Artificial Intelligence-Assisted Loop Mediated Isothermal Amplification (AI-LAMP) for Rapid Detection of SARS-CoV-2

 ,
,  ,
, 
 ,
,  , ,
, ,  , , , ,
, , , ,
Abstract
1. Introduction
2. Materials andMethods
2.1. Ethics Statement
2.2. Cells and Viruses
2.3. In Silico Nucleotide Sequence Comparisons and Primer Design
2.4. Cloning and In Vitro Transcription of RdRP Target Gene
2.5. Clinical Sample Processing and Spiking with miR-cel-miR-39-3p RNA
2.6. Real-Time Fluorescent-Based Quantitative PCR
2.7. AI-LAMP Assay Performance
2.8. Artificial Intelligence Based Test-Tube Colour Detection
2.9. Analytical Specificity and Sensitivity of the LAMP Assay
2.10. Quantitative Real Time PCR for miR-cel-miR-39-3p RNA
2.11. Statistical Analysis
3. Results
3.1. High Resolution Conversation Analysis of SARS-CoV-2 to Guide Oligo Design
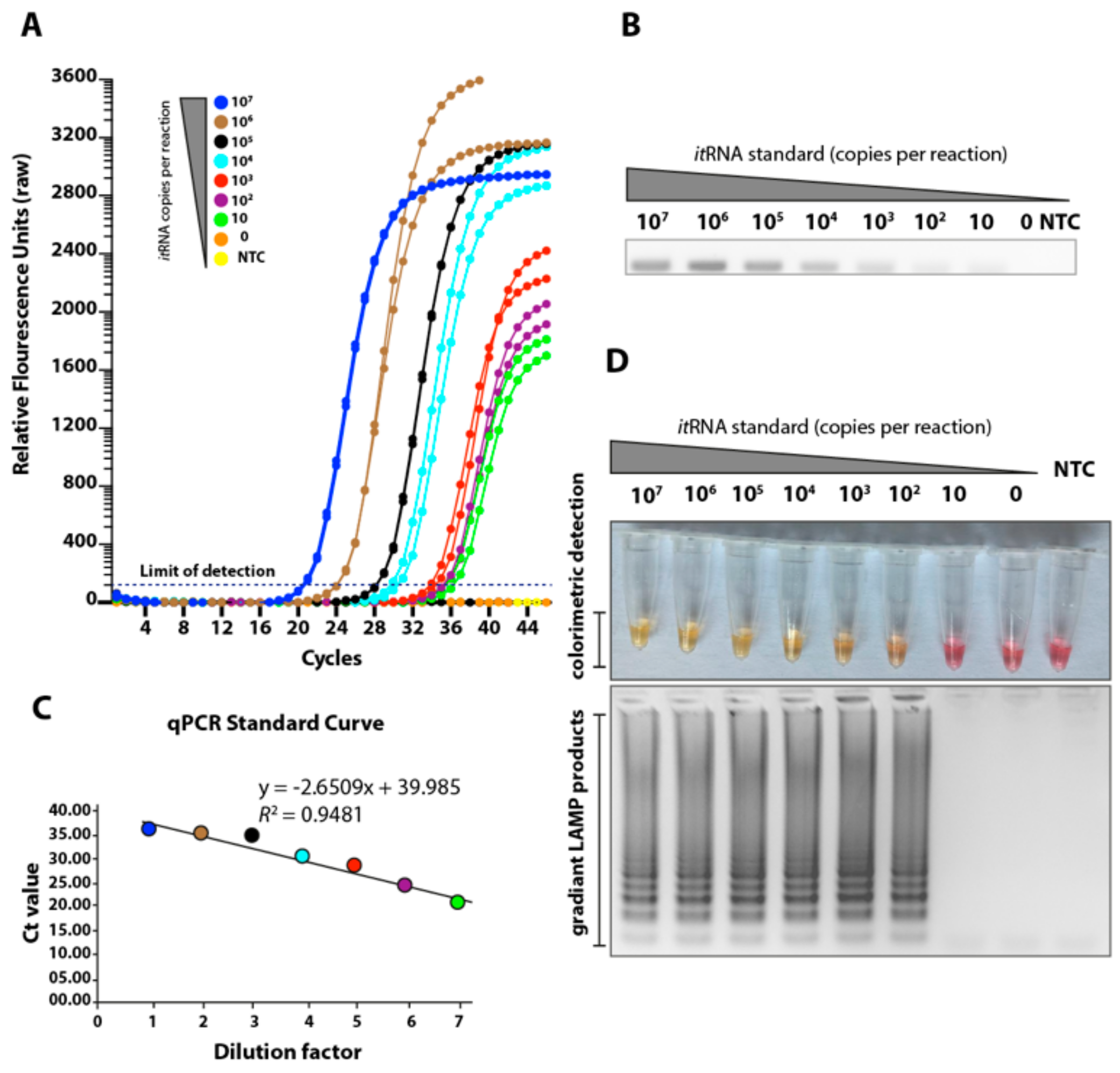
3.2. Determination of the Limit of Detection of the LAMP Assay Using Biochemically Synthesised RNA
3.3. Cross-Reactivity of the Novel LAMP Assay When Tested against Other Respiratory and Medically Important Viruses
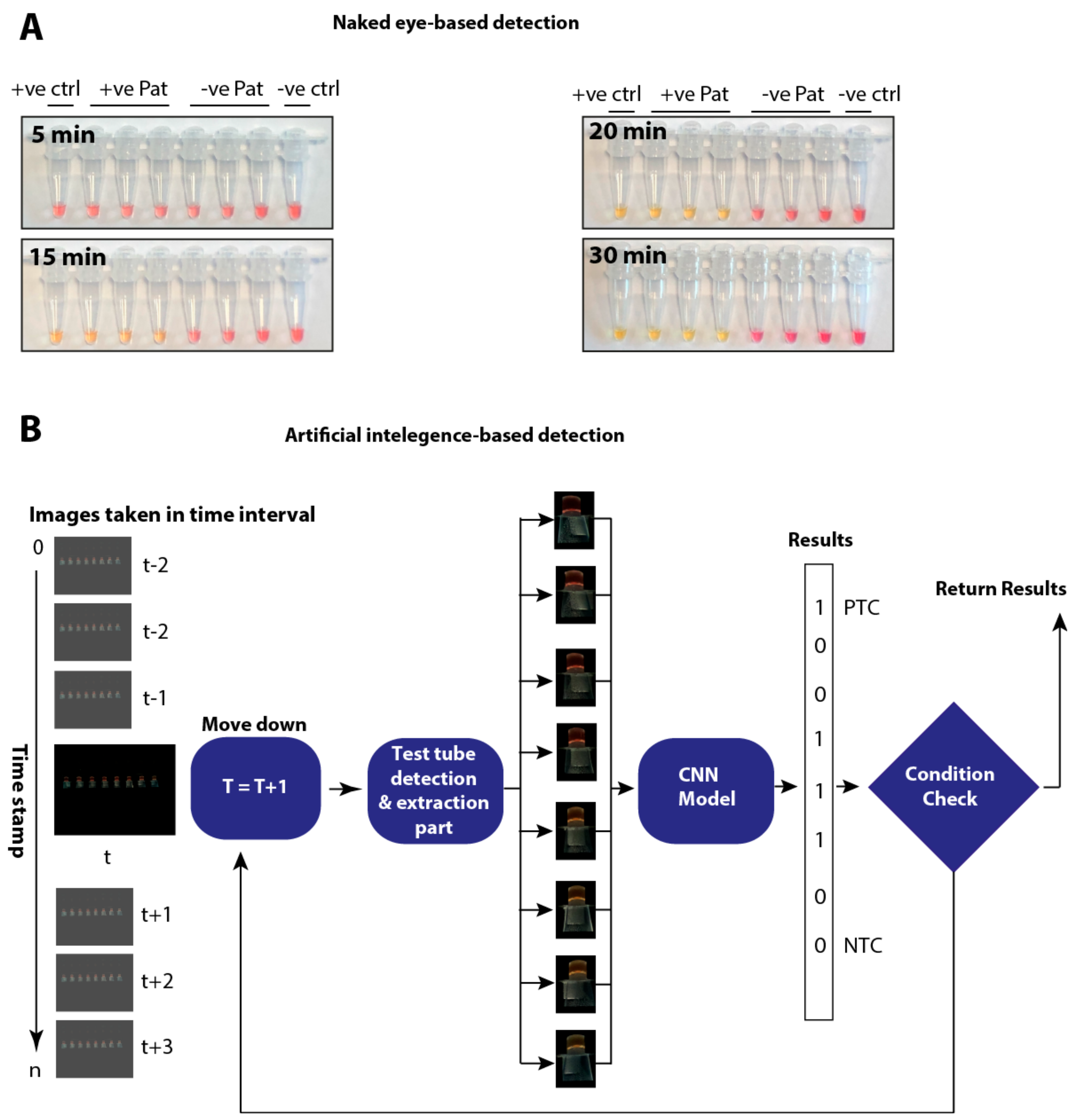
3.4. Temporal Investigations of the LAMP Assay and Its Impact on the Limit of Detection
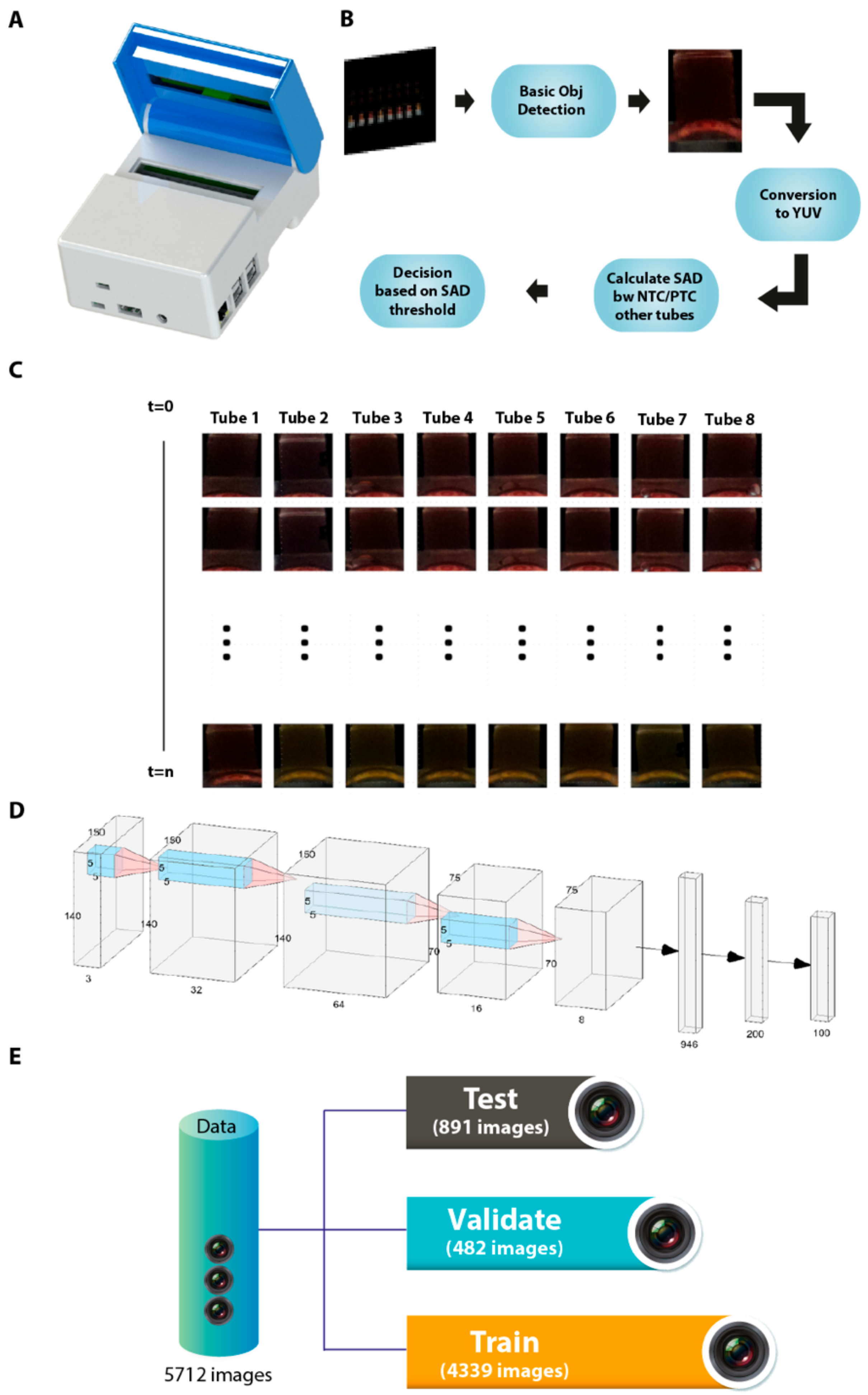
3.5. Manufacture of an Isothermal Nucleic Acid Amplification Device with Colorimetric Detection Features
3.6. Automated Image Acquisition and Processing through a Template Matching-Based Algorithm
3.7. Artificial Intelligence-Assisted Rapid Detection of Colour Changes Associated with the LAMP Reaction
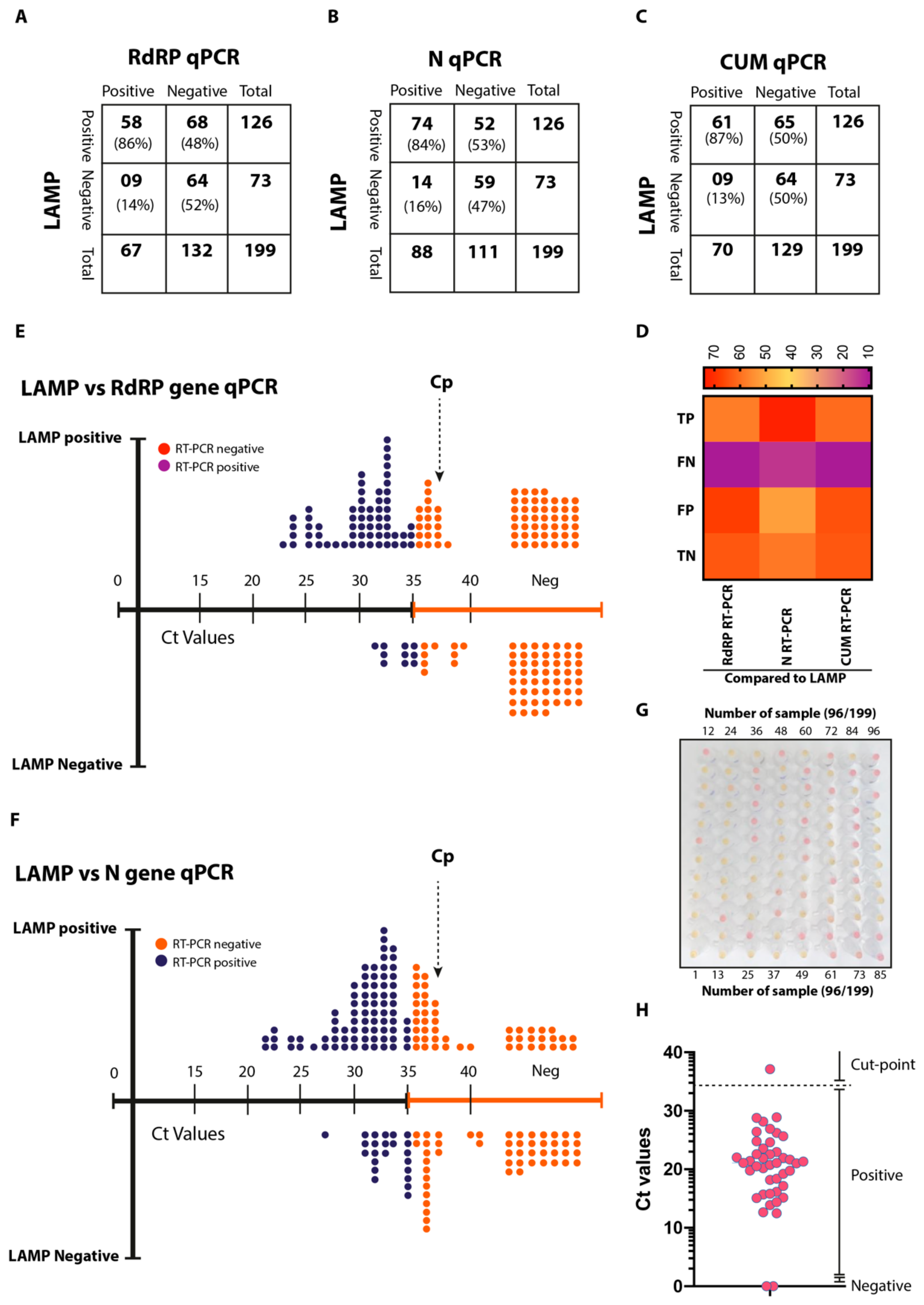
3.8. Validation of AI-LAMP and Comparative Performance in Clinical Settings
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Chan, J.F.; Yuan, S.; Kok, K.H.; To, K.K.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.; Poon, R.W.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Decaro, N.; Lorusso, A. Novel human coronavirus (SARS-CoV-2): A lesson from animal coronaviruses. Vet. Microbiol. 2020, 244, 108693. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.; Yang, J.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]
- Peiris, J.S.; Lai, S.T.; Poon, L.L.; Guan, Y.; Yam, L.Y.; Lim, W.; Nicholls, J.; Yee, W.K.; Yan, W.W.; Cheung, M.T.; et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003, 361, 1319–1325. [Google Scholar] [CrossRef]
- Woo, P.C.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin. Microbiol. Rev. 2007, 20, 660–694. [Google Scholar]
- Yuen, K.Y. Middle East respiratory syndrome coronavirus: Another zoonotic betacoronavirus causing SARS-like disease. Clin. Microbiol. Rev. 2015, 28, 465–522. [Google Scholar]
- Javaid, A.; Hussain, N. Mutational analysis of Forkhead box P3 gene in Pakistani Human Immunodeficient Virus Patients. Pak. J. Zool. 2019, 51, 1987–1990. [Google Scholar] [CrossRef]
- Afshan, G.; Zulfiqar, S.; Mehboob, S.; Khan, M.T.J.; Shakoori, A. Production of Antibodies against Hepatitis C Virus Envelope Glycoprotein E2- A Potential Vaccine Against HCV Infection. Pak. J. Zool. 2019, 51, 2311–2322. [Google Scholar] [CrossRef]
- Shahid, M.; Amin, I.; Afzal, S.; Fatima, Z.; Idrees, M. Comparative Analysis of Immunological and Genomic Outcomes of Dengue Virus Outbreak in Pakistan. Pak. J. Zool. 2019, 51, 1971–1974. [Google Scholar] [CrossRef]
- WHO. Novel Coronavirus—China. 2020. Available online: https://www.who.int/csr/don/12-january-2020-novel-coronavirus-china/en/ (accessed on 24 August 2020).
- Vogels, C.B.F.; Brito, A.F.; Wyllie, A.L.; Fauver, J.R.; Ott, I.M.; Kalinich, C.C.; Petrone, M.E.; Casanovas-Massana, A.; Muenker, M.C.; Moore, A.J.; et al. Analytical sensitivity and efficiency comparisons of SARS-COV-2 qRT-PCR assays. medRxiv. 2020. Available online: https://medrxiv.org/content/early/2020/04/01/2020.03.30.20048108 (accessed on 24 August 2020). [CrossRef]
- Kashir, J.; Yaqinuddin, A. Loop mediated isothermal amplification (LAMP) assays as a rapid diagnostic for COVID-19. Med. Hypotheses 2020, 141. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Cui, J.; Huang, L.; Du, B.; Chen, L.; Xue, G.; Li, S.; Zhang, W.; Zhao, L.; Sun, Y.; et al. Rapid and visual detection of 2019 novel coronavirus (SARS-CoV-2) by a reverse transcription loop-mediated isothermal amplification assay. Clin. Microbiol. Infect. 2020, 26, 773–779. [Google Scholar] [CrossRef]
- Mori, Y.; Notomi, T. Loop-mediated isothermal amplification (LAMP): Expansion of its practical application as a tool to achieve universal health coverage. J. Infect. Chemother. 2020, 26, 13–17. [Google Scholar] [CrossRef]
- Mahony, J.; Chong, S.; Bulir, D.; Ruyter, A.; Mwawasi, K.; Waltho, D. Development of a sensitive loop-mediated isothermal amplification assay that provides specimen-to-result diagnosis of respiratory syncytial virus infection in 30 minutes. J. Clin. Microbiol. 2013, 51, 2696–2701. [Google Scholar] [CrossRef][Green Version]
- Ganguli, A.; Ornob, A.; Yu, H.; Damhorst, G.L.; Chen, W.; Sun, F.; Bhuiya, A.; Cunningham, B.T.; Bashir, R. Hands-free smartphone-based diagnostics for simultaneous detection of Zika, Chikungunya, and Dengue at point-of-care. Biomed. Microdevices 2017, 19, 73. [Google Scholar] [CrossRef]
- Kaarj, K.; Akarapipad, P.; Yoon, J.Y. Simpler, Faster, and Sensitive Zika Virus Assay Using Smartphone Detection of Loop-mediated Isothermal Amplification on Paper Microfluidic Chips. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Yu, L.; Wu, S.; Hao, X.; Dong, X.; Mao, L.; Pelechano, V.; Chen, W.H.; Yin, X. Rapid Detection of COVID-19 Coronavirus Using a Reverse Transcriptional Loop-Mediated Isothermal Amplification (RT-LAMP) Diagnostic Platform. Clin. Chem. 2020, 66, 975–977. [Google Scholar] [CrossRef]
- Nguyen, T.; Bang, D.D.; Wolff, A. 2019 Novel coronavirus disease (COVID-19): Paving the road for rapid detection and point-of-care diagnostics. Micromachines 2020, 11, 306. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.; Yip, C.C.; To, K.K.; Tang, T.H.; Wong, S.C.; Leung, K.H.; Fung, A.Y.; Ng, A.C.; Zou, Z.; Tsoi, H.W.; et al. Improved molecular diagnosis of COVID-19 by the novel, highly sensitive and specific COVID-19-RdRp/Hel real-time reverse transcription-PCR assay validated in vitro and with clinical specimens. J. Clin. Microbiol. 2020, 58, e00310–e00320. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; RozewickI, J.; Yamada, K.D. MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Mao, X.; Hong, S.; Xu, W.; Gui, G. Template Matching-Based Method for Intelligent Invoice Information Identification. IEEE Access 2019, 7, 28392–28401. [Google Scholar] [CrossRef]
- Yang, G.; Li, H.; Zhang, L.; Cao, Y. Research on a skin color detection algorithm based on self-adaptive skin color model. In Proceedings of the 2010 International Conference on Communications and Intelligence Information Security, Nanning, China, 13–14 October 2010; pp. 266–270. [Google Scholar]
- Al-Tairi, Z.H.; Rahmat, R.W.O.; Saripan, M.I.; Sulaiman, P.S. Skin Segmentation Using YUV and RGB Color Spaces. J. Inf. Process Syst. 2014, 10, 283–299. [Google Scholar] [CrossRef]
- Sharif, M.; Attique Khan, M.; Rashid, M.; Yasmin, M.; Afza, F.; Tanik, U.J. Deep CNN and geometric features-based gastrointestinal tract diseases detection and classification from wireless capsule endoscopy images. J. Exp. Theor. Artif. Intell. 2019. [Google Scholar] [CrossRef]
- WHO. Coronavirus Disease (COVID-19) Pandemic. 2020. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 24 August 2020).
- Kurosaki, Y.; Grolla, A.; Fukuma, A.; Feldmann, H.; Yasuda, J. Development and evaluation of a simple assay for Marburg virus detection using a reverse transcription-loop-mediated isothermal amplification method. J. Clin. Microbiol. 2010, 48, 2330–2336. [Google Scholar] [CrossRef]
- Ge, Y.; Wu, B.; Qi, X.; Zhao, K.; Guo, X.; Zhu, Y.; Qi, Y.; Shi, Z.; Zhou, M.; Wang, H.; et al. Rapid and sensitive detection of novel avian-origin influenza A (H7N9) virus by reverse transcription loop-mediated isothermal amplification combined with a lateral-flow device. PLoS ONE. 2013, 8, e69941. [Google Scholar] [CrossRef]
- Kwallah, A.; Inoue, S.; Muigai, A.W.; Kubo, T.; Sang, R.; Morita, K.; Mwau, M. A real-time reverse transcription loop-mediated isothermal amplification assay for the rapid detection of yellow fever virus. J. Virol. Methods 2013, 193, 23–27. [Google Scholar] [CrossRef]
- Cao, Z.; Wang, H.; Wang, L.; Li, L.; Jin, H.; Xu, C.; Feng, N.; Wang, J.; Li, Q.; Zhao, Y.; et al. Visual detection of west nile virus using reverse transcription loop-mediated isothermal amplification combined with a vertical flow visualization strip. Front. Microbiol. 2016, 7, 554. [Google Scholar] [CrossRef]
- Xu, C.; Wang, H.; Jin, H.; Feng, N.; Zheng, X.; Cao, Z.; Li, L.; Wang, J.; Yan, F.; Wang, L.; et al. Visual detection of Ebola virus using reverse transcription loop-mediated isothermal amplification combined with nucleic acid strip detection. Arch. Virol. 2016, 161, 1125–1133. [Google Scholar] [CrossRef]
- Chotiwan, N.; Brewster, C.D.; Magalhaes, T.; Weger-Lucarelli, J.; Duggal, N.K.; Rückert, C.; Nguyen, C.; Garcia Luna, S.M.; Fauver, J.R.; Andre, B.; et al. Rapid and specific detection of Asian- and African- lineage Zika viruses. Sci. Transl. Med. 2017, 9, eaag0538. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, W.; Zhang, Q.; Xu, K.; Ye, G.; Wu, W.; Sun, Z.; Liu, F.; Wu, K.; Zhong, B.; et al. RNA based mNGS approach identifies a novel human coronavirus from two individual pneumonia cases in 2019 Wuhan outbreak. Emerg. Microbes Infect. 2020, 9, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Yang, W.; Dang, X.; Wang, Q.; Xu, M.; Zhao, Q.; Zhou, Y.; Zhao, H.; Wang, L.; Xu, Y.; Wang, J.; et al. Rapid Detection of SARS-CoV-2 Using Reverse transcription RT-LAMP method. medrRxiv 2020. [Google Scholar] [CrossRef]
- Wei, M.; Yuan, J.; Liu, Y.; Fu, T.; Yu, X.; Zhang, Z.J. Novel coronavirus infection in hospitalized infants under 1 year of age in China. JAMA 2020, 323, 1313–1314. [Google Scholar] [CrossRef]
- Sodi, R.; Eastwood, J.; Caslake, M.; Packard, C.J.; Denby, L. Relationship Between Circulating microRNA-30c With Total- And LDL-cholesterol, Their Circulatory Transportation and Effect of Statins. Clin. Chim. Acta 2017, 466, 13–19. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time in Minutes (after Start of the LAMP Reaction) | In Vitro Transcribed RNA Dilution (Copies/Reaction) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 107 | 106 | 105 | 104 | 103 | 102 | 10 | 0 | NTC | |
| 05 | − | − | − | − | − | − | − | − | − |
| 10 | − | − | − | − | − | − | − | − | − |
| 15 | + | + | + | + | − | − | − | − | − |
| 20 | + | + | + | + | + | + | − | − | − |
| 25 | + | + | + | + | + | + | − | − | − |
| 30 | + | + | + | + | + | + | − | − | − |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rohaim, M.A.; Clayton, E.; Sahin, I.; Vilela, J.; Khalifa, M.E.; Al-Natour, M.Q.; Bayoumi, M.; Poirier, A.C.; Branavan, M.; Tharmakulasingam, M.; et al. Artificial Intelligence-Assisted Loop Mediated Isothermal Amplification (AI-LAMP) for Rapid Detection of SARS-CoV-2. Viruses 2020, 12, 972. https://doi.org/10.3390/v12090972
Rohaim MA, Clayton E, Sahin I, Vilela J, Khalifa ME, Al-Natour MQ, Bayoumi M, Poirier AC, Branavan M, Tharmakulasingam M, et al. Artificial Intelligence-Assisted Loop Mediated Isothermal Amplification (AI-LAMP) for Rapid Detection of SARS-CoV-2. Viruses. 2020; 12(9):972. https://doi.org/10.3390/v12090972
Chicago/Turabian StyleRohaim, Mohammed A., Emily Clayton, Irem Sahin, Julianne Vilela, Manar E. Khalifa, Mohammad Q. Al-Natour, Mahmoud Bayoumi, Aurore C. Poirier, Manoharanehru Branavan, Mukunthan Tharmakulasingam, and et al. 2020. "Artificial Intelligence-Assisted Loop Mediated Isothermal Amplification (AI-LAMP) for Rapid Detection of SARS-CoV-2" Viruses 12, no. 9: 972. https://doi.org/10.3390/v12090972
APA StyleRohaim, M. A., Clayton, E., Sahin, I., Vilela, J., Khalifa, M. E., Al-Natour, M. Q., Bayoumi, M., Poirier, A. C., Branavan, M., Tharmakulasingam, M., Chaudhry, N. S., Sodi, R., Brown, A., Burkhart, P., Hacking, W., Botham, J., Boyce, J., Wilkinson, H., Williams, C., ... Munir, M. (2020). Artificial Intelligence-Assisted Loop Mediated Isothermal Amplification (AI-LAMP) for Rapid Detection of SARS-CoV-2. Viruses, 12(9), 972. https://doi.org/10.3390/v12090972