Efficient Non-Epigenetic Activation of HIV Latency through the T-Cell Receptor Signalosome




{kind=link}
{kind=link}
Abstract
1. Introduction
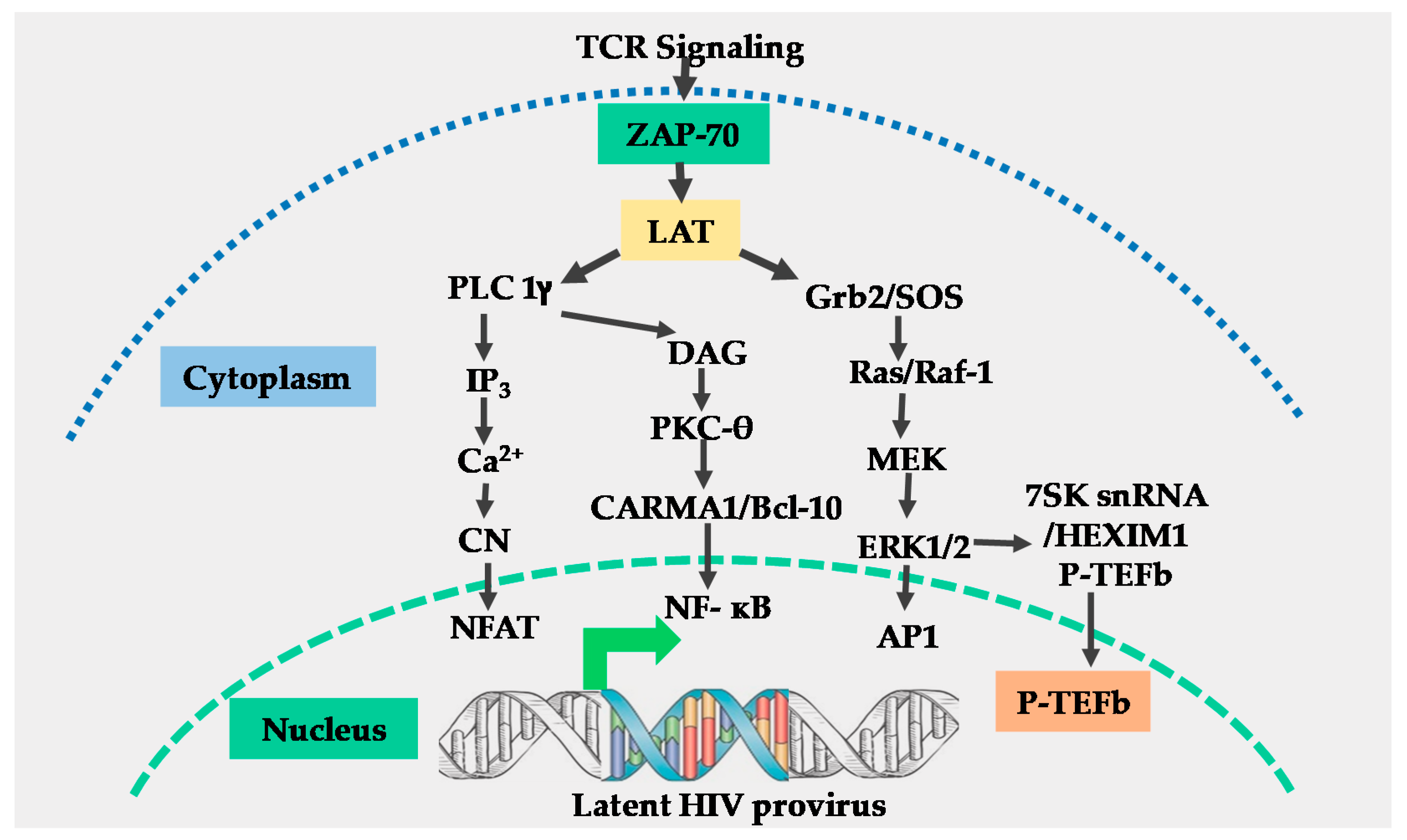
2. The T-Cell Receptor Signalosome
3. The NF-κB Transcription Factors
4. The NFAT Transcription Factors
5. The AP-1 Transcription Factors
6. Influence of TCR Activation on HIV Infection
7. Molecular Control of HIV Gene Expression
7.1. Role of NF-κB in HIV-1 Transcription
7.2. Role of NFAT in HIV-1 Transcription
7.3. Regulation of HIV-1 Transcription by MAPK-Activated AP-1
8. Role of HIV LTR Sequence Diversity on HIV Transcriptional Response
9. Conclusions and Prospects for Novel Drug Discovery
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Quinn, T.C. HIV epidemiology and the effects of antiviral therapy on long-term consequences. AIDS 2008, 22 (Suppl. 3), S7–S12. [Google Scholar] [CrossRef]
- Pau, A.K.; George, J.M. Antiretroviral therapy: Current drugs. Infect. Dis. Clin. N. Am. 2014, 28, 371–402. [Google Scholar] [CrossRef]
- Persaud, D.; Siberry, G.K.; Ahonkhai, A.; Kajdas, J.; Monie, D.; Hutton, N.; Watson, D.C.; Quinn, T.C.; Ray, S.C.; Siliciano, R.F. Continued production of drug-sensitive human immunodeficiency virus type 1 in children on combination antiretroviral therapy who have undetectable viral loads. J. Virol. 2004, 78, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Geeraert, L.; Kraus, G.; Pomerantz, R.J. Hide-and-seek: The challenge of viral persistence in HIV-1 infection. Annu. Rev. Med. 2008, 59, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Dornadula, G.; Zhang, H.; VanUitert, B.; Stern, J.; Livornese, L., Jr.; Ingerman, M.J.; Witek, J.; Kedanis, R.J.; Natkin, J.; DeSimone, J.; et al. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA 1999, 282, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Sahu, G.K. Potential implication of residual viremia in patients on effective antiretroviral therapy. AIDS Res. Hum. Retrovir. 2015, 31, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Hokello, J.; Sharma, A.L.; Dimri, M.; Tyagi, M. Insights into the HIV Latency and the Role of Cytokines. Pathogens 2019, 8, 137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ramratnam, B.; Tenner-Racz, K.; He, Y.; Vesanen, M.; Lewin, S.; Talal, A.; Racz, P.; Perelson, A.S.; Korber, B.T.; et al. Quantifying residual HIV-1 replication in patients receiving combination antiretroviral therapy. N. Engl. J. Med. 1999, 340, 1605–1613. [Google Scholar] [CrossRef]
- Vanhamel, J.; Bruggemans, A.; Debyser, Z. Establishment of latent HIV-1 reservoirs: What do we really know? J. Virus Erad. 2019, 5, 3–9. [Google Scholar] [CrossRef]
- Tyagi, M.; Pearson, R.J.; Karn, J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J. Virol. 2010, 84, 6425–6437. [Google Scholar] [CrossRef]
- Tyagi, M.; Bukrinsky, M. Human immunodeficiency virus (HIV) latency: The major hurdle in HIV eradication. Mol. Med. 2012, 18, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Ruelas, D.S.; Greene, W.C. An integrated overview of HIV-1 latency. Cell 2013, 155, 519–529. [Google Scholar] [CrossRef]
- Tyagi, M.; Iordanskiy, S.; Ammosova, T.; Kumari, N.; Smith, K.; Breuer, D.; Ilatovskiy, A.V.; Kont, Y.S.; Ivanov, A.; Uren, A.; et al. Reactivation of latent HIV-1 provirus via targeting protein phosphatase-1. Retrovirology 2015, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- Kabouridis, P.S. Lipid rafts in T cell receptor signalling. Mol. Membr. Biol. 2006, 23, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Tseng, S.Y.; Dustin, M.L. T-cell activation: A multidimensional signaling network. Curr. Opin. Cell Biol. 2002, 14, 575–580. [Google Scholar] [CrossRef]
- Harder, T. Lipid raft domains and protein networks in T-cell receptor signal transduction. Curr. Opin. Immunol. 2004, 16, 353–359. [Google Scholar] [CrossRef]
- Zhang, J.T.; Han, E.; Liu, Y. Role of the ribosome in sequence-specific regulation of membrane targeting and translocation of P-glycoprotein signal-anchor transmembrane segments. J. Cell Sci. 2000, 113 Pt 14, 2545–2555. [Google Scholar]
- Zhang, W.; Samelson, L.E. The role of membrane-associated adaptors in T cell receptor signalling. Semin. Immunol. 2000, 12, 35–41. [Google Scholar] [CrossRef]
- Kim, Y.K.; Mbonye, U.; Hokello, J.; Karn, J. T-cell receptor signaling enhances transcriptional elongation from latent HIV proviruses by activating P-TEFb through an ERK-dependent pathway. J. Mol. Biol. 2011, 410, 896–916. [Google Scholar] [CrossRef]
- Kane, L.P.; Shapiro, V.S.; Stokoe, D.; Weiss, A. Induction of NF-kappaB by the Akt/PKB kinase. Curr. Biol. CB 1999, 9, 601–604. [Google Scholar] [CrossRef]
- Narayan, P.; Holt, B.; Tosti, R.; Kane, L.P. CARMA1 is required for Akt-mediated NF-kappaB activation in T cells. Mol. Cell. Biol. 2006, 26, 2327–2336. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Abe, R. Signal Transduction Via Co-stimulatory and Co-inhibitory Receptors. Adv. Exp. Med. Biol. 2019, 1189, 85–133. [Google Scholar] [CrossRef] [PubMed]
- Harhaj, E.W.; Maggirwar, S.B.; Good, L.; Sun, S.C. CD28 mediates a potent costimulatory signal for rapid degradation of IkappaBbeta which is associated with accelerated activation of various NF-kappaB/Rel heterodimers. Mol. Cell. Biol. 1996, 16, 6736–6743. [Google Scholar] [CrossRef]
- Barat, C.; Tremblay, M.J. Engagement of CD43 enhances human immunodeficiency virus type 1 transcriptional activity and virus production that is induced upon TCR/CD3 stimulation. J. Biol. Chem. 2002, 277, 28714–28724. [Google Scholar] [CrossRef]
- Tardif, M.R.; Tremblay, M.J. Tetraspanin CD81 provides a costimulatory signal resulting in increased human immunodeficiency virus type 1 gene expression in primary CD4+ T lymphocytes through NF-kappaB, NFAT, and AP-1 transduction pathways. J. Virol. 2005, 79, 4316–4328. [Google Scholar] [CrossRef]
- Saito, T. Molecular Dynamics of Co-signal Molecules in T-Cell Activation. Adv. Exp. Med. Biol. 2019, 1189, 135–152. [Google Scholar] [CrossRef]
- Ormonde, J.V.S.; Li, Z.; Stegen, C.; Madrenas, J. TAOK3 Regulates Canonical TCR Signaling by Preventing Early SHP-1-Mediated Inactivation of LCK. J. Immunol. 2018, 201, 3431–3442. [Google Scholar] [CrossRef]
- Fenard, D.; Yonemoto, W.; de Noronha, C.; Cavrois, M.; Williams, S.A.; Greene, W.C. Nef is physically recruited into the immunological synapse and potentiates T cell activation early after TCR engagement. J. Immunol. 2005, 175, 6050–6057. [Google Scholar] [CrossRef]
- Neri, F.; Giolo, G.; Potesta, M.; Petrini, S.; Doria, M. The HIV-1 Nef protein has a dual role in T cell receptor signaling in infected CD4+ T lymphocytes. Virology 2011, 410, 316–326. [Google Scholar] [CrossRef]
- Fortin, J.F.; Barat, C.; Beausejour, Y.; Barbeau, B.; Tremblay, M.J. Hyper-responsiveness to stimulation of human immunodeficiency virus-infected CD4+ T cells requires Nef and Tat virus gene products and results from higher NFAT, NF-kappaB, and AP-1 induction. J. Biol. Chem. 2004, 279, 39520–39531. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. How NF-kappaB is activated: The role of the IkappaB kinase (IKK) complex. Oncogene 1999, 18, 6867–6874. [Google Scholar] [CrossRef] [PubMed]
- Serfling, E.; Berberich-Siebelt, F.; Avots, A.; Chuvpilo, S.; Klein-Hessling, S.; Jha, M.K.; Kondo, E.; Pagel, P.; Schulze-Luehrmann, J.; Palmetshofer, A. NFAT and NF-kappaB factors-the distant relatives. Int. J. Biochem. Cell Biol. 2004, 36, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef]
- Giffin, M.J.; Stroud, J.C.; Bates, D.L.; von Koenig, K.D.; Hardin, J.; Chen, L. Structure of NFAT1 bound as a dimer to the HIV-1 LTR kappa B element. Nat. Struct. Biol. 2003, 10, 800–806. [Google Scholar] [CrossRef]
- Amasaki, Y.; Masuda, E.S.; Imamura, R.; Arai, K.; Arai, N. Distinct NFAT family proteins are involved in the nuclear NFAT-DNA binding complexes from human thymocyte subsets. J. Immunol. 1998, 160, 2324–2333. [Google Scholar]
- McCaffrey, P.G.; Jain, J.; Jamieson, C.; Sen, R.; Rao, A. A T cell nuclear factor resembling NF-AT binds to an NF-kappa B site and to the conserved lymphokine promoter sequence “cytokine-1”. J. Biol. Chem. 1992, 267, 1864–1871. [Google Scholar]
- Hoey, T.; Sun, Y.L.; Williamson, K.; Xu, X. Isolation of two new members of the NF-AT gene family and functional characterization of the NF-AT proteins. Immunity 1995, 2, 461–472. [Google Scholar] [CrossRef]
- Lopez-Rodriguez, C.; Aramburu, J.; Rakeman, A.S.; Rao, A. NFAT5, a constitutively nuclear NFAT protein that does not cooperate with Fos and Jun. Proc. Natl. Acad. Sci. USA 1999, 96, 7214–7219. [Google Scholar] [CrossRef]
- Lopez-Rodriguez, C.; Aramburu, J.; Rakeman, A.S.; Copeland, N.G.; Gilbert, D.J.; Thomas, S.; Disteche, C.; Jenkins, N.A.; Rao, A. NF-AT5: The NF-AT family of transcription factors expands in a new direction. Cold Spring Harb. Symp. Quant. Biol. 1999, 64, 517–526. [Google Scholar] [CrossRef]
- Chuvpilo, S.; Avots, A.; Berberich-Siebelt, F.; Glockner, J.; Fischer, C.; Kerstan, A.; Escher, C.; Inashkina, I.; Hlubek, F.; Jankevics, E.; et al. Multiple NF-ATc isoforms with individual transcriptional properties are synthesized in T lymphocytes. J. Immunol. 1999, 162, 7294–7301. [Google Scholar] [PubMed]
- Quintana, A.; Griesemer, D.; Schwarz, E.C.; Hoth, M. Calcium-dependent activation of T-lymphocytes. Pflug. Arch. Eur. J. Physiol. 2005, 450, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Masuda, E.S.; Imamura, R.; Amasaki, Y.; Arai, K.; Arai, N. Signalling into the T-cell nucleus: NFAT regulation. Cell. Signal. 1998, 10, 599–611. [Google Scholar] [CrossRef]
- Roebuck, K.A.; Gu, D.S.; Kagnoff, M.F. Activating protein-1 cooperates with phorbol ester activation signals to increase HIV-1 expression. AIDS 1996, 10, 819–826. [Google Scholar] [CrossRef]
- Yang, X.; Chen, Y.; Gabuzda, D. ERK MAP kinase links cytokine signals to activation of latent HIV-1 infection by stimulating a cooperative interaction of AP-1 and NF-kappaB. J. Biol. Chem. 1999, 274, 27981–27988. [Google Scholar] [CrossRef]
- Rubinfeld, H.; Seger, R. The ERK cascade: A prototype of MAPK signaling. Mol. Biotechnol. 2005, 31, 151–174. [Google Scholar] [CrossRef]
- Strniskova, M.; Barancik, M.; Ravingerova, T. Mitogen-activated protein kinases and their role in regulation of cellular processes. Gen. Physiol. Biophys. 2002, 21, 231–255. [Google Scholar]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1995, 9, 726–735. [Google Scholar] [CrossRef]
- Herrera, R.; Agarwal, S.; Walton, K.; Satterberg, B.; Distel, R.J.; Goodman, R.; Spiegelman, B.M.; Roberts, T.M. A direct role for c-fos in AP-1-dependent gene transcription. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1990, 1, 483–490. [Google Scholar]
- Tong-Starkesen, S.E.; Luciw, P.A.; Peterlin, B.M. Signaling through T lymphocyte surface proteins, TCR/CD3 and CD28, activates the HIV-1 long terminal repeat. J. Immunol. 1989, 142, 702–707. [Google Scholar]
- Cleret-Buhot, A.; Zhang, Y.; Planas, D.; Goulet, J.P.; Monteiro, P.; Gosselin, A.; Wacleche, V.S.; Tremblay, C.L.; Jenabian, M.A.; Routy, J.P.; et al. Identification of novel HIV-1 dependency factors in primary CCR4(+)CCR6(+)Th17 cells via a genome-wide transcriptional approach. Retrovirology 2015, 12, 102. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, S.D.; Lu, H.K.; Chang, J.J.; Rhodes, A.; Lewin, S.R.; Cameron, P.U. The Pathway to Establishing HIV Latency Is Critical to How Latency Is Maintained and Reversed. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Young, J.A. HIV-1: Fifteen proteins and an RNA. Annu. Rev. Biochem. 1998, 67, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Kingsman, S.M.; Kingsman, A.J. The regulation of human immunodeficiency virus type-1 gene expression. Eur. J. Biochem. 1996, 240, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O. HIV-1 replication. Somat. Cell Mol. Genet. 2001, 26, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Bourgeois, C.F.; Pearson, R.; Tyagi, M.; West, M.J.; Wong, J.; Wu, S.Y.; Chiang, C.M.; Karn, J. Recruitment of TFIIH to the HIV LTR is a rate-limiting step in the emergence of HIV from latency. EMBO J. 2006, 25, 3596–3604. [Google Scholar] [CrossRef]
- Kumar, K.P.; Akoulitchev, S.; Reinberg, D. Promoter-proximal stalling results from the inability to recruit transcription factor IIH to the transcription complex and is a regulated event. Proc. Natl. Acad. Sci. USA 1998, 95, 9767–9772. [Google Scholar] [CrossRef]
- Karn, J. Tackling Tat. J. Mol. Biol. 1999, 293, 235–254. [Google Scholar] [CrossRef]
- Kim, Y.K.; Bourgeois, C.F.; Isel, C.; Churcher, M.J.; Karn, J. Phosphorylation of the RNA polymerase II carboxyl-terminal domain by CDK9 is directly responsible for human immunodeficiency virus type 1 Tat-activated transcriptional elongation. Mol. Cell. Biol. 2002, 22, 4622–4637. [Google Scholar] [CrossRef]
- Marciniak, R.A.; Sharp, P.A. HIV-1 Tat protein promotes formation of more-processive elongation complexes. EMBO J. 1991, 10, 4189–4196. [Google Scholar] [CrossRef]
- Tyagi, M.; Rusnati, M.; Presta, M.; Giacca, M. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J. Biol. Chem. 2001, 276, 3254–3261. [Google Scholar] [CrossRef] [PubMed]
- West, M.J.; Lowe, A.D.; Karn, J. Activation of human immunodeficiency virus transcription in T cells revisited: NF-kappaB p65 stimulates transcriptional elongation. J. Virol. 2001, 75, 8524–8537. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.; Kim, Y.K.; Hokello, J.; Lassen, K.; Friedman, J.; Tyagi, M.; Karn, J. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J. Virol. 2008, 82, 12291–12303. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, S.; Chen, B.K.; Kaneshima, H.; Nolan, G.P. Host control of HIV-1 parasitism in T cells by the nuclear factor of activated T cells. Cell 1998, 95, 595–604. [Google Scholar] [CrossRef]
- Kinoshita, S.; Su, L.; Amano, M.; Timmerman, L.A.; Kaneshima, H.; Nolan, G.P. The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity 1997, 6, 235–244. [Google Scholar] [CrossRef]
- Cron, R.Q.; Bartz, S.R.; Clausell, A.; Bort, S.J.; Klebanoff, S.J.; Lewis, D.B. NFAT1 enhances HIV-1 gene expression in primary human CD4 T cells. Clin. Immunol. 2000, 94, 179–191. [Google Scholar] [CrossRef]
- Markovitz, D.M.; Hannibal, M.C.; Smith, M.J.; Cossman, R.; Nabel, G.J. Activation of the human immunodeficiency virus type 1 enhancer is not dependent on NFAT-1. J. Virol. 1992, 66, 3961–3965. [Google Scholar] [CrossRef]
- Bosque, A.; Planelles, V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 2009, 113, 58–65. [Google Scholar] [CrossRef]
- Roebuck, K.A.; Brenner, D.A.; Kagnoff, M.F. Identification of c-fos-responsive elements downstream of TAR in the long terminal repeat of human immunodeficiency virus type-1. J. Clin. Investig. 1993, 92, 1336–1348. [Google Scholar] [CrossRef][Green Version]
- Van Lint, C.; Amella, C.A.; Emiliani, S.; John, M.; Jie, T.; Verdin, E. Transcription factor binding sites downstream of the human immunodeficiency virus type 1 transcription start site are important for virus infectivity. J. Virol. 1997, 71, 6113–6127. [Google Scholar] [CrossRef]
- Franza, B.R., Jr.; Rauscher, F.J., 3rd; Josephs, S.F.; Curran, T. The Fos complex and Fos-related antigens recognize sequence elements that contain AP-1 binding sites. Science 1988, 239, 1150–1153. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Gabuzda, D. Regulation of human immunodeficiency virus type 1 infectivity by the ERK mitogen-activated protein kinase signaling pathway. J. Virol. 1999, 73, 3460–3466. [Google Scholar] [CrossRef] [PubMed]
- Stein, B.; Baldwin, A.S., Jr.; Ballard, D.W.; Greene, W.C.; Angel, P.; Herrlich, P. Cross-coupling of the NF-kappa B p65 and Fos/Jun transcription factors produces potentiated biological function. EMBO J. 1993, 12, 3879–3891. [Google Scholar] [CrossRef] [PubMed]
- Mbonye, U.R.; Gokulrangan, G.; Datt, M.; Dobrowolski, C.; Cooper, M.; Chance, M.R.; Karn, J. Phosphorylation of CDK9 at Ser175 enhances HIV transcription and is a marker of activated P-TEFb in CD4(+) T lymphocytes. PLoS Pathog. 2013, 9, e1003338. [Google Scholar] [CrossRef]
- Rodriguez, M.A.; Shen, C.; Ratner, D.; Paranjape, R.S.; Kulkarni, S.S.; Chatterjee, R.; Gupta, P. Genetic and functional characterization of the LTR of HIV-1 subtypes A and C circulating in India. AIDS Res. Hum. Retrovir. 2007, 23, 1428–1433. [Google Scholar] [CrossRef]
- Neogi, U.; Sharma, Y.; Sood, V.; Wanchu, A.; Banerjea, A.C. Diversity of HIV type 1 long terminal repeat (LTR) sequences following mother-to-child transmission in North India. AIDS Res. Hum. Retrovir. 2010, 26, 1299–1305. [Google Scholar] [CrossRef]
- De Arellano, E.R.; Soriano, V.; Holguin, A. Genetic analysis of regulatory, promoter, and TAR regions of LTR sequences belonging to HIV type 1 Non-B subtypes. AIDS Res. Hum. Retrovir. 2005, 21, 949–954. [Google Scholar] [CrossRef]
- De Arellano, E.R.; Alcami, J.; Lopez, M.; Soriano, V.; Holguin, A. Drastic decrease of transcription activity due to hypermutated long terminal repeat (LTR) region in different HIV-1 subtypes and recombinants. Antivir. Res. 2010, 88, 152–159. [Google Scholar] [CrossRef]
- Jeeninga, R.E.; Hoogenkamp, M.; Armand-Ugon, M.; de Baar, M.; Verhoef, K.; Berkhout, B. Functional differences between the long terminal repeat transcriptional promoters of human immunodeficiency virus type 1 subtypes A through G. J. Virol. 2000, 74, 3740–3751. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hokello, J.; Sharma, A.L.; Tyagi, M. Efficient Non-Epigenetic Activation of HIV Latency through the T-Cell Receptor Signalosome. Viruses 2020, 12, 868. https://doi.org/10.3390/v12080868
Hokello J, Sharma AL, Tyagi M. Efficient Non-Epigenetic Activation of HIV Latency through the T-Cell Receptor Signalosome. Viruses. 2020; 12(8):868. https://doi.org/10.3390/v12080868
Chicago/Turabian StyleHokello, Joseph, Adhikarimayum Lakhikumar Sharma, and Mudit Tyagi. 2020. "Efficient Non-Epigenetic Activation of HIV Latency through the T-Cell Receptor Signalosome" Viruses 12, no. 8: 868. https://doi.org/10.3390/v12080868
APA StyleHokello, J., Sharma, A. L., & Tyagi, M. (2020). Efficient Non-Epigenetic Activation of HIV Latency through the T-Cell Receptor Signalosome. Viruses, 12(8), 868. https://doi.org/10.3390/v12080868