The Sialoside-Binding Pocket of SARS-CoV-2 Spike Glycoprotein Structurally Resembles MERS-CoV

 ,
,  , , ,
, , ,  ,
, 

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Phylogenetic Tree and Sequence Alignment
2.2. Structure Prediction
2.3. Molecular Docking and Simulations
3. Results and Discussion
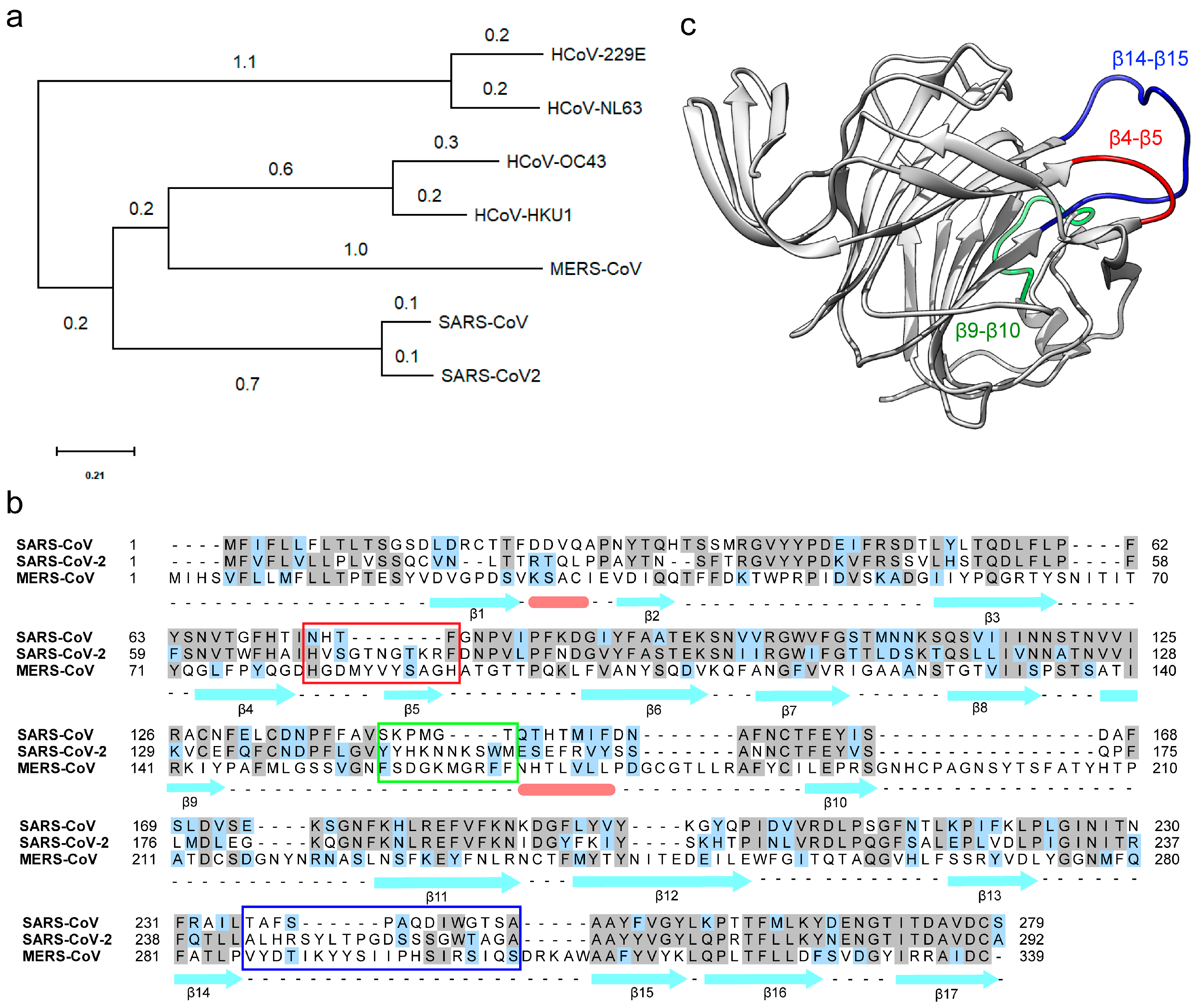
3.1. The N-Terminal Domain (NTD) of the SARS-CoV-2 Spike Protein Contains Divergent Loop Regions That Are Structurally Analogous to MERS-CoV
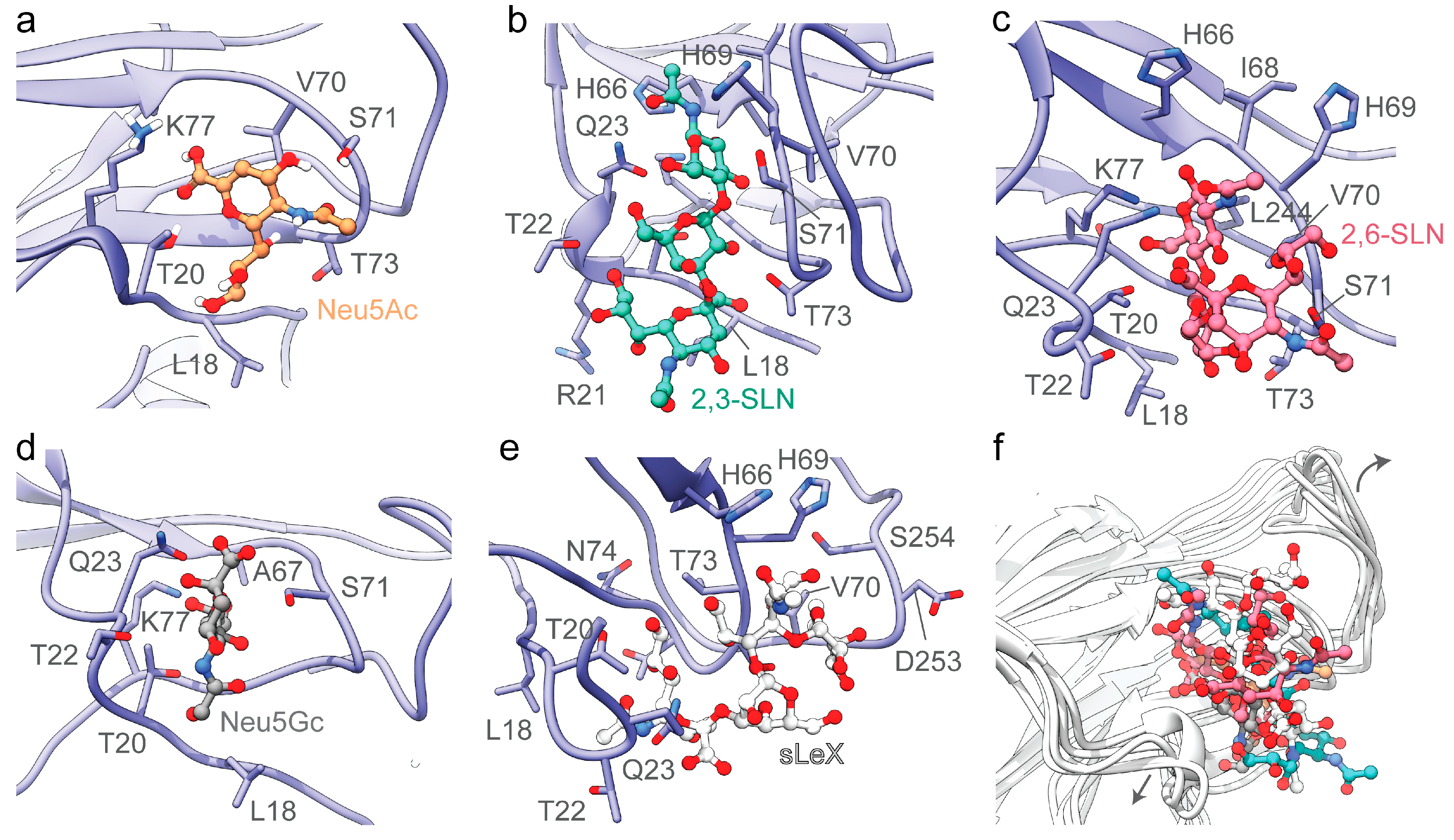
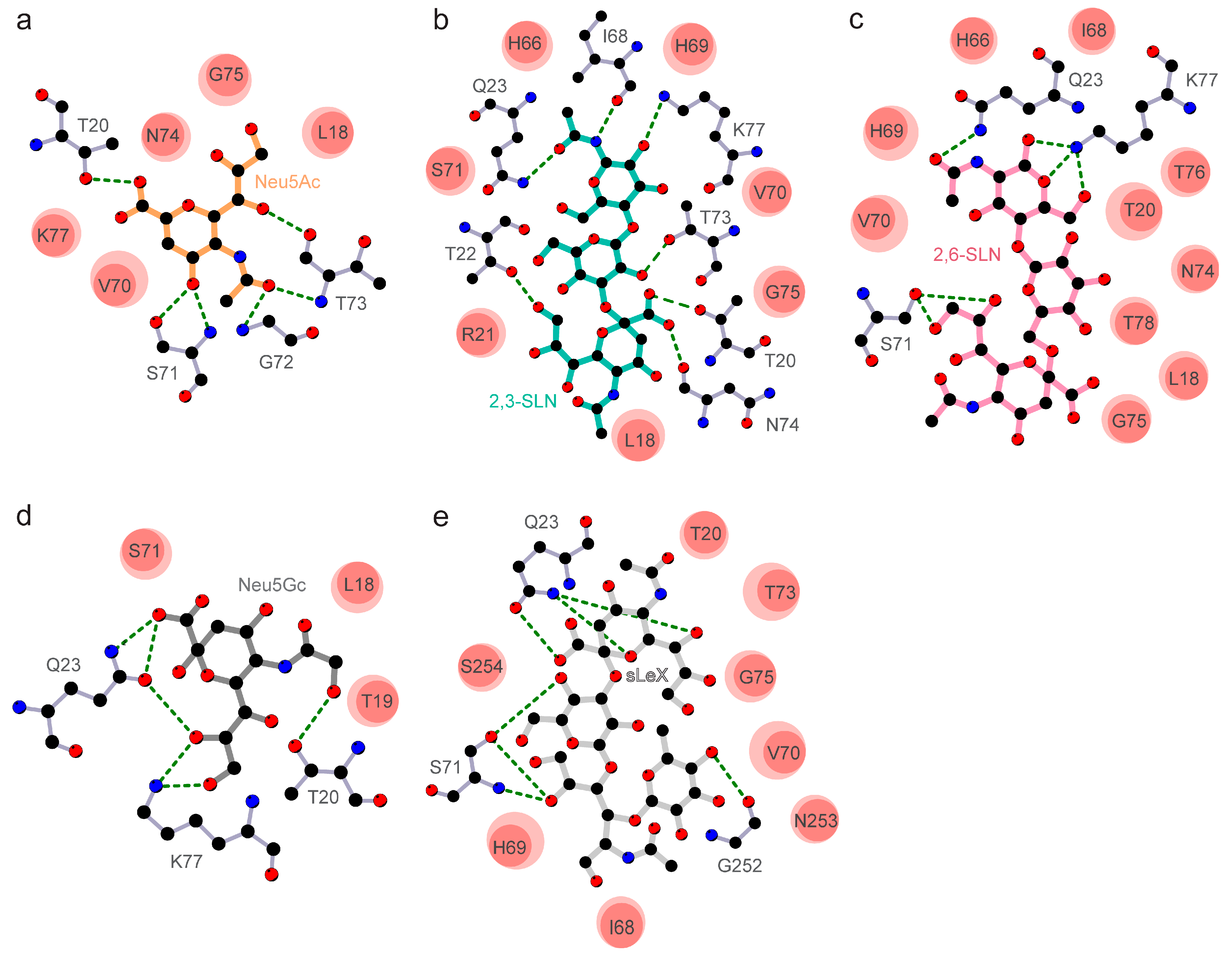
3.2. The Divergent Loop Regions within SARS-CoV-2 NTD Forms a Sialoside-Binding Pocket
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NTD | N-terminal domain |
| SARS | Severe acquired respiratory syndrome |
| MERS | Middle East respiratory syndrome |
| Neu5Ac | 5-N-acetyl neuraminic acid |
| Neu5Gc | 5-N-glycolyl neuraminic acid |
References
- Jiang, X.; Rayner, S.; Luo, M.H. Does SARS-CoV-2 Has a longer incubation period than SARS and MERS. J. Med. Virol. 2020, 92, 476–478. [Google Scholar] [CrossRef]
- Zumla, A.; Chan, J.F.; Azhar, E.I.; Hui, D.S.; Yuen, K.Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug. Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Qin, M.; Shen, B.; Cai, Y.; Liu, T.; Yang, F.; Gong, W.; Liu, X.; Liang, J.; Zhao, Q.; et al. Association of Cardiac Injury with Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol. 2020, 25, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yang, M.; Wan, C.; Yi, L.X.; Tang, F.; Zhu, H.Y.; Yi, F.; Yang, H.C.; Fogo, A.B.; Nie, X.; et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020, 98, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Othman, H.; Bouslama, Z.; Brandenburg, J.T.; da Rocha, J.; Hamdi, Y.; Ghedira, K.; Srairi-Abid, N.; Hazelhurst, S. Interaction of the spike protein RBD from SARS-CoV-2 with ACE2: Similarity with SARS-CoV, hot-spot analysis and effect of the receptor polymorphism. Biochem. Biophys. Res. Commun. 2020, 3, 702–708. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and antigenicity of the SARS-CoV-2 spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Hikmet, F.; Méar, L.; Uhlén, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. bioRxiv 2020. [Google Scholar] [CrossRef]
- Fung, T.S.; Liu, D.X. Human coronavirus: Host-pathogen interaction. Annu. Rev. Microbiol. 2019, 73, 529–557. [Google Scholar] [CrossRef]
- Park, Y.J.; Walls, A.C.; Wang, Z.; Sauer, M.M.; Li, W.; Tortorici, M.A.; Bosch, B.J.; DiMaio, F.; Veesler, D. Structures of MERS-CoV spike glycoprotein in complex with sialoside attachment receptors. Nat. Struct. Mol. Biol. 2019, 26, 1151–1157. [Google Scholar] [CrossRef]
- Behloul, N.; Baha, S.; Shi, R.; Meng, J. Role of the GTNGTKR motif in the N-terminal receptor-binding domain of the SARS-CoV-2 spike protein. Virus Res. 2020, 286, 198058. [Google Scholar] [CrossRef] [PubMed]
- Robson, B. Bioinformatics studies on a function of the SARS-CoV-2 spike glycoprotein as the binding of host sialic acid glycans. Comput. Biol. Med. 2020, 122, 103849. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.Y.; Pieper, U.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinform. 2016, 20. [Google Scholar] [CrossRef]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef]
- Gui, M.; Song, W.; Zhou, H.; Xu, J.; Chen, S.; Xiang, Y.; Wang, X. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 2017, 27, 119–129. [Google Scholar] [CrossRef]
- Pallesen, J.; Wang, N.; Corbett, K.S.; Wrapp, D.; Kirchdoerfer, R.N.; Turner, H.L.; Cottrell, C.A.; Becker, M.M.; Wang, L.; Shi, W.; et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. USA 2017, 114, E7348–E7357. [Google Scholar] [CrossRef]
- Shen, M.Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef]
- Ponder, J.W.; Case, D.A. Force fields for protein simulations. Adv. Protein Chem. 2003, 66, 27–85. [Google Scholar]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B., III; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Ravindranath, P.A.; Sanner, M.F. AutoSite: An automated approach for pseudo-ligands prediction-from ligand-binding sites identification to predicting key ligand atoms. Bioinformatics 2016, 32, 3142–3149. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.J.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Roussel, Y.; Giraud-Gatineau, A.; Jimeno, M.T.; Rolain, J.M.; Zandotti, C.; Colson, P.; Raoult, D. SARS-CoV-2: Fear versus data. Int. J. Antimicrob. Agents 2020, 55, 105947. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef]
- Qiang, X.L.; Xu, P.; Fang, G.; Liu, W.B.; Kou, Z. Using the spike protein feature to predict infection risk and monitor the evolutionary dynamic of coronavirus. Infect. Dis. Poverty 2020, 9, 33. [Google Scholar] [CrossRef]
- Zheng, J. SARS-CoV-2: An emerging coronavirus that causes a global threat. Int. J. Biol. Sci. 2020, 16, 1678–1685. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Li, F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef]
- Milanetti, E.; Miotto, M.; di Rienzo, L.; Monti, M.; Gosti, G.; Ruocco, G. In-silico evidence for two receptors based strategy of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Li, W.; Hulswit, R.J.G.; Widjaja, I.; Raj, V.S.; McBride, R.; Peng, W.; Widagdo, W.; Tortorici, M.A.; van Dieren, B.; Lang, Y.; et al. Identification of sialic acid-binding function for the Middle East respiratory syndrome coronavirus spike glycoprotein. Proc. Natl. Acad. Sci. USA 2017, 114, E8508–E8517. [Google Scholar] [CrossRef] [PubMed]
- Tortorici, M.A.; Walls, A.C.; Lang, Y.; Wang, C.; Li, Z.; Koerhuis, D.; Boons, G.J.; Bosch, B.J.; Rey, F.A.; de Groot, R.J.; et al. Structural basis for human coronavirus attachment to sialic acid receptors. Nat. Struct. Mol. Biol. 2019, 26, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Receptor recognition mechanisms of coronaviruses: A decade of structural studies. J. Virol. 2015, 89, 1954–1964. [Google Scholar] [CrossRef] [PubMed]
- Schwegmann-Wessels, C.; Herrler, G. Sialic acids as receptor determinants for coronaviruses. Glycoconj. J. 2006, 23, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Chiodo, F.; Bruijns, S.C.M.; Rodriguez, E.; Li, R.J.E.; Molinaro, A.; Silipo, A.; di Lorenzo, F.; Garcia-Rivera, D.; Valdes-Balbin, Y.; Verez-Bencomo, V.; et al. Novel ACE2-independent carbohydrate-binding of SARS-CoV-2 spike protein to host lectins and lung microbiota. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhao, J.; Yang, Y.; Huang, H.; Li, D.; Gu, D.; Lu, X.; Zhang, Z.; Liu, L.; Liu, T.; Liu, Y.; et al. Relationship between the ABO blood group and the COVID-19 susceptibility. medRxiv 2020. [Google Scholar] [CrossRef]
- Breiman, A.; Ruven-Clouet, N.; le Pendu, J. Harnessing the natural anti-glycan immune response to limit the transmission of enveloped viruses such as SARS-CoV-2. PLoS Pathog. 2020, 16, e1008556. [Google Scholar] [CrossRef]
- Cohen, M.; Hurtado-Ziola, N.; Varki, A. ABO blood group glycans modulate sialic acid recognition on erythrocytes. Blood 2009, 114, 3668–3676. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awasthi, M.; Gulati, S.; Sarkar, D.P.; Tiwari, S.; Kateriya, S.; Ranjan, P.; Verma, S.K. The Sialoside-Binding Pocket of SARS-CoV-2 Spike Glycoprotein Structurally Resembles MERS-CoV. Viruses 2020, 12, 909. https://doi.org/10.3390/v12090909
Awasthi M, Gulati S, Sarkar DP, Tiwari S, Kateriya S, Ranjan P, Verma SK. The Sialoside-Binding Pocket of SARS-CoV-2 Spike Glycoprotein Structurally Resembles MERS-CoV. Viruses. 2020; 12(9):909. https://doi.org/10.3390/v12090909
Chicago/Turabian StyleAwasthi, Mayanka, Sahil Gulati, Debi P. Sarkar, Swasti Tiwari, Suneel Kateriya, Peeyush Ranjan, and Santosh Kumar Verma. 2020. "The Sialoside-Binding Pocket of SARS-CoV-2 Spike Glycoprotein Structurally Resembles MERS-CoV" Viruses 12, no. 9: 909. https://doi.org/10.3390/v12090909
APA StyleAwasthi, M., Gulati, S., Sarkar, D. P., Tiwari, S., Kateriya, S., Ranjan, P., & Verma, S. K. (2020). The Sialoside-Binding Pocket of SARS-CoV-2 Spike Glycoprotein Structurally Resembles MERS-CoV. Viruses, 12(9), 909. https://doi.org/10.3390/v12090909