Abstract
Ion channels play key roles in almost all facets of cellular physiology and have emerged as key host cell factors for a multitude of viral infections. A catalogue of ion channel-blocking drugs have been shown to possess antiviral activity, some of which are in widespread human usage for ion channel-related diseases, highlighting new potential for drug repurposing. The emergence of ion channel–virus interactions has also revealed the intriguing possibility that channelopathies may explain some commonly observed virus induced pathologies. This field is rapidly evolving and an up-to-date summary of new discoveries can inform future perspectives. We herein discuss the role of ion channels during viral lifecycles, describe the recently identified ion channel drugs that can inhibit viral infections, and highlight the potential contribution of ion channels to virus-mediated disease.
1. Introduction
The human “channelome” contains over 300 known channels [1] that selectively and rapidly transport ions across biological membranes in response to specific stimuli. Ion channels are present on the plasma membranes and organelles of all cells, where they regulate organelle ion homeostasis, mitochondrial function, inflammasome activation, action potential firing, membrane potential, cell volume, and autophagy [2,3,4,5]. Given their importance, it follows that their dysfunctions leads to human diseases, termed channelopathies [6]. These include disease states of the nervous [2], musculoskeletal [5], cardiovascular [7], and immune systems [8]. This has motivated research on compounds that can modulate ion channel activity; ~19% of current FDA-approved drugs are ion channel modulators, second only to drugs targeting G-protein coupled receptors [9,10].
Many viruses encode their own ion channels [11,12,13] termed “viroporins,” highlighting the importance of ionic balance during viral infection. This field has spurred intense research and several drugs that target viroporins have emerged (reviewed in [11]). More recent evidence highlights how viruses can regulate and/or depend on the ion channels expressed by host cells, highlighting them as new host targets for therapeutic intervention (reviewed by Hover et al., 2017) [14]. Given recent and important advances in this field, we herein provide an up-to-date review of the virus–ion channel literature and discuss the future prospects of ion channel drugs as anti-viral agents. Firstly, we highlight recent evidence that suggests that viruses have adapted to take advantage of endolysosomal ionic balance as a cue for viral entry. We then discuss how intracellular ion channels contribute to the efficiency of viral replication. Finally, we describe how viral infections may result in ion channel dysfunction and lead to virus-induced channelopathies in infected individuals.
2. Ionic Balance in the Endosomal System
Most enveloped viruses enter cells by endocytosis [15]. The endolysosomal system is a dynamic series of intracellular membranous compartments that facilitate the uptake, degradation, and recycling of cellular cargoes and membrane proteins [16]. As endosomes mature, they undergo morphological and biological changes (reviewed in Scott et al., 2011) [17]. During early endosomal maturation, the intraluminal pH becomes more acidic, through the action of vacuolar-ATPases (v-ATPases). v-ATPases acidify intracellular vesicles through ATP-driven proton transport into the intraluminal space [18]. To mitigate the large positive charge within endosomes, chloride (Cl−) flows inwards through endosomal anion channels, accompanied by cation efflux via Na+K+/H+ exchangers [19,20,21,22,23,24]. Na+/K+-ATPases are present on early endosomes where they transport Na+ into the lumen to limit proton influx, and in turn, acidification [25]. Ca2+ plays a number of regulatory roles throughout the endosomal network, including the regulation of fusion and fission events, lipid trafficking, and lysosomal activity [26,27]. The role of K+ influx into endosomes is to-date uncharacterised, but an increase in luminal K+ occurs as endosomes mature [28]. Whilst the role of low pH in viral entry is well documented [29], the role of other endolysosomal ions is only beginning to be appreciated (Table 1).

Table 1.
Overview of ion channels implicated in viral entry.
3. Ion Channels Involved in Viral Entry
3.1. Ca2+ Channels and Viral Entry
The involvement of Ca2+ channels during viral entry is now well-documented [26]. Fujioka et al. showed that influenza virus (IAV) hemagglutinin (HA) triggers intracellular [Ca2+] oscillations that are required for viral infection [30]. The initial modulation of Ca2+ by IAV was demonstrated using Förster resonance energy transfer (FRET)-based imaging of the Ca2+ sensor Yellow Cameleon (YC3.60). These oscillations in Ca2+ were mediated by a specific voltage-gated Ca2+ channel (CaV1.2) identified through siRNA silencing approaches. Assays subsequently revealed that IAV directly binds to CaV1.2 via the interaction of HA and a sialylated site on CaV1.2. Accordingly, IAV entry could be inhibited by diltiazem, a clinically available Ca2+ blocker, highlighting the potential of these compounds for drug repurposing.
Ebola virus (EBOV) also requires Ca2+ channels for its entry into host cells. EBOV enters cells through endolysosomes positive for both Niemann–Pick C1 (NPC1) and two-pore Ca2+ channel 2 (TPC2) [32]. To further characterise this pathway, Penny et al. expanded the pharmacology of TPCs using a virtual screen of ~1500 FDA-approved drugs. All identified TPC modulators were cross-referenced with two recent anti-EBOV screens, with four dopamine receptor antagonists and five oestrogen receptor modulators identified. As such, it was reasoned that these drugs exert their inhibitory effects on EBOV through the blockade of TPCs (Figure 1E), subsequently confirmed through EBOV virus-like particle (VLP) assays [33]. Das et al. further characterised the role of Ca2+ in EBOV entry using single-molecule FRET (smFRET)-imaging. It was shown that Ca2+ and pH synergistically induce a conformational change in the EBOV glycoprotein GP2 (a key mediator of receptor binding and viral entry) to form a reversible intermediate state primed for NPC1 binding. NPC1 binding then further promotes the conformational transition into a fusion-ready “primed” state of invading EBOV virions [43].
Figure 1.
Ion channels implicated in viral entry since 2017. (A) Ifenprodil, glibenclamide, and TEA inhibit HIV through blockade of GIRK channels and KATP channels. (B) TEA and quinidine inhibit HAZV escape from EEs via blockade of an unknown channel. (C) Endosomal escape of SFTSV is inhibited by benidipine hydrochloride and nifedipine. (D) BUNV escape from late endosomes is inhibited by K2P blockade. (E) EBOV escape from lysosomes is TPC2-dependent and can be blocked by verapamil, tetrandrine, nicardipine, diltiazem, and fluphenazine. (F) MERS escape from endolysosomes is prevented by tetrandrine, fangchinoline, verapamil, nimodipine, and nicardipine blockade of TPCs. (G) MCPyV and (H) SV40 ER translocation is TPC2 mediated and can be inhibited by verapamil and tetrandrine. ER translocation of MCPyV is also susceptible to blockade of KV and T-type VGCCs by 4-AP and flunarizine respectively. (I) ER trafficking of BKPyV is CFTR dependent and susceptible to blockade by CFTR172 and glibenclamide. Key: PM: plasma membrane; EE: early endosome; LE: late endosome; EL: endolysosome. Ver: verapamil; Tet: tetrandrine; Nic: nicardipine; Dilt: diltiazem; Fluph: fluphenazine; Fang: fangchinoline; Nim: nimodipine; Nif: nifedipine; TEA: tetraethylammonium; Qd: quinidine; Ife: ifenprodil; Glib: glibenclamide; 4-AP: 4-aminopyridine.
Of importance to the current pandemic, it has been shown that Middle East respiratory syndrome coronavirus (MERS) [44] is dependent on TPCs to escape endosomes [34]. As an enveloped virus, MERS must fuse its envelope with host membranes to enter cells. Following receptor attachment, MERS particles can fuse at either the cell surface or intracellularly in the endosomal network. Fusion is mediated by the proteolytic cleavage of the viral spike (S) protein at its S1/S2 site. At the surface of the cell, this proteolytic event is facilitated by TMPRSS2, which in turn precludes exposure of the fusion loop and coalescence of host and viral membranes [45]. Alternatively, fusion can occur in late endosomes following translocation through the endocytic network and proteolytic processing by proprotein convertases, including furin, in a process regulated by Ca2+ [46,47]. In studies by Gunaratne et al., genetic silencing of TPC1 and TPC2 prevented the entry of pseudotyped MERS (Figure 1F). The dependence of MERS upon TPCs was further demonstrated through its inhibition by tetrandrine and fangchinoline (TPC inhibitors), which prevented a post-internalisation but pre-fusion entry event. The mechanism through which TPC blockade inhibited MERS was multi-faceted: TPC1 and TPC2 silencing impaired furin activity, whilst pharmacological and genetic inhibition of TPC1 impaired endosomal motility. Of note, the related SARS-CoV-2, the causative agent of COVID-19 [48,49], was similarly inhibited by TPC blockade. Specifically, treatment of cells with tetrandrine reduced the entry of a lentiviral vector pseudotyped with the SARS-CoV-2 S (spike) [36].
The bunyavirus severe fever with thrombocytopenia syndrome virus (SFTSV) is an emerging arbovirus with fatality rates of 12–50% and the potential to cause future pandemics [35]. Using a library of FDA-approved drugs, the Ca2+ channel blockers benidipine hydrochloride and nifedipine were shown to inhibit SFTSV infection (Figure 1C), with in vivo activity confirmed using C57BL/6 and humanised mouse models. Through a retrospective analysis of human SFTSV cases, clinical evidence of the efficacy of nifedipine as an anti-SFTSV therapeutic was also demonstrated. A cohort of patients receiving nifedipine prior to and during hospital admission showed enhanced viral clearance and reduced frequency of neurological syndromes, often associated with fatal outcomes [50,51]. The fatality rate of patients receiving nifedipine was reduced 5-fold compared to untreated patients, which corresponded to abnormal serum Ca2+ levels at admission. The viral processes through which SFTSV requires Ca2+ channels were subsequently shown to be during virus internalisation and genome replication.
3.2. K+ Channels and Viral Entry
The involvement of K+ channels in viral entry has been extensively characterised for the model bunyaviruses Bunyamwera orthobunyavirus (BUNV); and Hazara orthononairovirus (HAZV), a model for Crimean Congo haemorrhagic fever virus, which causes severe viral haemorrhagic fever outbreaks, with a case fatality rate of up to 40%. Initial work using known K+ channel pharmacology suggested that the blockade of two-pore K+ channels (K2P) inhibited the early stages of the BUNV lifecycle (Figure 1D) [37]. Subsequent work identified both acidic pH and K+ in endosomes as crucial biochemical cues for the endosomal escape of BUNV [28]. Similar studies in HAZV highlighted a dependence on K+ channels for infection, and that K+ primarily accumulated in cholesterol-rich endosomes (Figure 1B) [28,38,39]. The K+ dependence of HAZV involves the glycoprotein spikes; a change in K+ concentration triggers conformational changes in the glycoproteins, as revealed through cryo-electron tomography of HAZV virions incubated with K+ that “primed” them for insertion into target membranes (Figure 2A). Moreover, it was shown that both BUNV and HAZV could be “primed” in vitro in buffers containing high [K+], which expedited entry and subsequent viral gene expression. This phenomenon was analogous to earlier studies for IAV, in which acid bypass in the presence of K+ revealed that the exposure of IAV virions to low pH and high [K+] weakened interactions between the M1 matrix protein and ribonucleoprotein (RNP) bundles, a pre-requisite for genome release (Figure 2B). The exposure of IAV virions to K+ therefore drives viral uncoating and expedites IAV infection [31].
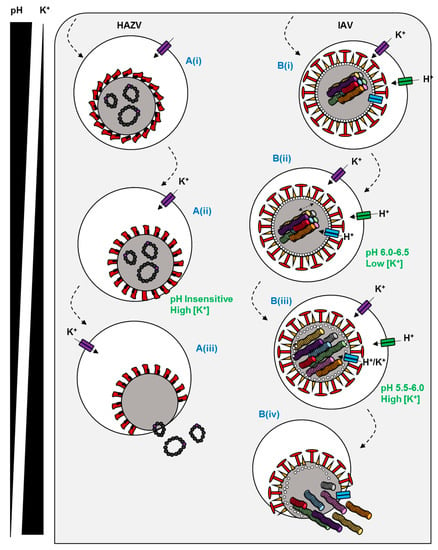
Figure 2.
Predicted mechanisms of ion channel dependence for two enveloped viruses. (A(i)) HAZV is endocytosed by an undefined mechanism. (A(ii)) Endosomal K+ influx triggers a conformational change in the HAZV glycoprotein spikes to a fusion-ready state. (A(iii)) Host and viral membranes fuse and RNPs are liberated into the cytosol. (B(i)) IAV is endocytosed via a clathrin-dependent or independent mechanism. (B(ii)) The virus traffics to late endosomes where the M2 viroporin is activated by acidic pH. (B(iii)) The influx of K+ and H+ destabilises matrix-RNP interactions in the core. (B(iv)) At low pH, a conformational change in HA promotes fusion and RNP release.
Recent work also highlights a requirement for K+ channels during human immunodeficiency virus (HIV) infection. Using pharmacological approaches (Figure 1A) [40] HIV entry could be blocked with ifenprodil and the broad spectrum K+ channel blocker tetraethylammonium (TEA). Khan et al. also showed that the pharmacological activation of the endolysosome-resident transient receptor potential mucolipin 1 channel (TRPML1) enhanced the degradation of HIV-Tat (a multi-function viral protein involved in transcription, splicing, capping, and translation), which in turn reduced the transition from viral latency [52]. TRPML1 activates large conductance Ca2+-activated potassium (BK) channels in endosomes [53], implying that this channel is required for HIV infection.
The reliance of viruses upon ion channels is not restricted to RNA viruses. It was recently shown that both K+ and Ca2+ channels are important host factors for polyomavirus infection [41]. Using a panel of ion channel modulators, the entry of Merkel cell polyomavirus (MCPyV), the causative agent of Merkel cell carcinoma (MCC), was shown to be sensitive to 4-aminopyridine (4-AP), a blocker of voltage-gated K+ (KV) channels (Figure 1G). Moreover, both MCPyV and Simian virus 40 (SV40) (Figure 1H) were sensitive to verapamil, a broad-spectrum Ca2+ channel blocker. The identities of the Ca2+ channels involved in polyomavirus entry were further explored, revealing a requirement for transient (T-type, low-voltage activated) channel family members in MCPyV infection but not SV40. Tetrandrine, a TPC blocker, restricted both viruses. The role of TPCs was found to be during endoplasmic reticulum (ER) disassembly and/or ER docking for SV40 [54], which may be explained by the recent demonstration that Ca2+ ions mediate the stabilization of SV40 capsids and contribute to its disassembly [55].
3.3. Cl− Channels and Viral Entry
BK polyomavirus (BKPyV) is a potentially fatal pathogen in patients undergoing solid organ transplantation. Panou et al. demonstrated that the pharmacological and genetic disruption of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel could reduce BKPyV infection in primary kidney cell models (Figure 1I) [42]. Time of addition assays using the CFTR inhibitors CFTR172 and glibenclamide, combined with the assessment of exposure of VP2/VP3 minor capsid proteins, indicated a role for CFTR in the trafficking of BKPyV to the ER. Whilst the mechanism of CFTR involvement in BKPyV ER trafficking remains unclear, it is hypothesised that the channel may be important in the acidification and ER docking of BKPyV-containing endosomes. CFTR has been implicated in the fusion of endosomes [56], and co-localises with vacuolar ATPase (vATPase) to provide the counter-charge during endosomal acidification [57].
4. Ion Channels in Viral Replication
Once viral genomes are released inside the host cell, replication can commence. Recent evidence suggests that this process is partially controlled by ion concentrations and can therefore be targeted by ion channel drugs.
4.1. Ca2+ Channels and Viral Replication
Flaviviruses establish replication complexes in modified intracellular membranes, often derived from the ER. The ER stores the majority of intracellular Ca2+, and so it is perhaps unsurprising that an array of flaviviruses depend on intracellular Ca2+ ion channels for their replication (Table 2). Japanese encephalitis virus (JEV) is an arthropod-borne virus linked to acute encephalitis. Wang et al. performed a screen of FDA-approved drugs to assess in vitro activity against JEV [58]. Within the screen, three of the five most potent inhibitors were blockers of voltage-gated Ca2+ channels (VGCCs), including manidipine, cilnidipine, and benidipine hydrochloride. Time of addition assays suggested that these three drugs did not act during viral entry, nor were they virucidal, but specifically inhibited virus replication. The potential of these drugs as broad-acting anti-flavivirus treatments was further assessed and each led to a concentration-dependent inhibition of Dengue virus DENV), West Nile virus, and Zika virus (ZIKV) replication. Yellow fever virus (YFV) was, however, insensitive to manidipine. Interestingly, the in vitro selection of a manidipine-resistant JEV identified a Q130R mutation in the non-structural protein NS4B. Sequence alignments confirmed that the Q130 site was conserved in each of the flaviviruses susceptible to manidipine, but not YFV. The efficacy of manidipine against JEV was further confirmed in in vivo mouse models, with manidipine-treated mice exhibiting significantly higher survival rates compared to untreated mice challenged with JEV.
Table 2.
Overview of ion channels implicated in viral replication.
A role for Ca2+ channels during DENV replication was further revealed by Dionicio et al. [59]. DENV was shown to inhibit intracellular Ca2+ release from the ER, which in turn activated the store-operated Ca2+ (SOCE) pathway. In addition, specialised Ca2+ release activated Ca2+ channels (CRACs) were identified as a requirement for DENV replication. Small molecule blockers of these channels resulted in a 70% reduction in virus yield. Additionally, yeast two-hybrid screens identified the Ca2+-permeable non-selective transient receptor potential vanilloid 4 (TRPV4) in complex with the DEAD-box helicase (DDX3X) as a key regulator of viral mRNA translation for ZIKV [60].
The hepatitis B virus (HBV) X protein is an oncoprotein that regulates cytosolic [Ca2+] [61]. Recently, Yao et al. characterised this mechanism, revealing its interaction with ORAI1, a critical component of the SOCE pathway [62]. HBV replication was also dependent on K+ in studies by Chakraborty et al. [63]. They reported that K+-dependent nucleolytic activity in the presence of HBV RNA mediates the self-cleavage of a 53 nt oligomer with ribozyme activity that is required for viral replication.
4.2. Cl− Channels or Other Anions and Vials Replication
Chikungunya virus (CHIKV) is a re-emerging arbovirus associated with long-term complications and high morbidity. Using siRNA silencing of the Cl− intracellular channels (CLIC) 1 and 4, Müller et al. demonstrated a requirement for both channels during the replication of a CHIKV sub-genomic replicon in mammalian and invertebrate cells [65]. The voltage-dependent anion channel 1 (VDAC1) is also implicated in viral replication; it is upregulated by infectious bursal disease virus (IBDV). Han et al. showed that knockdown of VDAC1 inhibited IBDV replication through the reduction of viral polymerase activity, and that the overexpression of VDAC1 promotes polymerase activity. Immunoprecipitation (IP) experiments showed that VDAC1 interacts with IBDV VP1 and VP3, components of RNPs, indicating a role for this channel in RNP formation [64].
5. Viruses as Causative Agents of Acquired Channelopathies
Viral pathologies are becoming increasingly linked to the dysregulation of host ion channels (Table 3). This reveals an interesting and new avenue for ion channel drugs, as the pharmacological manipulation of virus-targeted channels may not only impair viral infection at the cellular level, but may circumvent virus-induced channelopathies.
Table 3.
Overview of ion channels implicated in virus-mediated disease.
5.1. Viral Channelopathies and Ca2+ Channels
Recent studies have linked viral infection to neuronal pathologies through the dysregulation of Ca2+ signalling. The FDA-approved Alzheimer’s drug memantine protected against neuronal cell death induced by ZIKV infection [66]. Memantine acts upon the N-methyl-d-aspartate receptor (NMDAR), which mediates Ca2+ signalling to govern synaptic plasticity [67]. The overstimulation of NMDAR is linked to neurodegeneration, a pathology commonly associated with ZIKV. Upon challenge with memantine, ZIKV replication was unaffected, but antagonism of NMDAR invoked a neuroprotective effect in vivo. Whilst the exact mechanism(s) of ZIKV neuropathologies are unknown, it is predicted that the virus hyper-stimulates NMDAR to upregulate Ca2+ signalling to the point of Ca2+ overload and postsynaptic neuronal death, a process termed “glutamate excitotoxicity.”
The reactivation of herpes simplex virus 1 (HSV-1) can lead to cranial nerve disorders and severe pain. Zhang et al. revealed that HSV-1 disrupts the expression of T-type Ca2+ channels in differentiated sensory-like neurons, as a means to disrupt pain responses [68,69]. Proteomics and transcriptomics showed that HSV-1 decreased the expression of the CaV3.2 T-type Ca2+ channel subunit at the protein level, despite increasing CaV3.2 mRNA synthesis. This upregulation of CaV3.2 mRNA synthesis was postulated to be a compensatory response to decreased expression of the channel subunit. The loss of CaV3.2 initially led to reduced pain transmission in infected neurons; however, the release of interleukin-6 (IL-6) in response to viral infection was subsequently shown to restore CaV3.2 current density and pain responses.
Rotavirus (RV) dysregulates cellular Ca2+ homeostasis through the depletion of ER stores. Using genetically-encoded Ca2+ indicators in infected cells, cytosolic Ca2+ increased in distinct peaks [70,71,72], which was mediated by the non-structural protein NSP4. RV-NSP4 acts as a Ca2+-permeable viroporin to release Ca2+ from the ER, which in turn activates the ER Ca2+ sensor STIM1, subsequently leading to SOCE activation. RV-infected cells then secrete a cleavage product of NSP4 (eNSP4), which elicits an inward inositol triphosphate (IP3)-dependent Ca2+ signal, which in turn stimulates Cl− release through Ca2+-activated Cl− channels (CaCCs) [73]. The efflux of Cl− from cells is a known causative factor of diarrhoea in vivo, thereby identifying NSP4 as the first viral enterotoxin. This multi-faceted control of Ca2+ signalling suggests a crucial role for Ca2+ channels in the pathophysiology of RV. In this regard, it was shown that the blockade of CaCCs, including TMEM16A, reduces intestinal motility and fluid loss in vivo with no direct effects on the levels of virus infection [74].
5.2. Viral Channelopathies Associated with K+ Channels
Coxsackie virus B3 (CVB3), amongst other enteroviruses, is associated with cardiomyopathies and sudden cardiac death [76]. KCNQ1 is a KV channel (KV7.1) that mediates a delayed, slow rectifying K+ current in ventricular tissue to regulate contractility. Kv7.1 trafficking and activity are regulated by the serine/threonine kinase SGK1 [80], which is upregulated by CVB3. As such, KCNQ1 currents are elevated to 125% in CVB infected cells, whilst hERG1 (KV11.1) and CaV1.2 activity decrease by 59% and 83% respectively. These results corroborated with localisation studies of each channel and demonstrated that the surface expression of KCNQ1 increased, whilst hERG1 and CaV1.2 expression decreased in infected cells. Inherited mutations in each of these three ion channels are associated with heart rhythm disorders. Importantly, decreased hERG1 expression increases the risk of drug-induced arrhythmias; the additional inhibition or reduced trafficking of this channel in cells targeted by small therapeutic molecules reduces the number of redundant repolarisation currents, in turn depleting the overall repolarisation reserve. Together, these data suggest that CVB3 re-programmes ion channel expression in cardiac tissue, leading to an increased risk of arrhythmia. This highlights these channels as therapeutic targets to prevent the sudden cardiac death that results from CVB3 infection.
5.3. Viral Channelopathies Associated with Na+ Channels
A number of viruses that primarily infect the airway system have been shown to dysregulate airway epithelial Na+ transport. Human respiratory syncytial virus (HRSV) primarily infects airway epithelial cells, and dysregulates epithelial Na+ channels (ENaC) to disrupt Na+ flux in the airways [81]. ENaCs are a critical mediator of osmotic fluid absorption across airway epithelia, through the selective transport of Na+. Electrochemical balance is maintained through apical Cl− channels, which include CFTR. Clinical studies of infants diagnosed with HRSV showed a negative correlation between ENaC mRNA expression and the severity of HRSV bronchiolitis [82]. HRSV has been shown to manipulate airway ion flux through upregulation of channels involved in the cough reflex, namely, transient receptor cation channel, subfamily A, member 1 (TRPA1), and the acid sensing ion channel receptor 3 (ASIC3), a member of the ENaC family of Na+ channels [77]. HRSV induced a 30-fold increase in ASIC3 mRNA in normal bronchial epithelial cells, compared to the 3-fold increase observed in cells challenged with measles virus (MeV). Interestingly, UV-inactivated HRSV and MeV maintained their ability to upregulate ASIC3, suggesting these effects were independent of genome replication. The virion-induced upregulation of IL-6 and IL-8 was subsequently shown to inhibit TRP receptor activity, identifying these receptors as potential targets for virus-induced cough symptoms.
ENaC and CFTR channel expression are also modulated by IAV. From single-cell recordings in intact lung epithelial cells, Brand et al. described a reduction in ENaC and CFTR activity upon IAV-infection, through reduced surface expression. The mechanisms governing this downregulation were not characterised, but it is thought that IAV promote ER stress, known to cause deficits in ENaC abundance [83]. The loss of ENaC and CFTR surface expression was accompanied by a reduction in airway surface liquid (ASL), a known contributor to cystic fibrosis. Importantly, treatment with the FDA-approved CFTR corrector lumacaftor could restore IAV-mediated ASL perturbations, highlighting how virus-induced pathologies can be treated by therapies targeting host ion channels [78].
The altered activity of Na+ channels was observed in cells latently infected HSV-1 dorsal root ganglion neurones. Upon acute lytic infection, HSV-1 was found to reduce functional voltage-gated sodium channel (VGSC) expression within 24 h and abolish VGSC activity within 3 days, whilst latent HSV-1 infection led to a recovery of these currents and increased NaV1.7 channel expression. Furthermore, HSV-1 reactivation from a dormant state decreased VGSC activity. It is known that VGSCs play a role in the transmission of pain signals [75]. Similarly, post-herpetic neuralgia associated with varicella-zoster virus is associated with an increase in Na+ current amplitude through the activity of NaV1.6 and NaV1.7 [84]. NaV1.7 dysregulation has also been associated with hereditary pain disorders. Gain-of-function mutations in SCN9A, the gene encoding NaV1.7, are causative of primary erythromelalgia (PE), a rare neuropathy characterised by recurring pain, warmth, and redness of the extremities. Research into the management of PE identified two novel selective NaV1.7 blockers, PF-05089771 and TV-45070, which may hold promise in ameliorating pain symptoms associated with PE [85] and viral-induced neuropathies.
5.4. Viral Channelopathies Associated with Cl− Channels
Stakaitytė et al., used a proteomics approach to identify changes in the host channelome in response to the overexpression of MCPyV small tumour antigen (ST). The analysis revealed a role for two CLIC proteins [79], CLIC1 and CLIC4 (4-fold and 5-fold up regulation in cells overexpressing ST vs. control cells, respectively). Pharmacological and genetic inhibition of these channels reduced ST-induced motility and migration, implicating their function in MCPyV, ST-induced metastatic processes [86]. These data were reinforced by the finding that MCPyV-positive MCC tumours showed enhanced CLIC1 and CLIC4 expression (2.5-fold and 3.5-fold increase respectively), implicating their involvement in MCC tumorigenesis. These findings align with earlier evidence that CLIC1 and CLIC4 are involved in the metastatic progression of specific tumour types, through switching of cellular localisation and function to integral transmembrane proteins as active anion channels and signal transducers [87].
6. Conclusions and Further Perspectives
It is now clear that host cell ion channels play an important role during viral infection at the cellular level, and as causative factors of disease states in infected tissues. Ion channels have been linked to multiple stages of viral lifecycles, in which viruses are either passively dependent upon or actively modulate channel functionality. Given this knowledge, evidence is emerging that ion channel inhibitors represent a new antiviral strategy. Whilst toxicity profiles for ion channel inhibitors are only available in the context of those used to treat hereditary channelopathies, in vivo evidence is emerging that these drugs can be efficacious against viruses. Moreover, overlapping mechanisms of acquired and hereditary channelopathies may underpin the efficacy of ion channel modulators in treating virally-induced pathophysiologies. The manipulation of host ion homeostasis presents an attractive target for the treatment of many clinically important viruses and their associated pathologies, whilst circumventing the risks of resistance associated with direct-acting antiviral drugs. As such, continued studies of host-virus interactions may guide future antiviral approaches.
Funding
J.M. and J.F. are supported by the Academic Fellow scheme at the University of Leeds.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Yu, F.H.; Yarov-Yarovoy, V.; Gutman, G.A.; Catterall, W.A. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev. 2005, 57, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Hanna, M.G. Genetic neurological channelopathies. Nat. Clin. Pract. Neurol. 2006, 2, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Netter, R.C.; Amberg, S.M.; Balliet, J.W.; Biscone, M.J.; Vermeulen, A.; Earp, L.J.; White, J.M.; Bates, P. Heptad repeat 2-based peptides inhibit avian sarcoma and leukosis virus subgroup a infection and identify a fusion intermediate. J. Virol. 2004, 78, 13430–13439. [Google Scholar] [CrossRef]
- Verkman, A.S.; Galietta, L. Chloride channels as drug targets. Nat. Rev. Drug Discov. 2008, 8, 153–171. [Google Scholar] [CrossRef]
- Phillips, L.; Trivedi, J.R. Skeletal muscle channelopathies. Neurotherapeutics 2018, 15, 954–965. [Google Scholar] [CrossRef]
- Kim, J.-B. Channelopathies. Korean J. Pediatrics 2014, 57, 1–18. [Google Scholar] [CrossRef]
- Garcia-Elias, A.; Benito, B. Ion channel disorders and sudden cardiac death. Int. J. Mol. Sci. 2018, 19, 692. [Google Scholar] [CrossRef]
- Vaeth, M.; Feske, S. Ion channelopathies of the immune system. Curr. Opin. Immunol. 2018, 52, 39–50. [Google Scholar] [CrossRef]
- Farre, C.; Fertig, N. New strategies in ion channel screening for drug discovery: Are there ways to improve its productivity? Expert Opin. Drug Discov. 2014, 9, 1103–1107. [Google Scholar] [CrossRef]
- McManus, O.B. HTS assays for developing the molecular pharmacology of ion channels. Curr. Opin. Pharmacol. 2014, 15, 91–96. [Google Scholar] [CrossRef]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Genet. 2012, 10, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Nieva, J.L.; Carrasco, L. Viroporins: Structures and functions beyond cell membrane permeabilization. Viruses 2015, 7, 5169–5171. [Google Scholar] [CrossRef] [PubMed]
- Royle, J.; Dobson, S.J.; Müller, M.; Macdonald, A. Emerging roles of viroporins encoded by DNA viruses: Novel targets for antivirals? Viruses 2015, 7, 5375–5387. [Google Scholar] [CrossRef] [PubMed]
- Hover, S.; Foster, B.; Barr, J.N.; Mankouri, J. Viral dependence on cellular ion channels—An emerging antiviral target? J. Gen. Virol. 2017, 98, 345–351. [Google Scholar] [CrossRef]
- Helenius, A.; Kartenbeck, J.; Simons, K.; Fries, E. On the entry of semliki forest virus into BHK-21 cells. J. Cell Biol. 1980, 84, 404–420. [Google Scholar] [CrossRef]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef]
- Scott, C.C.; Gruenberg, J. Ion flux and the function of endosomes and lysosomes: PH is just the start. BioEssays 2010, 33, 103–110. [Google Scholar] [CrossRef]
- Pamarthy, S.; Kulshrestha, A.; Katara, G.K.; Beaman, K.D. The curious case of vacuolar ATPase: Regulation of signaling pathways. Mol. Cancer 2018, 17, 1–9. [Google Scholar] [CrossRef]
- Stauber, T.; Jentsch, T.J. Sorting motifs of the endosomal/lysosomal CLC chloride transporters. J. Biol. Chem. J. Biol. Chem. 2010, 285, 34537–34548. [Google Scholar] [CrossRef]
- Scheel, O.; Zdebik, A.A.; Lourdel, S.; Jentsch, T.J. Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature 2005, 436, 424–427. [Google Scholar] [CrossRef]
- Steinberg, B.E.; Huynh, K.K.; Brodovitch, A.; Jabs, S.; Stauber, T.; Jentsch, T.J.; Grinstein, S. A cation counterflux supports lysosomal acidification. J. Cell Biol. 2010, 189, 1171–1186. [Google Scholar] [CrossRef] [PubMed]
- Brett, C.L.; Tukaye, D.N.; Mukherjee, S.; Rao, R. The yeast endosomal Na+(K+)/H+ exchanger Nhx1 regulates cellular pH to control vesicle trafficking. Mol. Biol. Cell 2005, 16, 1396–1405. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.K.; Grinstein, S. Regulation of vacuolar PH and its modulation by some microbial species. Microbiol. Mol. Biol. Rev. 2007, 71, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Van Dyke, R.W. Na+/H+ exchange modulates acidification of early rat liver endocytic vesicles. Am. J. Physiol. Cell Physiol. 2005, 269, C943–C954. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, R.; Schmid, S.; Mellman, I. A possible role for Na+,K+-ATPase in regulating ATP-dependent endosome acidification. Proc. Natl. Acad. Sci. USA 1989, 86, 539–543. [Google Scholar] [CrossRef]
- Luzio, J.P.; Bright, N.; Pryor, P. The role of calcium and other ions in sorting and delivery in the late endocytic pathway. Biochem. Soc. Trans. 2007, 35, 1088–1091. [Google Scholar] [CrossRef]
- Gerasimenko, O.V.; Tepikin, A.V.; Petersen, O.H.; Gerasimenko, O.V. Calcium uptake via endocytosis with rapid release from acidifying endosomes. Curr. Biol. 1998, 8, 1335–1338. [Google Scholar] [CrossRef]
- Hover, S.; Foster, B.; Fontana, J.; Kohl, A.; Goldstein, S.A.; Barr, J.N.; Mankouri, J. Bunyavirus requirement for endosomal K+ reveals new roles of cellular ion channels during infection. PLoS Pathog. 2018, 14, e1006845. [Google Scholar] [CrossRef]
- Staring, J.; Raaben, M.; Brummelkamp, T.R. Viral escape from endosomes and host detection at a glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Fujioka, Y.; Nishide, S.; Ose, T.; Suzuki, T.; Kato, I.; Fukuhara, H.; Fujioka, M.; Horiuchi, K.; Satoh, A.O.; Nepal, P.; et al. A sialylated voltage-dependent Ca2+ channel binds hemagglutinin and mediates influenza a virus entry into mammalian cells. Cell Host Microbe 2018, 23, 809–818. [Google Scholar] [CrossRef]
- Stauffer, S.; Feng, Y.; Nebioglu, F.; Heilig, R.; Picotti, P.; Helenius, A. Stepwise priming by acidic pH and a high K+ concentration is required for efficient uncoating of influenza a virus cores after penetration. J. Virol. 2014, 88, 13029–13046. [Google Scholar] [CrossRef] [PubMed]
- Simmons, J.A.; D’Souza, R.S.; Ruas, M.; Galione, A.; Casanova, J.E.; White, J.M. Ebolavirus glycoprotein directs fusion through NPC1+ endolysosomes. J. Virol. 2015, 90, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Penny, C.J.; Vassileva, K.; Jha, A.; Yuan, Y.; Chee, X.; Yates, E.; Mazzon, M.; Kilpatrick, B.S.; Muallem, S.; Marsh, M.; et al. Mining of Ebola virus entry inhibitors identifies approved drugs as two-pore channel pore blockers. Biochim. Biophys. Acta Mol. Cell Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gunaratne, G.S.; Yang, Y.; Li, F.; Walseth, T.F.; Marchant, J.S. NAADP-dependent Ca2+ signaling regulates Middle East respiratory syndrome-Coronavirus pseudovirus translocation through the endolysosomal system. Cell Calcium 2018, 75, 30–41. [Google Scholar] [CrossRef]
- Li, H.; Zhang, L.K.; Li, S.F.; Zhang, S.F.; Wan, W.W.; Zhang, Y.L.; Xin, Q.L.; Dai, K.; Hu, Y.Y.; Wang, Z.B.; et al. Calcium channel blockers reduce severe fever with thrombocytopenia syndrome virus (SFTSV) related fatality. Cell Res. 2019, 29, 739–753. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Hover, S.; King, B.; Hall, B.; Loundras, E.A.; Taqi, H.; Daly, J.; Dallas, M.; Peers, C.; Schnettler, E.; McKimmie, C.; et al. Modulation of potassium channels inhibits bunyavirus infection. J. Biol. Chem. 2016, 291, 3411–3422. [Google Scholar] [CrossRef]
- Punch, E.K.; Hover, S.; Blest, H.T.; Fuller, J.; Hewson, R.; Fontana, J.; Mankouri, J.; Barr, J.N. Potassium is a trigger for conformational change in the fusion spike of an enveloped RNA virus. J. Biol. Chem. 2018, 293, 9937–9944. [Google Scholar] [CrossRef]
- Charlton, F.W.; Hover, S.; Fuller, J.; Hewson, R.; Fontana, J.; Barr, J.N.; Mankouri, J. Cellular cholesterol abundance regulates potassium accumulation within endosomes and is an important determinant in bunyavirus entry. J. Biol. Chem. 2019, 294, 7335–7347. [Google Scholar] [CrossRef]
- Dubey, R.C.; Mishra, N.; Gaur, R. G protein-coupled and ATP-sensitive inwardly rectifying potassium ion channels are essential for HIV entry. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Dobson, S.J.; Mankouri, J.; Whitehouse, A. Identification of potassium and calcium channel inhibitors as modulators of polyomavirus endosomal trafficking. Antiviral Res. 2020, 104819. [Google Scholar] [CrossRef] [PubMed]
- Panou, M.M.; Antoni, M.; Morgan, E.L.; Loundras, E.A.; Wasson, C.W.; Welberry-Smith, M.; Mankouri, J.; Macdonald, A. Glibenclamide inhibits BK polyomavirus infection in kidney cells through CFTR blockade. Antiviral Res. 2020, 178, 104778. [Google Scholar] [CrossRef] [PubMed]
- Das, D.K.; Bulow, U.; Diehl, W.E.; Durham, N.D.; Senjobe, F.; Chandran, K.; Luban, J.; Munro, J.B. Conformational changes in the Ebola virus membrane fusion machine induced by pH, Ca2+, and receptor binding. PLoS Biol. 2020, 18, e3000626. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Hui, D.S.; Perlman, S. Middle east respiratory syndrome. Lancet 2015, 386, 995–1007. [Google Scholar] [CrossRef]
- Park, J.E.; Li, K.; Barlan, A.; Fehr, A.R.; Perlman, S.; McCray, P.B.; Gallagher, T. Proteolytic processing of middle east respiratory syndrome coronavirus spikes expands virus tropism. Proc. Natl. Acad. Sci. USA 2016, 113, 12262–12267. [Google Scholar] [CrossRef]
- Mille, J.K.; Whittaker, G.R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA 2014, 111, 15214–15219. [Google Scholar] [CrossRef]
- Ruas, M.; Rietdorf, K.; Arredouani, A.; Davis, L.C.; Lloyd-Evans, E.; Koegel, H.; Funnell, T.M.; Morgan, A.J.; Ward, J.A.; Watanabe, K.; et al. Purified TPC isoforms form NAADP receptors with distinct roles for Ca2+ signaling and endolysosomal trafficking. Curr. Biol. 2010, 20, 703–709. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Li, H.; Lu, Q.B.; Xing, B.; Zhang, S.F.; Liu, K.; Du, J.; Li, X.K.; Cui, N.; Yang, Z.D.; Wang, L.Y.; et al. Epidemiological and clinical features of laboratory-diagnosed severe fever with thrombocytopenia syndrome in China, 2011–17: A prospective observational study. Lancet Infect. Dis. 2018, 18, 1127–1137. [Google Scholar] [CrossRef]
- Bao, C.J.; Guo, X.L.; Qi, X.; Hu, J.L.; Zhou, M.H.; Varma, J.K.; Cui, L.B.; Yang, H.T.; Jiao, Y.J.; Klena, J.D.; et al. A family cluster of infections by a newly recognized bunyavirus in Eastern China, 2007: Further evidence of person-to-person transmission. Clin. Infect. Dis. 2011, 53, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Lakpa, K.L.; Halcrow, P.W.; Afghah, Z.; Miller, N.M.; Geiger, J.D.; Chen, X. BK channels regulate extracellular Tat-mediated HIV-1 LTR transactivation. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Zhong, X.Z.; Zou, Y.; Zhang, Z.; Toro, L.; Dong, X.P. BK channels alleviate lysosomal storage diseases by providing positive feedback regulation of lysosomal Ca2+ release. Dev. Cell 2015, 33, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Kolokoltsov, A.A.; Chen, C.C.; Tidwell, M.W.; Bauta, W.E.; Klugbauer, N.; Grimm, C.; Wahl-Schott, C.; Biel, M.; Davey, R.A. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 2015, 347, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.A.; Xing, L.; Tsukamoto, H.; Inoue, T.; Handa, H.; Cheng, R.H. Calcium bridge triggers capsid disassembly in the cell entry process of simian virus 40. J. Biol. Chem. 2009, 284, 34703–34712. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, N.A. Intracellular CFTR: Localization and function. Physiol. Rev. 1999, 79, S175–S191. [Google Scholar] [CrossRef]
- Collaco, A.M.; Geibel, P.; Lee, B.S.; Geibel, J.P.; Ameen, N.A. Functional vacuolar ATPase (V-ATPase) proton pumps traffic to the enterocyte brush border membrane and require CFTR. Am. J. Physiol. Cell Physiol. 2013, 305, C981–C996. [Google Scholar] [CrossRef]
- Wang, S.; Liu, Y.; Guo, J.; Wang, P.; Zhang, L.; Xiao, G.; Wang, W. Screening of FDA-approved drugs for inhibitors of Japanese Encephalitis virus infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Dionicio, C.L.; Pena, F.; Constantino-Jonapa, L.A.; Vazquez, C.; Yocupicio-Monroy, M.; Rosales, R.; Zambrano, J.L.; Ruiz, M.C.; Del Angel, R.M.; Ludert, J.E. Dengue virus induced changes in Ca2+ homeostasis in human hepatic cells that favor the viral replicative cycle. Virus Res. 2018, 245, 17–28. [Google Scholar] [CrossRef]
- Doñate-Macián, P.; Jungfleisch, J.; Pérez-Vilaró, G.; Rubio-Moscardo, F.; Perálvarez-Marín, A.; Diez, J.; Valverde, M.A. The TRPV4 channel links calcium influx to DDX3X activity and viral infectivity. Nat. Commun. 2018, 9, 2307. [Google Scholar] [CrossRef]
- Bouchard, M.J.; Wang, L.H.; Schneider, R.J. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science 2001, 294, 2376–2378. [Google Scholar] [CrossRef]
- Yao, J.-H.; Liu, Z.-J.; Yi, J.-H.; Wang, J.; Liu, Y.-N. Hepatitis B virus X protein upregulates intracellular calcium signaling by binding C-terminal of orail protein. Curr. Med Sci. 2018, 38, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Ghosh, S. The epsilon motif of hepatitis B virus RNA exhibits a potassium-dependent ribonucleolytic activity. FEBS J. 2017, 284, 1184–1203. [Google Scholar] [CrossRef]
- Han, C.; Zeng, X.; Yao, S.; Gao, L.; Zhang, L.; Qi, X.; Duan, Y.; Yang, B.; Gao, Y.; Liu, C.; et al. Voltage-dependent anion channel 1 interacts with ribonucleoprotein complexes to enhance infectious bursal disease virus polymerase activity. J. Virol. 2017, 91, e00584-17. [Google Scholar] [CrossRef]
- Müller, M.; Slivinski, N.; Todd, E.J.; Khalid, H.; Li, R.; Karwatka, M.; Merits, A.; Mankouri, J.; Tuplin, A. Chikungunya virus requires cellular chloride channels for efficient genome replication. PLoS Negl. Trop. Dis. 2019, 13, e0007703. [Google Scholar] [CrossRef]
- Costa, V.V.; Del Sarto, J.L.; Rocha, R.F.; Silva, F.R.; Doria, J.G.; Olmo, I.G.; Marques, R.E.; Queiroz-Junior, C.M.; Foureaux, G.; Araújo, J.M.S.; et al. N-methyl-D-aspartate (NMDA) receptor blockade prevents neuronal death induced by Zika virus infection. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Sirohi, D.; Kuhn, R.J. Can an FDA-approved Alzheimer’s drug be repurposed for alleviating neuronal symptoms of zika virus? mBio 2017, 8, e00916-17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hsia, S.C.; Martin-Caraballo, M. Regulation of T-type Ca2+ channel expression by interleukin-6 in sensory-like ND7/23 cells post-herpes simplex virus (HSV-1) infection. J. Neurochem. 2019, 151, 238–254. [Google Scholar] [CrossRef]
- Zhang, Q.; Hsia, S.C.; Martin-Caraballo, M. Regulation of T-type Ca2+ channel expression by herpes simplex virus-1 infection in sensory-like ND7 cells. J. Neurovirol. 2017, 23, 657–670. [Google Scholar] [CrossRef]
- Chang-Graham, A.L.; Perry, J.L.; Strtak, A.C.; Ramachandran, N.K.; Criglar, J.M.; Philip, A.A.; Patton, J.T.; Estes, M.K.; Hyser, J.M. Rotavirus calcium dysregulation manifests as dynamic calcium signaling in the cytoplasm and endoplasmic reticulum. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Suzuki, J.; Kanemaru, K.; Ishii, K.; Ohkura, M.; Okubo, Y.; Iino, M. Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 2014, 5, 4153. [Google Scholar] [CrossRef]
- Perry, J.L.; Ramachandran, N.K.; Utama, B.; Hyser, J.M. Use of genetically-encoded calcium indicators for live cell calcium imaging and localization in virus-infected cells. Methods 2015, 90, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zeng, C.Q.-Y.; Morris, A.P.; Estes, M.K. A functional NSP4 enterotoxin peptide secreted from rotavirus-infected cells. J. Virol. 2000, 74, 11663–11670. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yu, B.; Yang, H.; Ma, T. Shikonin inhibits intestinal calcium-activated chloride channels and prevents rotaviral diarrhea. Front. Pharmacol. 2016, 7, 270. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, F.; Martin-Caraballo, M.; Hsia, S. Modulation of voltage-gated sodium channel (VGSC) activity in human dorsal root ganglion (DRG) neurons by herpesvirus quiescent infection. J. Virol. 2019, 714691. [Google Scholar] [CrossRef]
- Peischard, S.; Ho, H.T.; Theiss, C.; Strutz-Seebohm, N.; Seebohm, G. A kidnapping story: How coxsackievirus B3 and its host cell interact. Cell. Physiol. Biochem. 2019, 53, 121–140. [Google Scholar]
- Omar, S.; Clarke, R.; Abdullah, H.; Brady, C.; Corry, J.; Winter, H.; Touzelet, O.; Power, U.F.; Lundy, F.; McGarvey, L.P.; et al. Respiratory virus infection up-regulates TRPV1, TRPA1 and ASICS3 receptors on airway cells. PLoS ONE 2017, 12, e0171681. [Google Scholar] [CrossRef]
- Brand, J.D.; Lazrak, A.; Trombley, J.E.; Shei, R.J.; Adewale, A.T.; Tipper, J.L.; Yu, Z.; Ashtekar, A.R.; Rowe, S.M.; Matalon, S.; et al. Influenza-mediated reduction of lung epithelial ion channel activity leads to dysregulated pulmonary fluid homeostasis. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Stakaitytė, G.; Nwogu, N.; Lippiat, J.D.; Blair, G.E.; Poterlowicz, K.; Boyne, J.R.; Macdonald, A.; Mankouri, J.; Whitehouse, A. The cellular chloride channels CLIC1 and CLIC4 contribute to virus-mediated cell motility. J. Biol. Chem. 2018, 293, 4582–4590. [Google Scholar] [CrossRef]
- Seebohm, G.; Strutz-Seebohm, N.; Ureche, O.N.; Henrion, U.; Baltaev, R.; Mack, A.F.; Korniychuk, G.; Steinke, K.; Tapken, D.; Pfeufer, A.; et al. Long QT syndrome—Associated mutations in KCNQ1 and KCNE1 subunits disrupt normal endosomal recycling of IKs channels. Circ. Res. 2008, 103, 1451–1457. [Google Scholar] [CrossRef]
- Chen, L.; Song, W.; Davis, I.C.; Shrestha, K.; Schwiebert, E.; Sullender, W.M.; Matalon, S. Inhibition of Na+ transport in lung epithelial cells by respiratory syncytial virus infection. Am. J. Respir. Cell Mol. Biol. 2009, 40, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Kaskinen, A.; Alexandersson, A.; Andersson, S.; Saxén, H.; Peltola, V.; Kolho, K.L.; Helve, O. Decreased airway epithelial ion transport was associated with the severity of the respiratory syncytial virus infection and complications in infants. Acta Paediatr. 2020, 15311. [Google Scholar] [CrossRef] [PubMed]
- Crambert, G.; Ernandez, T.; Lamouroux, C.; Roth, I.; Dizin, E.; Martin, P.Y.; Féraille, E.; Hasler, U. Epithelial sodium channel abundance is decreased by an unfolded protein response induced by hyperosmolality. Physiol. Rep. 2014, 2. [Google Scholar] [CrossRef]
- Kennedy, P.G.; Montague, P.; Scott, F.; Grinfeld, E.; Ashrafi, G.H.; Breuer, J.; Rowan, E.G. Varicella-zoster viruses associated with post-herpetic neuralgia induce sodium current density increases in the ND7-23 Nav-1.8 Neuroblastoma cell line. PLoS ONE 2013, 8, e51570. [Google Scholar] [CrossRef]
- Tang, Z.; Chen, Z.; Tang, B.; Jiang, H. Primary erythromelalgia: A review. Orphanet J. Rare Dis. 2015, 10, 127. [Google Scholar] [CrossRef]
- Knight, L.M.; Stakaityte, G.; Jennifer, J.W.; Abdul-Sada, H.; Griffiths, D.A.; Howell, G.J.; Wheat, R.; Blair, G.E.; Steven, N.M.; Macdonald, A.; et al. Merkel cell polyomavirus small T antigen mediates microtubule destabilization to promote cell motility and migration. J. Virol. 2015, 89, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Peretti, M.; Angelini, M.; Savalli, N.; Florio, T.; Yuspa, S.H.; Mazzanti, M. Chloride channels in cancer: Focus on chloride intracellular channel 1 and 4 (CLIC1 AND CLIC4) proteins in tumor development and as novel therapeutic targets. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2523–2531. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).