. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef]
- Bosch, B.J.; Martina, B.E.; Van Der Zee, R.; Lepault, J.; Haijema, B.J.; Versluis, C.; Heck, A.J.; De Groot, R.; Osterhaus, A.D.; Rottier, P.J. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 8455–8460. [Google Scholar] [CrossRef]
- Broer, R.; Boson, B.; Spaan, W.; Cosset, F.L.; Corver, J. Important role for the transmembrane domain of severe acute respiratory syndrome coronavirus spike protein during entry. J. Virol. 2006, 80, 1302–1310. [Google Scholar] [CrossRef]
- Modis, Y. Class II fusion proteins. Adv. Exp. Med. Biol. 2013, 790, 150–166. [Google Scholar]
- Zeng, X.; Herndon, A.M.; Hu, J.C. Buried asparagines determine the dimerization specificities of leucine zipper mutants. Proc. Natl. Acad. Sci. USA 1997, 94, 3673. [Google Scholar] [CrossRef]
- Yoshida, H.; Oku, M.; Suzuki, M.; Mori, K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J. Cell. Biol. 2006, 172, 565–575. [Google Scholar] [CrossRef]
- Xia, X. Beyond Trees: Regulons and Regulatory Motif Characterization. Genes 2020, 11, 995. [Google Scholar] [CrossRef]
- Chambers, P.; Pringle, C.R.; Easton, A.J. Heptad repeat sequences are located adjacent to hydrophobic regions in several types of virus fusion glycoproteins. J. Gen. Virol. 1990, 71, 3075–3080. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Hao, X.; Chen, A.; Chrétien, M.; Basak, A. Structural and biochemical investigation of heptad repeat derived peptides of human SARS corona virus (hSARS-CoV) spike protein. Protein Pept. Lett. 2008, 15, 874–886. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y.; et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 2017, 8, 15092. [Google Scholar] [CrossRef]
- Ni, L.; Zhu, J.; Zhang, J.; Yan, M.; Gao, G.F.; Tien, P. Design of recombinant protein-based SARS-CoV entry inhibitors targeting the heptad-repeat regions of the spike protein S2 domain. Biochem. Biophys. Res. Commun. 2005, 330, 39–45. [Google Scholar] [CrossRef]
- Pallesen, J.; Wang, N.; Corbett, K.S.; Wrapp, D.; Kirchdoerfer, R.N.; Turner, H.L.; Cottrell, C.A.; Becker, M.M.; Wang, L.; Shi, W.; et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. USA 2017, 114, E7348. [Google Scholar] [CrossRef]
- Corver, J.; Broer, R.; van Kasteren, P.; Spaan, W. Mutagenesis of the transmembrane domain of the SARS coronavirus spike glycoprotein: Refinement of the requirements for SARS coronavirus cell entry. Virol. J. 2009, 6, 230. [Google Scholar] [CrossRef]
- Liao, Y.; Zhang, S.M.; Neo, T.L.; Tam, J.P. Tryptophan-dependent membrane interaction and heteromerization with the internal fusion peptide by the membrane proximal external region of SARS-CoV spike protein. Biochemistry 2015, 54, 1819–1830. [Google Scholar] [CrossRef]
- Petit, C.M.; Chouljenko, V.N.; Iyer, A.; Colgrove, R.; Farzan, M.; Knipe, D.M.; Kousoulas, K.G. Palmitoylation of the cysteine-rich endodomain of the SARS-coronavirus spike glycoprotein is important for spike-mediated cell fusion. Virology 2007, 360, 264–274. [Google Scholar] [CrossRef]
- Petit, C.M.; Melancon, J.M.; Chouljenko, V.N.; Colgrove, R.; Farzan, M.; Knipe, D.M.; Kousoulas, K.G. Genetic analysis of the SARS-coronavirus spike glycoprotein functional domains involved in cell-surface expression and cell-to-cell fusion. Virology 2005, 341, 215–230. [Google Scholar] [CrossRef]
- Millet, J.K.; Kien, F.; Cheung, C.Y.; Siu, Y.L.; Chan, W.L.; Li, H.; Leung, H.L.; Jaume, M.; Bruzzone, R.; Peiris, J.S.; et al. Ezrin interacts with the SARS coronavirus Spike protein and restrains infection at the entry stage. PLoS ONE 2012, 7, e49566. [Google Scholar]
- Anderson, E.J.; Rouphael, N.G.; Widge, A.T.; Jackson, L.A.; Roberts, P.C.; Makhene, M.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; Pruijssers, A.J.; et al. Safety and Immunogenicity of SARS-CoV-2 mRNA-1273 Vaccine in Older Adults. N. Engl. J. Med. 2020, 383, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA Vaccine against SARS-CoV-2—Preliminary Report. N. Engl. J. Med. 2020, 383, 1920–1931. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.R.; Graham, R.L.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses: An RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011, 8, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Ferron, F.; Subissi, L.; Silveira De Morais, A.T.; Le, N.T.T.; Sevajol, M.; Gluais, L.; Decroly, E.; Vonrhein, C.; Bricogne, G.; Canard, B.; et al. Structural and molecular basis of mismatch correction and ribavirin excision from coronavirus RNA. Proc. Natl. Acad. Sci. USA 2018, 115, E162–E171. [Google Scholar] [CrossRef] [PubMed]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.-L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e819. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.e738. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, X.; Hu, T.; Li, J.; Song, H.; Liu, Y.; Wang, P.; Liu, D.; Yang, J.; Holmes, E.C.; et al. A Novel Bat Coronavirus Closely Related to SARS-CoV-2 Contains Natural Insertions at the S1/S2 Cleavage Site of the Spike Protein. Curr. Biol. 2020, 30, 2196–2203.e3. [Google Scholar] [CrossRef]
- Reinke, L.M.; Spiegel, M.; Plegge, T.; Hartleib, A.; Nehlmeier, I.; Gierer, S.; Hoffmann, M.; Hofmann-Winkler, H.; Winkler, M.; Pöhlmann, S. Different residues in the SARS-CoV spike protein determine cleavage and activation by the host cell protease TMPRSS2. PLoS ONE 2017, 12, e0179177. [Google Scholar]
- Yao, Y.X.; Ren, J.; Heinen, P.; Zambon, M.; Jones, I.M. Cleavage and Serum Reactivity of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein. J. Infect. Dis. 2004, 190, 91–98. [Google Scholar] [CrossRef]
- Xia, X. Extreme genomic CpG deficiency in SARS-CoV-2 and evasion of host antiviral defense. Mol. Biol. Evol. 2020, 37, 2699–2705. [Google Scholar] [CrossRef]
- Takata, M.A.; Gonçalves-Carneiro, D.; Zang, T.M.; Soll, S.J.; York, A.; Blanco-Melo, D.; Bieniasz, P.D. CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature 2017, 550, 124–127. [Google Scholar] [CrossRef]
- Lobry, J.R. Origin of replication of Mycoplasma genitalium. Science 1996, 272, 745–746. [Google Scholar] [CrossRef]
- Kleine-Weber, H.; Pöhlmann, S.; Hoffmann, M. Spike proteins of novel MERS-coronavirus isolates from North- and West-African dromedary camels mediate robust viral entry into human target cells. Virology 2019, 535, 261–265. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Name | Length | ICpG | NCG | NGC | Ln (NCG/NGC) |
|---|---|---|---|---|---|
| NC_045512_SARS-CoV-2 | 3819 | 0.2179 | 29 | 137 | −1.5527 |
| MN996532_Bat_RaTG13 | 3807 | 0.2753 | 37 | 140 | −1.3307 |
| pangolin/EPI_ISL_410721/2019 | 3795 | 0.2857 | 38 | 139 | −1.2969 |
| MG772933_Bat_SARS-like | 3738 | 0.3618 | 50 | 148 | −1.0852 |
| MG772934_Bat_SARS-like | 3735 | 0.3697 | 50 | 142 | −1.0438 |
| NC_004718_SARS | 3765 | 0.3673 | 52 | 174 | −1.2078 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, X. Domains and Functions of Spike Protein in SARS-Cov-2 in the Context of Vaccine Design. Viruses 2021, 13, 109. https://doi.org/10.3390/v13010109
Xia X. Domains and Functions of Spike Protein in SARS-Cov-2 in the Context of Vaccine Design. Viruses. 2021; 13(1):109. https://doi.org/10.3390/v13010109
Chicago/Turabian StyleXia, Xuhua. 2021. "Domains and Functions of Spike Protein in SARS-Cov-2 in the Context of Vaccine Design" Viruses 13, no. 1: 109. https://doi.org/10.3390/v13010109
APA StyleXia, X. (2021). Domains and Functions of Spike Protein in SARS-Cov-2 in the Context of Vaccine Design. Viruses, 13(1), 109. https://doi.org/10.3390/v13010109