Wide Diversity of Coronaviruses in Frugivorous and Insectivorous Bat Species: A Pilot Study in Guinea, West Africa

 , , , , , and
, , , , , and 
Abstract
1. Introduction
2. Materials and Methods
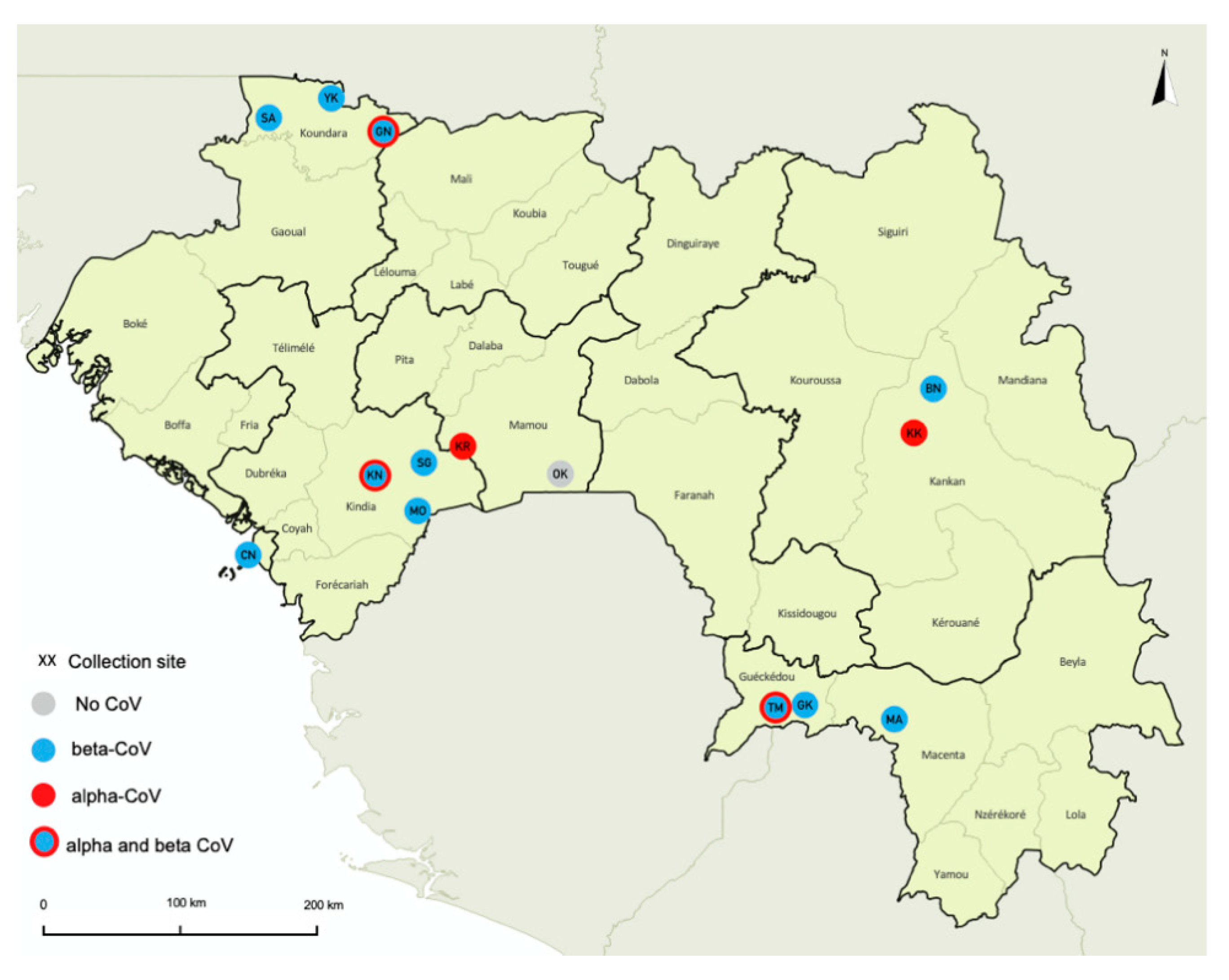
2.1. Study Sites, Sample Collection and Ethical Permits
2.2. Nucleic Acid Extraction
2.3. Molecular Confirmation of Bat Species
2.4. RT–PCR Screening for Detection of Coronavirus RNA
2.5. Phylogenetic Analyses
2.6. Statistical Analyse
3. Results
3.1. Bat Species and Sampling
3.2. PCR Screening and Presence of α and β Coronaviruses
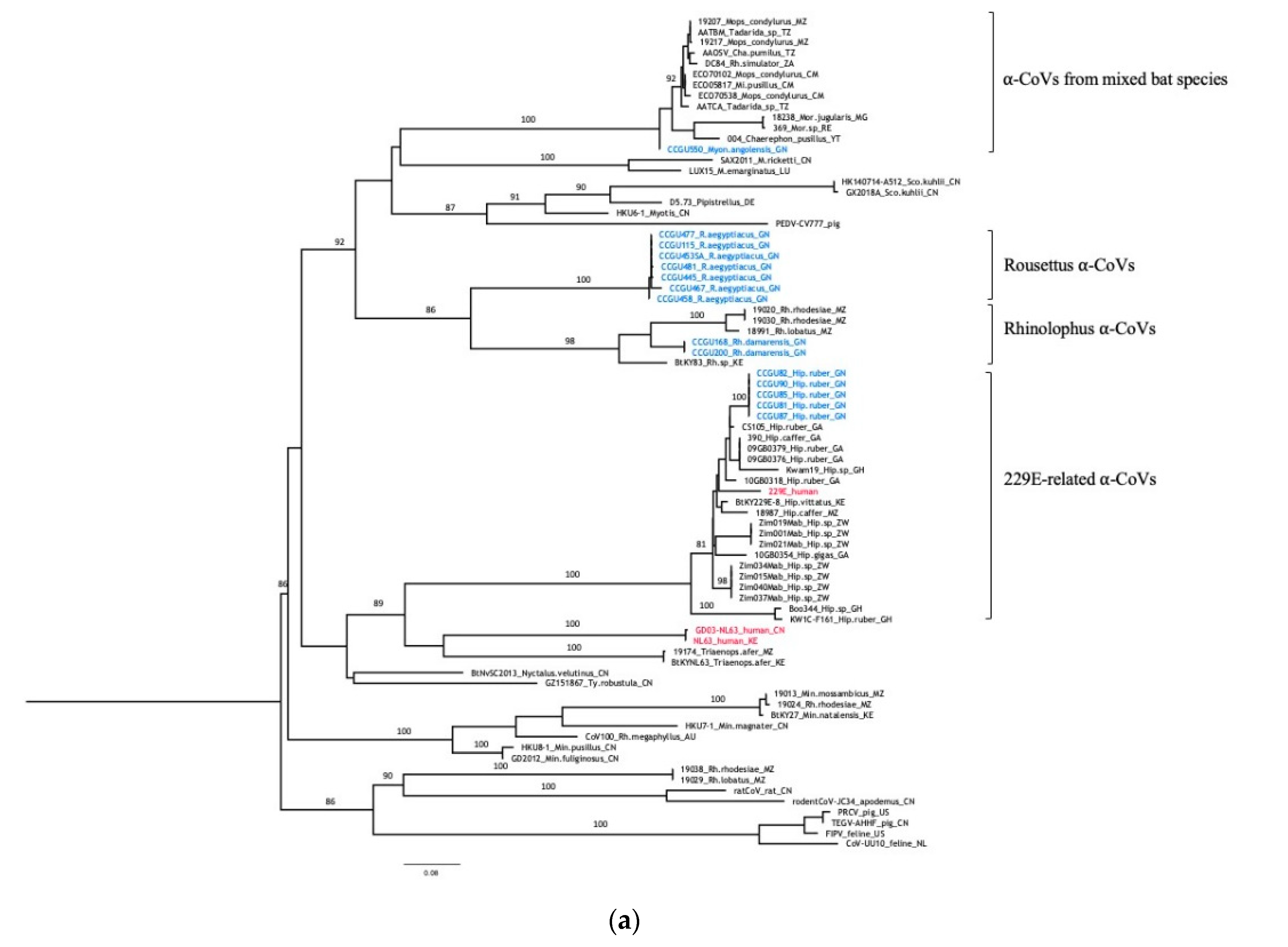
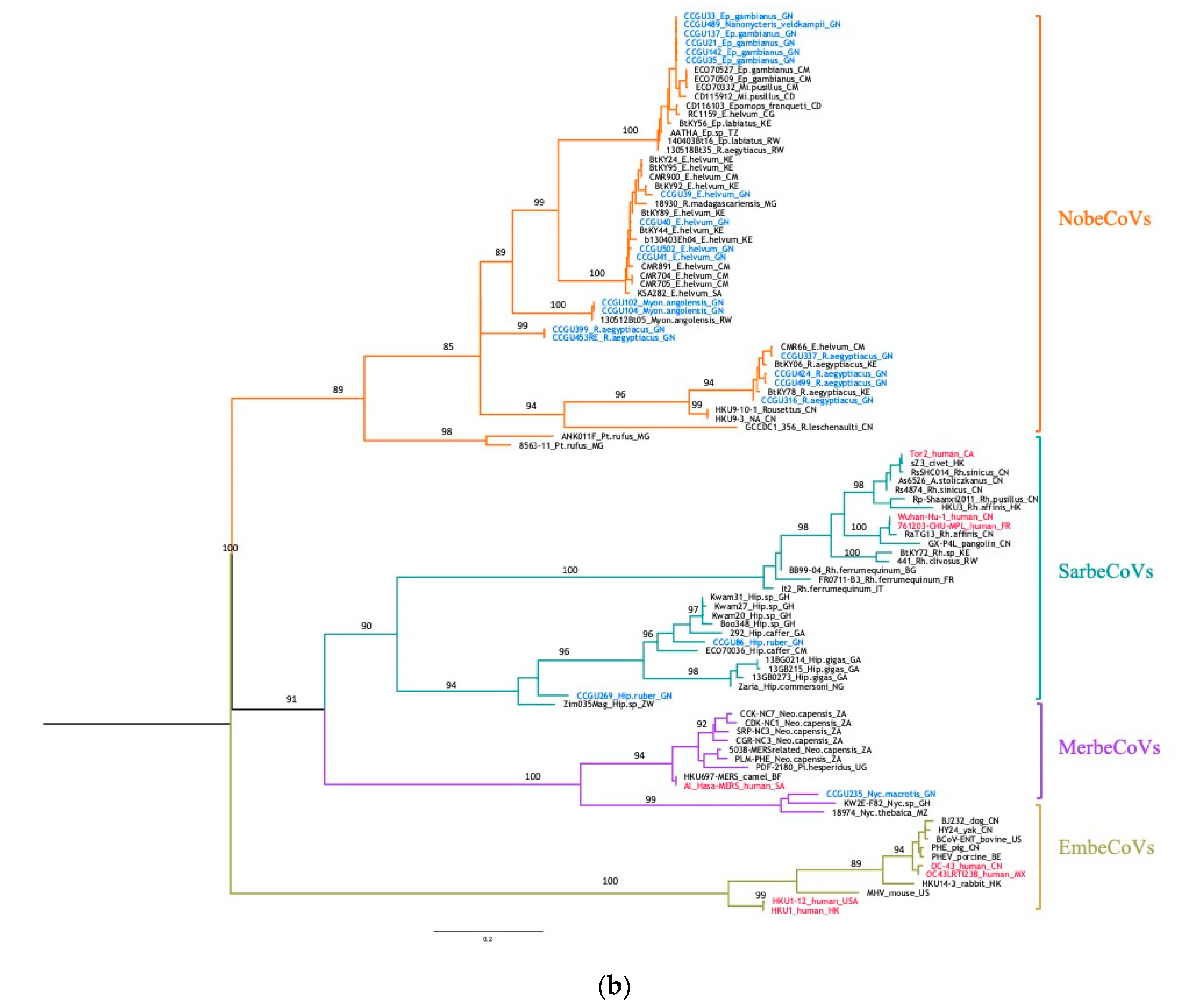
3.3. Genetic Diversity of Bat Coronaviruses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Wolfe, N.D.; Dunavan, C.P.; Diamond, J. Origins of major human infectious diseases. Nature 2007, 447, 279–283. [Google Scholar] [CrossRef]
- Cunningham, A.A.; Daszak, P.; Wood, J.L.N. One Health, emerging infectious diseases and wildlife: Two decades of progress? Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160167. [Google Scholar] [CrossRef]
- Altizer, S.; Ostfeld, R.S.; Johnson, P.T.J.; Kutz, S.; Harvell, C.D. Climate Change and Infectious Diseases: From Evidence to a Predictive Framework. Science 2013, 341, 514–519. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Coronavirus Disease (COVID-19) Situation Report 129; World Health Organization: Geneva, Switzerland, 2020; Available online: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200528-covid-19-sitrep-129.pdf?sfvrsn=5b154880_2 (accessed on 29 May 2020).
- Ye, Z.-W.; Yuan, S.; Yuen, K.-S.; Fung, S.-Y.; Chan, C.-P.; Jin, D.-Y. Zoonotic origins of human coronaviruses. Int. J. Biol. Sci. 2020, 16, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.M.; Yuen, K.Y.; Osterhaus, A.D.M.E.; Stöhr, K. The Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 349, 2431–2441. [Google Scholar] [CrossRef]
- Peiris, J.S.M.; Lai, S.T.; Poon, L.L.M.; Guan, Y.; Yam, L.Y.C.; Lim, W.; Nicholls, J.; Yee, W.K.S.; Yan, W.W.; Cheung, M.T.; et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003, 361, 1319–1325. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Hui, D.S.; Perlman, S. Middle East respiratory syndrome. Lancet 2015, 386, 995–1007. [Google Scholar] [CrossRef]
- World Health Organization OMS. Coronavirus du Syndrome Respiratoire du Moyen-Orient (MERS-CoV)—Qatar. Available online: http://www.who.int/csr/don/12-march-2020-mers-qatar/fr/ (accessed on 27 May 2020).
- He, X.; Lau, E.H.; Wu, P.; Deng, X.; Wang, J.; Hao, X.; Lau, Y.C.; Wong, J.Y.; Guan, Y.; Tan, X.; et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. medRxiv 2020, 26, 672–675. [Google Scholar] [CrossRef] [PubMed]
- Vijaykrishna, D.; Smith, G.J.D.; Zhang, J.X.; Peiris, J.S.M.; Chen, H.; Guan, Y. Evolutionary insights into the ecology of coronaviruses. J. Virol. 2007, 81, 4012–4020. [Google Scholar] [CrossRef] [PubMed]
- Pfefferle, S.; Oppong, S.; Drexler, J.F.; Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Müller, M.A.; Annan, A.; Vallo, P.; Adu-Sarkodie, Y.; et al. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in Bats, Ghana. Emerg. Infect. Dis 2009, 15, 1377–1384. [Google Scholar] [CrossRef]
- Huynh, J.; Li, S.; Yount, B.; Smith, A.; Sturges, L.; Olsen, J.C.; Nagel, J.; Johnson, J.B.; Agnihothram, S.; Gates, J.E.; et al. Evidence supporting a zoonotic origin of human coronavirus strain NL63. J. Virol. 2012, 86, 12816–12825. [Google Scholar] [CrossRef]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and sources of endemic human coronaviruses. Adv. Virus Res. 2018, 100, 163–188. [Google Scholar] [CrossRef]
- Guan, Y.; Zheng, B.J.; He, Y.Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Guan, Y.J.; et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Woo, P.C.Y.; Li, K.S.M.; Huang, Y.; Tsoi, H.-W.; Wong, B.H.L.; Wong, S.S.Y.; Leung, S.-Y.; Chan, K.-H.; Yuen, K.-Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. PNAS 2005, 102, 14040–14045. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-Like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.W.; Lau, S.K.P.; To, K.K.W.; Cheng, V.C.C.; Woo, P.C.Y.; Yuen, K.-Y. Middle East respiratory syndrome coronavirus: Another zoonotic betacoronavirus causing SARS-like disease. Clin. Microbiol. Rev. 2015, 28, 465–522. [Google Scholar] [CrossRef]
- Luo, C.-M.; Wang, N.; Yang, X.-L.; Liu, H.-Z.; Zhang, W.; Li, B.; Hu, B.; Peng, C.; Geng, Q.-B.; Zhu, G.-J.; et al. Discovery of novel bat coronaviruses in south china that use the same receptor as middle east respiratory syndrome coronavirus. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Corman, V.M.; Ithete, N.L.; Richards, L.R.; Schoeman, M.C.; Preiser, W.; Drosten, C.; Drexler, J.F. Rooting the phylogenetic tree of Middle East respiratory syndrome coronavirus by characterization of a conspecific virus from an African bat. J. Virol. 2014, 88, 11297–11303. [Google Scholar] [CrossRef]
- Lam, T.T.-Y.; Shum, M.H.-H.; Zhu, H.-C.; Tong, Y.-G.; Ni, X.-B.; Liao, Y.-S.; Wei, W.; Cheung, W.Y.-M.; Li, W.-J.; Li, L.-F.; et al. Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature 2020, 1–6. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020. [Google Scholar] [CrossRef]
- Banerjee, A.; Kulcsar, K.; Misra, V.; Frieman, M.; Mossman, K. Bats and coronaviruses. Viruses 2019, 11, 41. [Google Scholar] [CrossRef]
- Teeling, E.C.; Springer, M.S.; Madsen, O.; Bates, P.; O’brien, S.J.; Murphy, W.J. A molecular phylogeny for bats illuminates biogeography and the fossil record. Science 2005, 307, 580–584. [Google Scholar] [CrossRef]
- Wolfe, N.D.; Daszak, P.; Kilpatrick, A.M.; Burke, D.S. Bushmeat hunting, deforestation, and prediction of zoonotic disease. Emerg. Infect. Dis. 2005, 11, 1822. [Google Scholar] [CrossRef] [PubMed]
- Kamins, A.O.; Restif, O.; Ntiamoa-Baidu, Y.; Suu-Ire, R.; Hayman, D.T.S.; Cunningham, A.A.; Wood, J.L.N.; Rowcliffe, J.M. Uncovering the fruit bat bushmeat commodity chain and the true extent of fruit bat hunting in Ghana, West Africa. Biol. Conserv. 2011, 144, 3000–3008. [Google Scholar] [CrossRef]
- Lee, T.M.; Sigouin, A.; Pinedo-Vasquez, M.; Nasi, R. The Harvest of Wildlife for Bushmeat and Traditional Medicine in East, South and Southeast Asia: Current Knowledge Base, Challenges, Opportunities and Areas for Future Research. Available online: https://www.cifor.org/knowledge/publication/5135/ (accessed on 15 May 2020).
- Mickleburgh, S.; Waylen, K.; Racey, P. Bats as bushmeat: A global review. Oryx 2009, 43, 217–234. [Google Scholar] [CrossRef]
- Frick, W.F.; Kingston, T.; Flanders, J. A review of the major threats and challenges to global bat conservation. Ann. N. Y. Acad. Sci. 2019. [Google Scholar] [CrossRef]
- Mildenstein, T.; Tanshi, I.; Racey, P.A. Exploitation of bats for bushmeat and medicine. In Bats in the Anthropocene: Conservation of Bats in a Changing World; Voigt, C.C., Kingston, T., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 325–375. ISBN 978-3-319-25220-9. [Google Scholar]
- Tong, S.; Conrardy, C.; Ruone, S.; Kuzmin, I.V.; Guo, X.; Tao, Y.; Niezgoda, M.; Haynes, L.; Agwanda, B.; Breiman, R.F.; et al. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis. 2009, 15, 482–485. [Google Scholar] [CrossRef]
- Quan, P.-L.; Firth, C.; Street, C.; Henriquez, J.A.; Petrosov, A.; Tashmukhamedova, A.; Hutchison, S.K.; Egholm, M.; Osinubi, M.O.V.; Niezgoda, M.; et al. Identification of a severe acute respiratory syndrome coronavirus-like virus in a leaf-nosed bat in Nigeria. mBio 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Ithete, N.L.; Stoffberg, S.; Corman, V.M.; Cottontail, V.M.; Richards, L.R.; Schoeman, M.C.; Drosten, C.; Drexler, J.F.; Preiser, W. Close Relative of human middle east respiratory syndrome coronavirus in bat, South Africa. Emerg. Infect. Dis. 2013, 19, 1697–1699. [Google Scholar] [CrossRef] [PubMed]
- Annan, A.; Baldwin, H.J.; Corman, V.M.; Klose, S.M.; Owusu, M.; Nkrumah, E.E.; Badu, E.K.; Anti, P.; Agbenyega, O.; Meyer, B.; et al. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg. Infect. Dis. 2013, 19, 456–459. [Google Scholar] [CrossRef]
- Razanajatovo, N.H.; Nomenjanahary, L.A.; Wilkinson, D.A.; Razafimanahaka, J.H.; Goodman, S.M.; Jenkins, R.K.; Jones, J.P.; Heraud, J.-M. Detection of new genetic variants of betacoronaviruses in endemic frugivorous bats of Madagascar. Virol. J. 2015, 12, 42. [Google Scholar] [CrossRef]
- Bourgarel, M.; Pfukenyi, D.M.; Boué, V.; Talignani, L.; Chiweshe, N.; Diop, F.; Caron, A.; Matope, G.; Missé, D.; Liégeois, F. Circulation of alphacoronavirus, betacoronavirus and paramyxovirus in hipposideros bat species in Zimbabwe. Infect. Genet. Evol. 2018, 58, 253–257. [Google Scholar] [CrossRef]
- Joffrin, L.; Goodman, S.M.; Wilkinson, D.A.; Ramasindrazana, B.; Lagadec, E.; Gomard, Y.; Le Minter, G.; Dos Santos, A.; Corrie Schoeman, M.; Sookhareea, R.; et al. Bat coronavirus phylogeography in the Western Indian Ocean. Sci. Rep. 2020, 10, 6873. [Google Scholar] [CrossRef]
- Maganga, G.D.; Pinto, A.; Mombo, I.M.; Madjitobaye, M.; Mbeang Beyeme, A.M.; Boundenga, L.; Ar Gouilh, M.; N’Dilimabaka, N.; Drexler, J.F.; Drosten, C.; et al. Genetic diversity and ecology of coronaviruses hosted by cave-dwelling bats in Gabon. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Nziza, J.; Goldstein, T.; Cranfield, M.; Webala, P.; Nsengimana, O.; Nyatanyi, T.; Mudakikwa, A.; Tremeau-Bravard, A.; Byarugaba, D.; Tumushime, J.C.; et al. Coronaviruses detected in bats in close contact with humans in Rwanda. Ecohealth 2020, 17, 152–159. [Google Scholar] [CrossRef]
- Anthony, S.J.; Gilardi, K.; Menachery, V.D.; Goldstein, T.; Ssebide, B.; Mbabazi, R.; Navarrete-Macias, I.; Liang, E.; Wells, H.; Hicks, A.; et al. Further evidence for bats as the evolutionary source of Middle East respiratory syndrome coronavirus. MBio 2017, 8. [Google Scholar] [CrossRef]
- De Nys, H.M.; Kingebeni, P.M.; Keita, A.K.; Butel, C.; Thaurignac, G.; Villabona-Arenas, C.-J.; Lemarcis, T.; Geraerts, M.; Vidal, N.; Esteban, A.; et al. Survey of ebola viruses in frugivorous and insectivorous bats in Guinea, Cameroon, and the Democratic Republic of the Congo, 2015–2017. Emerg. Infect. Dis. 2018, 24. [Google Scholar] [CrossRef]
- Irwin, D.M.; Kocher, T.D.; Wilson, A.C. Evolution of the cytochromeb gene of mammals. J. Mol. Evol. 1991, 32, 128–144. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.K.W.; Leung, C.Y.H.; Gilbert, M.; Joyner, P.H.; Ng, E.M.; Tse, T.M.; Guan, Y.; Peiris, J.S.M.; Poon, L.L.M. Avian coronavirus in wild aquatic birds. J. Virol. 2011, 85, 12815–12820. [Google Scholar] [CrossRef] [PubMed]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Nesi, N.; Nakouné, E.; Cruaud, C.; Hassanin, A. DNA barcoding of African fruit bats (Mammalia, Pteropodidae). The mitochondrial genome does not provide a reliable discrimination between Epomophorus gambianus and Micropteropus pusillus. C. R. Biol. 2011, 334, 544–554. [Google Scholar] [CrossRef]
- Wong, A.C.P.; Li, X.; Lau, S.K.P.; Woo, P.C.Y. Global epidemiology of bat coronaviruses. Viruses 2019, 11, 174. [Google Scholar] [CrossRef]
- Tao, Y.; Shi, M.; Chommanard, C.; Queen, K.; Zhang, J.; Markotter, W.; Kuzmin, I.V.; Holmes, E.C.; Tong, S. Surveillance of bat coronaviruses in kenya identifies relatives of human coronaviruses NL63 and 229E and their recombination history. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Geldenhuys, M.; Weyer, J.; Nel, L.H.; Markotter, W. Coronaviruses in South African bats. Vector Borne Zoonotic Dis. 2013, 13, 516–519. [Google Scholar] [CrossRef]
- Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Göttsche, M.; Panning, M.; Drexler, J.F.; Petersen, N.; Annan, A.; Grywna, K.; Müller, M.; et al. Detection and prevalence patterns of group i coronaviruses in bats, Northern Germany. Emerg. Infect. Dis. 2008, 14, 626–631. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Wegner, T.; Tateno, A.F.; Zerbinati, R.M.; Gloza-Rausch, F.; Seebens, A.; Müller, M.A.; Drosten, C. Amplification of emerging viruses in a bat colony. Emerg. Infect. Dis. 2011, 17, 449–456. [Google Scholar] [CrossRef]
- Wacharapluesadee, S.; Duengkae, P.; Chaiyes, A.; Kaewpom, T.; Rodpan, A.; Yingsakmongkon, S.; Petcharat, S.; Phengsakul, P.; Maneeorn, P.; Hemachudha, T. Longitudinal study of age-specific pattern of coronavirus infection in Lyle’s flying fox (Pteropus lylei) in Thailand. Virol. J. 2018, 15, 38. [Google Scholar] [CrossRef]
- Anthony, S.J.; Johnson, C.K.; Greig, D.J.; Kramer, S.; Che, X.; Wells, H.; Hicks, A.L.; Joly, D.O.; Wolfe, N.D.; Daszak, P.; et al. Global patterns in coronavirus diversity. Virus Evol. 2017, 3, vex012. [Google Scholar] [CrossRef] [PubMed]
- Kock, D.; Arm, Z.; Mickleburgh, A.M.; Bergmans, W.; Aulagnier, S. IUCN Red List of Threatened Species: Sundevall’s Roundleaf Bat. Available online: https://www.iucnredlist.org/en (accessed on 26 May 2020).
- Hutson, A.; Mickleburgh, S.; Bergmans, W.; Fahr, J.; Swaziland, A.M. University of IUCN Red List of Threatened Species: Noack’s Roundleaf Bat. Available online: https://www.iucnredlist.org/en (accessed on 26 May 2020).
- Maganga, G.D.; Bourgarel, M.; Vallo, P.; Dallo, T.D.; Ngoagouni, C.; Drexler, J.F.; Drosten, C.; Nakouné, E.R.; Leroy, E.M.; Morand, S. Bat distribution size or shape as determinant of viral richness in African bats. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Ge, X.-Y.; Wang, N.; Zhang, W.; Hu, B.; Li, B.; Zhang, Y.-Z.; Zhou, J.-H.; Luo, C.-M.; Yang, X.-L.; Wu, L.-J.; et al. Coexistence of multiple coronaviruses in several bat colonies in an abandoned mineshaft. Virol. Sin. 2016, 31, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Yang, L.; Ren, X.; Zhang, J.; Yang, F.; Zhang, S.; Jin, Q. ORF8-related genetic evidence for Chinese horseshoe bats as the source of human severe acute respiratory syndrome coronavirus. J. Infect. Dis. 2016, 213, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Valitutto, M.T.; Aung, O.; Tun, K.Y.N.; Vodzak, M.E.; Zimmerman, D.; Yu, J.H.; Win, Y.T.; Maw, M.T.; Thein, W.Z.; Win, H.H.; et al. Detection of novel coronaviruses in bats in Myanmar. PLoS ONE 2020, 15, e0230802. [Google Scholar] [CrossRef]
- Chu, D.K.W.; Peiris, J.S.M.; Chen, H.; Guan, Y.; Poon, L.L.M. Genomic characterizations of bat coronaviruses (1A, 1B and HKU8) and evidence for co-infections in Miniopterus bats. J. Gen. Virol. 2008, 89, 1282–1287. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Li, K.S.M.; Huang, Y.; Shek, C.-T.; Tse, H.; Wang, M.; Choi, G.K.Y.; Xu, H.; Lam, C.S.F.; Guo, R.; et al. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J. Virol. 2010, 84, 2808–2819. [Google Scholar] [CrossRef]
- Johnson, C.K.; Hitchens, P.L.; Smiley Evans, T.; Goldstein, T.; Thomas, K.; Clements, A.; Joly, D.O.; Wolfe, N.D.; Daszak, P.; Karesh, W.B.; et al. Spillover and pandemic properties of zoonotic viruses with high host plasticity. Sci. Rep. 2015, 5, 14830. [Google Scholar] [CrossRef]
- Leopardi, S.; Holmes, E.C.; Gastaldelli, M.; Tassoni, L.; Priori, P.; Scaravelli, D.; Zamperin, G.; De Benedictis, P. Interplay between co-divergence and cross-species transmission in the evolutionary history of bat coronaviruses. Infect. Genet. Evol. 2018, 58, 279–289. [Google Scholar] [CrossRef]
- Fenton, M.B.; Simmons, N.B. Bats: A World of Science and Mystery; University of Chicago Press: Chicago, IL, USA, 2015; ISBN 978-0-226-06512-0. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Prefecture | Site | Number |
|---|---|---|
| Conakry | CN | 59 |
| Subtotal | 59 | |
| Gueckedou | GK | 6 |
| TM | 41 | |
| Subtotal | 47 | |
| Kankan | BN | 8 |
| KK | 12 | |
| Subtotal | 20 | |
| Kindia | KN | 13 |
| MO | 54 | |
| SG | 35 | |
| Subtotal | 102 | |
| Koundara | GN | 35 |
| SA | 15 | |
| YK | 6 | |
| Subtotal | 56 | |
| Macenta | MA | 9 |
| Subtotal | 9 | |
| Mamou | KR | 15 |
| OK | 11 | |
| Subtotal | 26 | |
| Total | 319 |
| Family Species | Conakry | Kankan | Kindia | Koundara | Gueckedou | Macenta | Mamou | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CN | BN | KK | KN | MO | SG | YK | SA | GN | GK | TM | MA | OK | KR | Total | % pos | |
| Insectivorous Bats | ||||||||||||||||
| Hipposideridae | 0/1 | 1/4 | 0/1 | 6/13 | 0/1 | - | 0/1 | - | - | - | - | - | - | - | 7/21 | 33.3% |
| Hipposideros sp. | 0/1 | 1/4 | 0/1 | 6/13 | 0/1 | - | 0/1 | - | - | - | - | - | - | - | 7/21 | 33.3% |
| Molossidae | - | - | - | - | 0/3 | - | - | - | 0/10 | - | - | - | - | - | 0/13 | 0.0% |
| Chaerephon sp. | - | - | - | - | 0/3 | - | - | - | - | - | - | - | - | - | 0/3 | 0.0% |
| Mops condylurus | - | - | - | - | - | - | - | - | 0/10 | - | - | - | - | - | 0/10 | 0.0% |
| Nycteridae | - | 1/2 | - | - | - | - | - | - | 0/1 | - | - | - | - | - | 1/3 | 33.3% |
| Nycteris sp. | - | 1/2 | - | - | - | - | - | - | 0/1 | - | - | - | - | - | 1/3 | 33.3% |
| Rhinolophidae | - | - | 2/5 | - | - | - | - | - | - | - | - | - | - | - | 2/5 | 40.0% |
| Rhinolophus sp. | - | - | 2/5 | - | - | - | - | - | - | - | - | - | - | - | 2/5 | 40.0% |
| Vespertilionidae | - | - | - | - | 0/2 | - | - | - | 0/1 | - | - | - | - | - | 0/3 | 0.0% |
| Scotophilus leucogaster | - | - | - | - | 0/2 | - | - | - | 0/1 | - | - | - | - | - | 0/3 | 0.0% |
| Fruit Bats | ||||||||||||||||
| Pteropodidae | 6/58 | 0/2 | 0/6 | - | 2/48 | 2/35 | 1/5 | 1/15 | 3/23 | 1/6 | 6/41 | 2/9 | 0/11 | 1/15 | 25/274 | 9.1% |
| Eidolon helvum | 3/6 | - | - | - | - | - | - | 0/1 | - | - | - | 1/2 | - | - | 4/9 | 44.4% |
| Epomophorus gambianus | 3/48 | 0/2 | 0/5 | - | 0/2 | - | 1/3 | 1/14 | 0/2 | - | - | - | 0/11 | - | 5/87 | 5.7% |
| Epomops buettikoferi | - | - | - | - | - | - | - | - | - | 0/3 | - | - | - | - | 0/3 | 0.0% |
| Hypsignathus monstrosus | - | - | - | - | - | - | - | - | - | 0/1 | - | 0/5 | - | - | 0/6 | 0.0% |
| Lissonycteris angolensis | - | - | - | - | 0/8 | 0/6 | - | - | 2/13 | - | - | - | - | 1/15 | 3/42 | 7.1% |
| Micropteropus pusillus | 0/4 | - | 0/1 | - | - | - | - | - | 0/1 | - | - | - | - | - | 0/6 | 0.0% |
| Nanonycteris veldkampii | - | - | - | - | - | - | - | - | - | 1/1 | - | - | - | - | 1/1 | 100% |
| Rousettus aegyptiacus | - | - | - | - | 2/38 | 2/29 | 0/2 | - | 1/7 | 0/1 | 6/41 | 1/2 | - | - | 12/120 | 10.0% |
| Total | 6/59 | 2/8 | 2/12 | 6/13 | 2/54 | 2/35 | 1/6 | 1/15 | 3/35 | 1/6 | 6/41 | 2/9 | 0/11 | 1/15 | 35/319 | 11.0% |
| % pos per site | 10.2% | 25.0% | 16.7% | 46.2% | 3.7% | 5.7% | 16.7% | 6.7% | 8.6% | 16.7% | 14.6% | 22.2% | 0.0% | 6.7% | ||
| Sample | Collection Date | Prefecture | Site | Environment | Species | Type of Sample | Accession Number | ||
|---|---|---|---|---|---|---|---|---|---|
| Rectal Swab | Feces | Oral Swab | |||||||
| CCGU00021 | 18 February 2016 | Conakry | CN | city garden | Epomophorus gambianus | β-CoV | na | – | MT586830 |
| CCGU00033 | 21 February 2016 | Conakry | CN | city garden | Epomophorus gambianus | β-CoV | na | – | MT586831 |
| CCGU00035 | 21 February 2016 | Conakry | CN | city garden | Epomophorus gambianus | – | β-CoV | – | MT586832 |
| CCGU00039 | 20 April 2016 | Conakry | CN | city garden | Eidolon helvum | β-CoV | na | – | MT586833 |
| CCGU00040 | 20 April 2016 | Conakry | CN | city garden | Eidolon helvum | β-CoV | na | – | MT586834 |
| CCGU00041 | 20 April 2016 | Conakry | CN | city garden | Eidolon helvum | β-CoV | na | – | MT586835 |
| CCGU00081 | 31 May 2016 | Kindia | KN | cave/village | Hipposideros ruber | na | α-CoV | – | MT586836 |
| CCGU00082 | 31 May 2016 | Kindia | KN | cave/village | Hipposideros ruber | α-CoV | na | – | MT586837 |
| CCGU00085 1 | 31 May 2016 | Kindia | KN | cave/village | Hipposideros ruber | na | α-CoV | α-CoV | MT586838; MT586839 |
| CCGU00086 | 31 May 2016 | Kindia | KN | cave/village | Hipposideros ruber | na | β-CoV | – | MT586840 |
| CCGU00087 1 | 31 May 2016 | Kindia | KN | cave/village | Hipposideros ruber | α-CoV | na | α-CoV | MT58684; MT586842 |
| CCGU00090 | 31 May 2016 | Kindia | KN | cave/village | Hipposideros ruber | α-CoV | na | – | MT586843 |
| CCGU00102 | 21 July 2016 | Koundara | GN | cave/village | Lissonycteris angolensis | β-CoV | na | – | MT586844 |
| CCGU00104 | 21 July 2016 | Koundara | GN | cave/village | Lissonycteris angolensis | β-CoV | na | – | MT586845 |
| CCGU00115 | 22 July 2016 | Koundara | GN | village | Rousettus aegyptiacus | – | na | α-CoV | MT586846 |
| CCGU00137 | 24 July 2016 | Koundara | YK | village | Epomophorus gambianus | β-CoV | na | – | MT586847 |
| CCGU00142 | 27 July 2016 | Koundara | SA | village | Epomophorus gambianus | β-CoV | na | – | MT586848 |
| CCGU00168 | 22 September 2016 | Kankan | KK | cave/forest | Rhinolophus darlingi | na | α-CoV | – | MT586849 |
| CCGU00200 | 25 September 2016 | Kankan | KK | cave/forest | Rhinolophus darlingi | na | α-CoV | – | MT586850 |
| CCGU00235 | 28 September 2016 | Kankan | BN | cave | Nycteris macrotis | na | β-CoV | – | MT586851 |
| CCGU00269 | 29 September 2016 | Kankan | BN | cave | Hipposideros ruber | na | β-CoV | – | MT586852 |
| CCGU00316 | 27 October 2016 | Kindia | MO | cave/forest | Rousettus aegyptiacus | β-CoV | na | – | MT586853 |
| CCGU00337 | 29 October 2016 | Kindia | MO | plantation | Rousettus aegyptiacus | β-CoV | na | – | MT586854 |
| CCGU00399 | 2 November 2016 | Kindia | SG | cave/forest | Rousettus aegyptiacus | β-CoV | na | – | MT586855 |
| CCGU00424 | 3 November 2016 | Kindia | SG | forest | Rousettus aegyptiacus | β-CoV | na | – | MT586856 |
| CCGU00445 | 5 December 2016 | Gueckedou | TM | cave/forest | Rousettus aegyptiacus | – | na | α-CoV | MT586857 |
| CCGU00453 | 6 December 2016 | Gueckedou | TM | village | Rousettus aegyptiacus | β-CoV | na | α-CoV | MT586858-586859 |
| CCGU00458 | 6 December 2016 | Gueckedou | TM | village | Rousettus aegyptiacus | – | na | α-CoV | MT586860 |
| CCGU00467 | 7 December 2016 | Gueckedou | TM | village | Rousettus aegyptiacus | – | na | α-CoV | MT586861 |
| CCGU00477 | 7 December 2016 | Gueckedou | TM | village | Rousettus aegyptiacus | α-CoV | na | – | MT586862 |
| CCGU00481 | 7 December 2016 | Gueckedou | TM | village | Rousettus aegyptiacus | – | na | α-CoV | MT586863 |
| CCGU00489 | 10 December 2016 | Gueckedou | GK | village | Nanonycteris veldkampii | β-CoV | na | – | MT586864 |
| CCGU00499 | 12 December 2016 | Macenta | MA | city | Rousettus aegyptiacus | β-CoV | na | – | MT586865 |
| CCGU00502 | 12 December 2016 | Macenta | MA | city | Eidolon helvum | β-CoV | na | – | MT586866 |
| CCGU00550 | 9 January 2017 | Mamou | KR | cave/forest | Lissonycteris angolensis | α-CoV | na | – | MT586867 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lacroix, A.; Vidal, N.; Keita, A.K.; Thaurignac, G.; Esteban, A.; De Nys, H.; Diallo, R.; Toure, A.; Goumou, S.; Soumah, A.K.; et al. Wide Diversity of Coronaviruses in Frugivorous and Insectivorous Bat Species: A Pilot Study in Guinea, West Africa. Viruses 2020, 12, 855. https://doi.org/10.3390/v12080855
Lacroix A, Vidal N, Keita AK, Thaurignac G, Esteban A, De Nys H, Diallo R, Toure A, Goumou S, Soumah AK, et al. Wide Diversity of Coronaviruses in Frugivorous and Insectivorous Bat Species: A Pilot Study in Guinea, West Africa. Viruses. 2020; 12(8):855. https://doi.org/10.3390/v12080855
Chicago/Turabian StyleLacroix, Audrey, Nicole Vidal, Alpha K. Keita, Guillaume Thaurignac, Amandine Esteban, Hélène De Nys, Ramadan Diallo, Abdoulaye Toure, Souana Goumou, Abdoul Karim Soumah, and et al. 2020. "Wide Diversity of Coronaviruses in Frugivorous and Insectivorous Bat Species: A Pilot Study in Guinea, West Africa" Viruses 12, no. 8: 855. https://doi.org/10.3390/v12080855
APA StyleLacroix, A., Vidal, N., Keita, A. K., Thaurignac, G., Esteban, A., De Nys, H., Diallo, R., Toure, A., Goumou, S., Soumah, A. K., Povogui, M., Koivogui, J., Monemou, J.-L., Raulino, R., Nkuba, A., Foulongne, V., Delaporte, E., Ayouba, A., & Peeters, M. (2020). Wide Diversity of Coronaviruses in Frugivorous and Insectivorous Bat Species: A Pilot Study in Guinea, West Africa. Viruses, 12(8), 855. https://doi.org/10.3390/v12080855