Genetic Characterizations and Molecular Evolution of VP7 Gene in Human Group A Rotavirus G1

Abstract
1. Introduction
2. Materials and Methods
2.1. Strain Selection
2.2. Genetic Diversity of RVA G1 VP7 Gene
2.3. Selection Pressure Analysis
2.4. Root-to-Tip Analysis
2.5. Accumulation Pattern of Amino Acid Substitutions
2.6. Evolutionary Dynamics Analysis
3. Results
3.1. Genetic Characterizations of RVA G1 VP7 Gene
3.2. Root-to-Tip Divergence Analysis
3.3. Accumulation Pattern of Amino Acid Substitutions
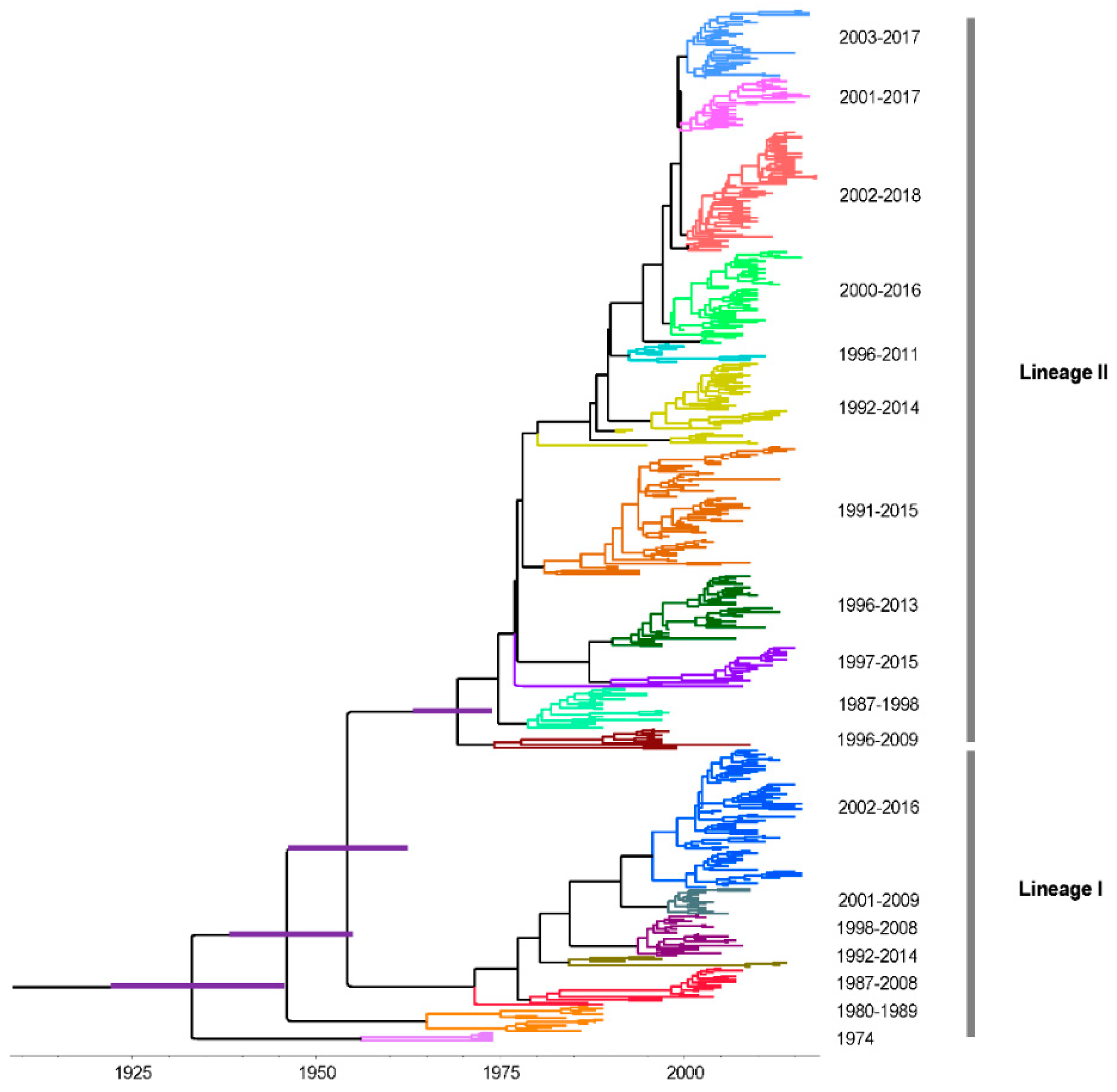
3.4. Time-Scaled Evolutionary Analysis
3.5. Phylodynamics of RVA G1 VP7 Gene
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Troeger, C.; Khalil, I.A.; Rao, P.C.; Cao, S.; Blacker, B.F.; Ahmed, T.; Armah, G.; Bines, J.E.; Brewer, T.G.; Colombara, D.V.; et al. Rotavirus vaccination and the global burden of rotavirus diarrhea among children younger than 5 Years. JAMA Pediatr. 2018, 172, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Estes, M.K.; Kapikian, Z.A. Rotaviruses, Fields Virology, 5th ed.; Lippincott Williams & Wilkens: Philadelphia, PA, USA, 2007; pp. 19171–19974. [Google Scholar]
- Prasad, B.V.V.; Wang, G.J.; Clerx, J.P.M.; Chiu, W. Three-dimensional structure of rotavirus. J. Mol. Biol. 1988, 199, 269–275. [Google Scholar] [CrossRef]
- Hoque, S.A.; Khandoker, N.; Thongprachum, A.; Khamrin, P.; Takanashi, S.; Okitsu, S.; Nishimura, S.; Kikuta, H.; Yamamoto, A.; Sugita, K.; et al. Distribution of rotavirus genotypes in Japan from 2015 to 2018: Diversity in genotypes before and after introduction of rotavirus vaccines. Vaccine 2020, 38, 3980–3986. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Hagbom, M.; Svensson, L.; Nordgren, J. The impact of human genetic polymorphisms on rotavirus susceptibility, epidemiology, and vaccine take. Viruses 2020, 12, 324. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Vaccine in national Immunization Programme Update. Available online: www.who.int/immunization/monitoring_surveillance/VaccineIntroStatus.pptx?ua=1 (accessed on 15 March 2020).
- Guillermo, M.R.P.; Irene, P.S.; F Raúl, V.; Hector, A.; Thomas, B.; SueAnn, C.C.; Brigitte, C.; Felix, E.; Paul, G.; Bruce, L.I.; et al. Safety and efficacy of an attenuated vaccine against severe rotavirus gastroenteritis. N. Engl. J. Med. 2006, 354, 11–22. [Google Scholar]
- Timo, V.; David, O.M.; Penelope, D.; Pierre, V.D.; Mathuram, S.; Zoe, R.; Michael, J.D.; Joseph, F.H.; Michelle, G.G.; Steven, B.B.; et al. Safety and efficacy of a pentavalent human-bovine (WC3) reassortant rotavirus vaccine. N. Engl. J. Med. 2006, 354, 23–33. [Google Scholar]
- Roczo-Farkas, S.; Kirkwood, C.D.; Cowley, D.; Barnes, G.L.; Bishop, R.F.; Bogdanovic-Sakran, N.; Boniface, K.; Donato, C.M.; Bines, J.E. The impact of rotavirus vaccines on genotype diversity: A comprehensive analysis of 2 decades of Australian surveillance data. J. Infect. Dis. 2018, 218, 546–554. [Google Scholar] [CrossRef]
- Luchs, A.; da Costa, A.C.; Cilli, A.; Komninakis, S.C.V.; Carmona, R.C.C.; Boen, L.; Morillo, S.G.; Sabino, E.C.; Timenetsky, M. Spread of the emerging equine-like G3P[8] DS-1-like genetic backbone rotavirus strain in Brazil and identification of potential genetic variants. J. Gen. Virol. 2019, 100, 7–25. [Google Scholar] [CrossRef]
- Yandle, Z.; Coughlan, S.; Dean, J.; Tuite, G.; Conroy, A.; De Gascun, C.F. Group A rotavirus detection and genotype distribution before and after introduction of a national immunisation programme in Ireland: 2015–2019. Pathogens 2020, 9, 449. [Google Scholar] [CrossRef]
- Damtie, D.; Melku, M.; Tessema, B.; Vlasova, A.N. Prevalence and genetic diversity of rotaviruses among under-five children in Ethiopia: A systematic review and meta-analysis. Viruses 2020, 12, 62. [Google Scholar] [CrossRef]
- Letsa, V.; Damanka, S.; Dennis, F.; Lartey, B.; Armah, G.E.; Betrapally, N.; Gautam, R.; Esona, M.D.; Bowen, M.D.; Quaye, O. Distribution of rotavirus genotypes in the postvaccine introduction era in Ashaiman, Greater Accra Region, Ghana, 20142–016. J. Med. Virol. 2019, 91, 2025–2028. [Google Scholar] [CrossRef] [PubMed]
- Nayak, M.K.; Banerjee, A.; Sarkar, R.; Mitra, S.; Dutta, K.; Ganguly, N.; Ghosh, C.; Girish Kumar, C.P.; Niyogi, P.; Panda, S.; et al. Genetic characterization of group-A rotaviruses among children in eastern India during 20142–016: Phylodynamics of co-circulating genotypes. Vaccine 2019, 37, 6842–6856. [Google Scholar] [CrossRef] [PubMed]
- Markkula, J.; Hemming-Harlo, M.; Vesikari, T. Shedding of oral pentavalent bovine-human reassortant rotavirus vaccine indicates high uptake rate of vaccine and prominence of G-type G1. Vaccine 2020, 38, 1378–1383. [Google Scholar] [CrossRef] [PubMed]
- Yuki, H.; Masaru, I.; Yu, M.; Ayumi, K.; Sayaka, Y.; Hiroyuki, H.; Ryota, S.; Masafumi, M.; Hiroki, M.; Satoshi, K.; et al. Monitoring shedding of five genotypes of RotaTeq vaccine viruses by genotype-specific real-time reverse transcription-PCR assays. J. Clin. Microbiol. 2018, 56, e00035-18. [Google Scholar]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit_a user-friendly biological sequence alignment editor and analysis program for Windows 95_98_NT. Nucleic Acids Res. Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Shih, A.C.; Lee, D.T.; Peng, C.L.; Wu, Y.W. Phylo-mLogo: An interactive and hierarchical multiple-logo visualization tool for alignment of many sequences. BMC Bioinform. 2007, 8, 63. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Zhou, L.; Wang, B. Molecular evolution of classic human astrovirus, as revealed by the analysis of the capsid protein gene. Viruses 2019, 11, 707. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Novikova, N.A.; Sashina, T.A.; Epifanova, N.V.; Kashnikov, A.U.; Morozova, O.V. Long-term monitoring of G1P[8] rotaviruses circulating without vaccine pressure in Nizhny Novgorod, Russia, 1984–2019. Arch. Virol. 2020, 165, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Doan, Y.H.; Suzuki, Y.; Nakagomi, T.; Nakagomi, O.; Katayama, K. Study of complete genome sequences of rotavirus a epidemics and evolution in Japan in 2012–2014. Front. Microbiol. 2019, 10, 38. [Google Scholar] [CrossRef]
- Santos, F.S.; Sousa Junior, E.C.; Guerra, S.F.S.; Lobo, P.S.; Penha Junior, E.T.; Lima, A.B.F.; Vinente, C.B.G.; Chagas, E.H.N.; Justino, M.C.A.; Linhares, A.C.; et al. G1P[8] Rotavirus in children with severe diarrhea in the post-vaccine introduction era in Brazil: Evidence of reassortments and structural modifications of the antigenic VP7 and VP4 regions. Infect. Genet. Evol. 2019, 69, 255–266. [Google Scholar] [CrossRef]
- Damanka, S.A.; Agbemabiese, C.A.; Dennis, F.E.; Lartey, B.L.; Adiku, T.K.; Enweronu-Laryea, C.C.; Armah, G.E. Genetic analysis of Ghanaian G1P[8] and G9P[8] rotavirus A strains reveals the impact of P[8] VP4 gene polymorphism on P-genotyping. PLoS ONE 2019, 14, e0218790. [Google Scholar] [CrossRef]
- Phan, T.G.; Khamrin, P.; Quang, T.D.; Dey, S.K.; Takanashi, S.; Okitsu, S.; Maneekarn, N.; Ushijima, H. Detection and genetic characterization of group A rotavirus strains circulating among children with acute gastroenteritis in Japan. J. Virol. 2007, 81, 4645–4653. [Google Scholar] [CrossRef]
- Dyall-Smith, M.L.; Lazdins, I.; Tregear, G.W.; Holmes, I.H. Location of the major antigenic sites involved in rotavirus serotype-specific neutralization. Proc. Natl. Acad. Sci. USA 1986, 83, 3465–3468. [Google Scholar] [CrossRef]
- Kobayashi, N.; Taniguchi, K.; Urasawa, T.; Urasawa, S. Analysis of the neutralization epitopes on human rotavirus VP7 recognized by monotype-specific monoclonal antibodies. J. Gen. Virol. 1991, 72, 1855–1861. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K.; Hoshino, Y.; Taniguchi, K.; Green, K.Y.; Greenberg, H.B.; Kapikian, A.Z.; Chanock, R.M.; Gorziglia, M. Rotavirus VP7 neutralization epitopes of serotype 3 strains. Virology 1989, 171, 503–515. [Google Scholar] [CrossRef]
- Gunn, P.R.; Sato, F.; Powell, K.F.; Bellamy, A.R.; Napier, J.R.; Harding, D.R.; Hancock, W.S.; Siegman, L.J.; Both, G.W. Rotavirus neutralizing protein VP7: Antigenic determinants investigated by sequence analysis and peptide synthesis. J. Virol. 1985, 54, 791–797. [Google Scholar] [CrossRef]
- Taniguchi, K.; Hoshino, Y.; Nishikawa, K.; Green, K.Y.; Maloy, W.L.; Morita, Y.; Urasawa, S.; Kapikian, A.Z.; Chanock, R.M.; Gorziglia, M. Cross-reactive and serotype-specific neutralization epitopes on VP7 of human rotavirus: Nucleotide sequence analysis of antigenic mutants selected with monoclonal antibodies. J. Virol. 1988, 62, 1870–1874. [Google Scholar] [CrossRef]
- Coulson, B.C.; Kirkwood, C. Relation of VP7 amino acid sequence to monoclonal antibody neutralization of rotavirus and rotavirus monotype. J. Virol. 1991, 65, 5968–5974. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.Y.; Settembre, E.C.; Trask, S.D.; Greenberg, H.B.; Harrison, S.C.; Dormitzer, P.R. Structure of rotavirus outer-layer protein VP7 bound with a neutralizing Fab. Science 2009, 324, 1444–1447. [Google Scholar] [CrossRef]
- Parra, G.I.; Squires, R.B.; Karangwa, C.K.; Johnson, J.A.; Lepore, C.J.; Sosnovtsev, S.V.; Green, K.Y. Static and evolving norovirus genotypes: Implications for epidemiology and immunity. PLoS Pathog. 2017, 13, e1006136. [Google Scholar] [CrossRef]
- Dennis, A.F.; McDonald, S.M.; Payne, D.C.; Mijatovic-Rustempasic, S.; Esona, M.D.; Edwards, K.M.; Chappell, J.D.; Patton, J.T. Molecular epidemiology of contemporary G2P[4] human rotaviruses cocirculating in a single U.S. community: Footprints of a globally transitioning genotype. J. Virol. 2014, 88, 3789–3801. [Google Scholar] [CrossRef]
- Magagula, N.B.; Esona, M.D.; Nyaga, M.M.; Stucker, K.M.; Halpin, R.A.; Stockwell, T.B.; Seheri, M.L.; Steele, A.D.; Wentworth, D.E.; Mphahlele, M.J. Whole genome analyses of G1P[8] rotavirus strains from vaccinated and non-vaccinated South African children presenting with diarrhea. J. Med. Virol. 2015, 87, 79–101. [Google Scholar] [CrossRef]
- McDonald, S.M.; Matthijnssens, J.; McAllen, J.K.; Hine, E.; Overton, L.; Wang, S.; Lemey, P.; Zeller, M.; Van Ranst, M.; Spiro, D.J.; et al. Evolutionary dynamics of human rotaviruses: Balancing reassortment with preferred genome constellations. PLoS Pathog. 2009, 5, e1000634. [Google Scholar] [CrossRef]
- Zhang, S.; McDonald, P.W.; Thompson, T.A.; Dennis, A.F.; Akopov, A.; Kirkness, E.F.; Patton, J.F.; McDonald, S.M. Analysis of human rotaviruses from a single location over an 18-year time span suggests that protein coadaption influences gene constellations. J. Virol. 2014, 88, 9842–9863. [Google Scholar] [CrossRef] [PubMed]
- Zeller, M.; Donato, C.; Trovao, N.S.; Cowley, D.; Heylen, E.; Donker, N.C.; McAllen, J.K.; Akopov, A.; Kirkness, E.F.; Lemey, P.; et al. Genome-wide evolutionary analyses of G1P[8] strains isolated before and after rotavirus vaccine introduction. Genome Biol. Evol. 2015, 7, 2473–2483. [Google Scholar] [CrossRef]
- Afrad, M.H.; Matthijnssens, J.; Afroz, S.F.; Rudra, P.; Nahar, L.; Rahman, R.; Hossain, M.E.; Rahman, S.R.; Azim, T.; Rahman, M. Differences in lineage replacement dynamics of G1 and G2 rotavirus strains versus G9 strain over a period of 22 years in Bangladesh. Infect. Genet. Evol. 2014, 28, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 7–1a | 7–1b | 7–2 | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 87 | 91 | 94 | 96 | 97 | 98 | 99 | 100 | 104 | 123 | 125 | 129 | 130 | 291 | 201 | 211 | 212 | 213 | 238 | 242 | 145 | 146 | 147 | 148 | 190 | 217 | 221 | 264 | ||
| RotaRix | T | T | N | G | E | W | K | D | Q | S | V | V | D | K | Q | N | V | D | N | T | D | Q | N | L | S | M | N | G | |
| RotaTeq | T | T | N | G | D | W | K | D | Q | S | V | V | D | K | Q | N | V | D | N | T | D | Q | S | L | S | M | N | G | |
| Lineage I | T | T | N | G | E | W | K | D | Q | S | V | V | D | K | Q | N | V | D | N | T | D | Q | N | L | S | M | N | G | |
| Lineage II | T | T | S | G | E | W | K | D | Q | N | V | V | D | R | Q | N | V | D | N | T | D | Q | N | L | S | T | N | G | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, N.; Zhou, L.; Wang, B. Genetic Characterizations and Molecular Evolution of VP7 Gene in Human Group A Rotavirus G1. Viruses 2020, 12, 831. https://doi.org/10.3390/v12080831
Zhou N, Zhou L, Wang B. Genetic Characterizations and Molecular Evolution of VP7 Gene in Human Group A Rotavirus G1. Viruses. 2020; 12(8):831. https://doi.org/10.3390/v12080831
Chicago/Turabian StyleZhou, Nan, Lu Zhou, and Bei Wang. 2020. "Genetic Characterizations and Molecular Evolution of VP7 Gene in Human Group A Rotavirus G1" Viruses 12, no. 8: 831. https://doi.org/10.3390/v12080831
APA StyleZhou, N., Zhou, L., & Wang, B. (2020). Genetic Characterizations and Molecular Evolution of VP7 Gene in Human Group A Rotavirus G1. Viruses, 12(8), 831. https://doi.org/10.3390/v12080831