Viral Metagenomic Content Reflects Seawater Ecological Quality in the Coastal Zone

 , ,
, ,  ,
,  , ,
, , 
Abstract
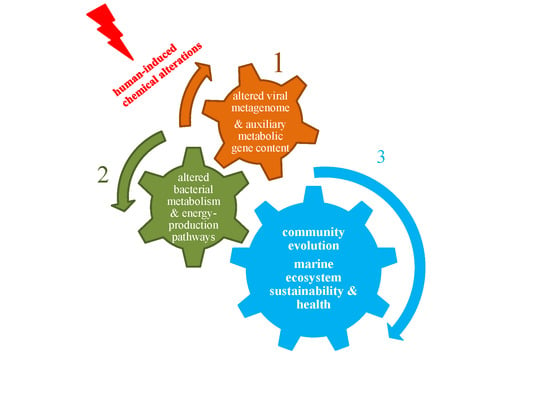
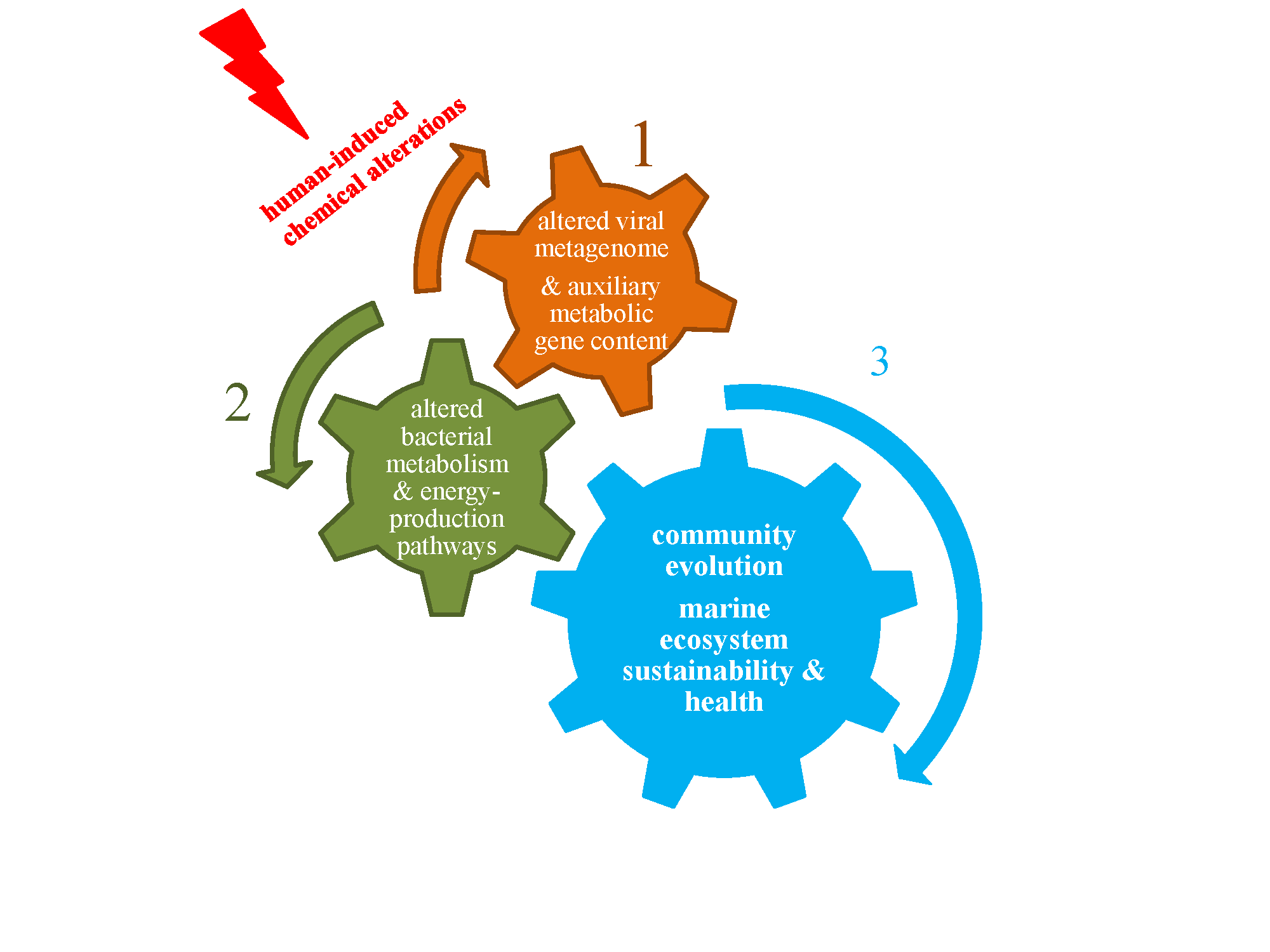
1. Introduction
2. Materials and Methods
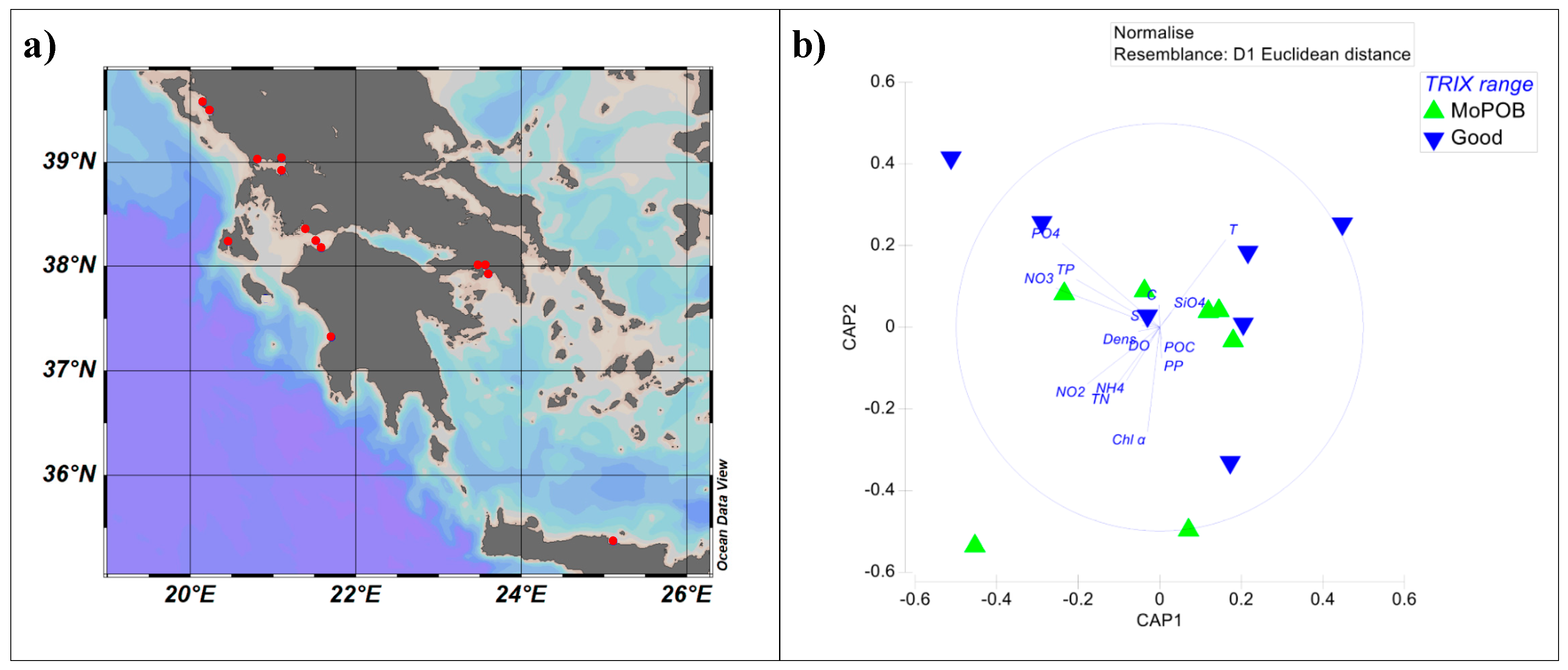
2.1. Sampling Design and Sample-Grouping Substantiation
2.2. Determination of Physicochemical and Biological Parameters
2.3. DNA Extraction, Amplification, Sequencing, and Sequence Processing
2.4. Statistics
3. Results
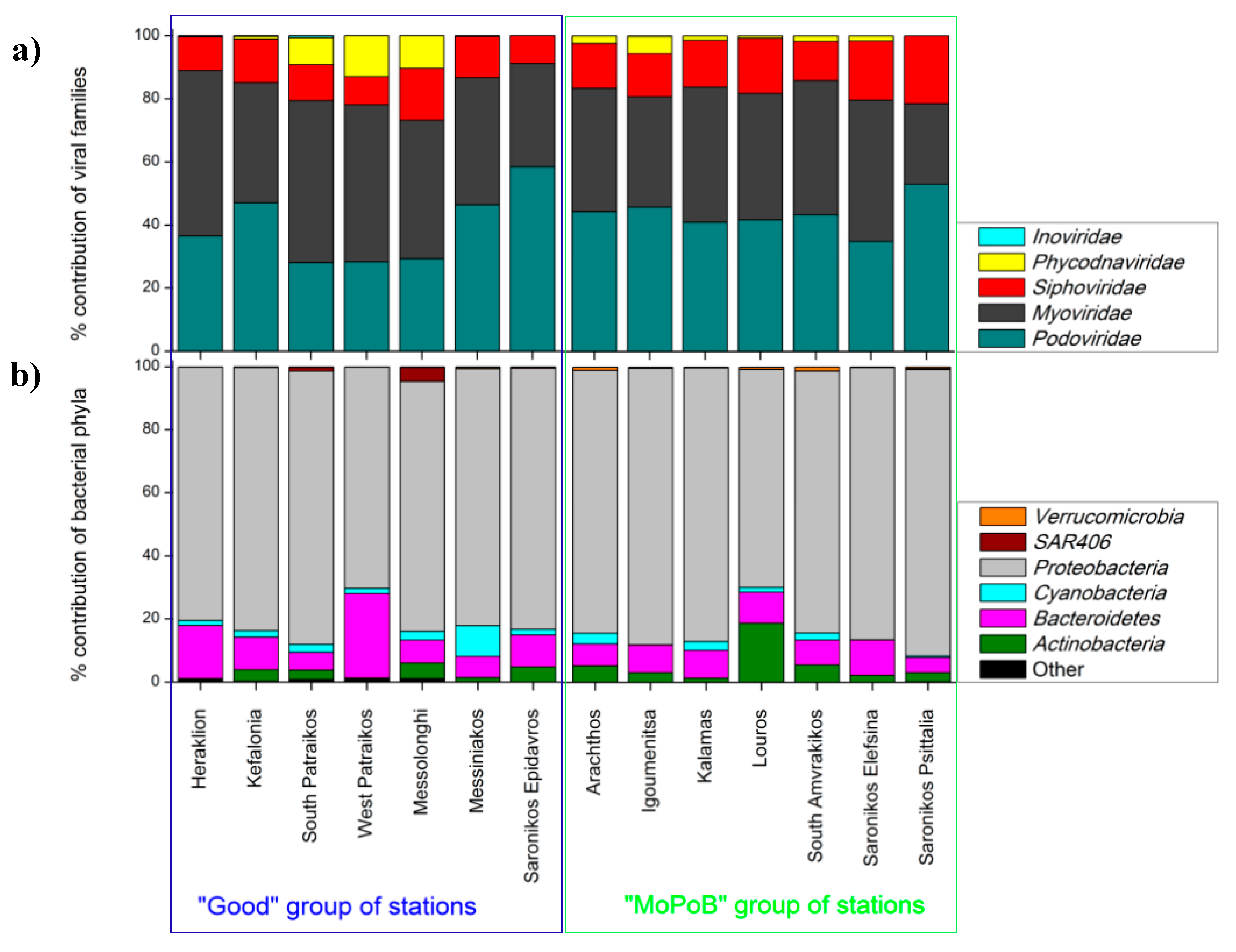
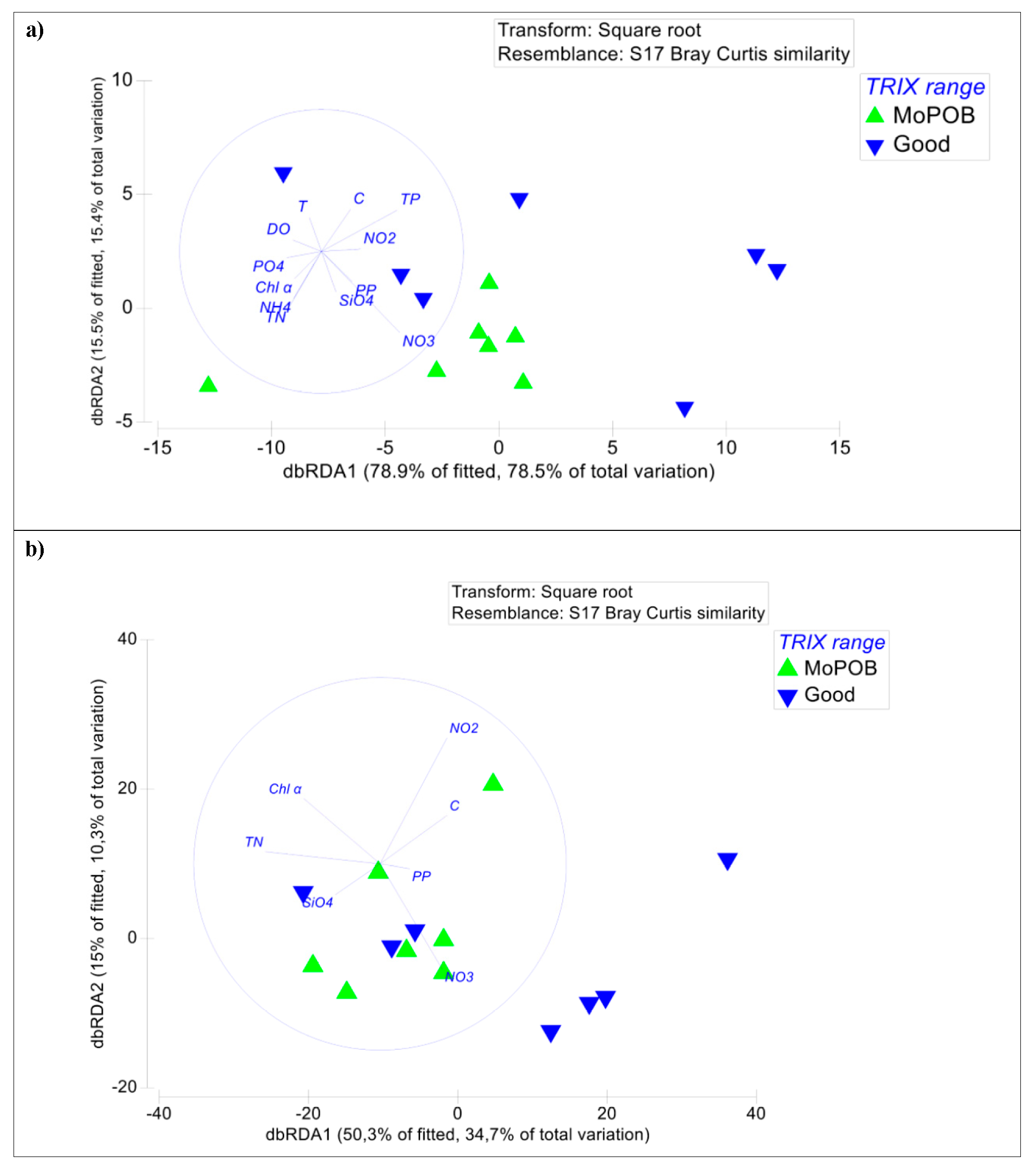
3.1. Viral Community Composition Does Not Differ Based on Ecological Quality (TRIX Range)
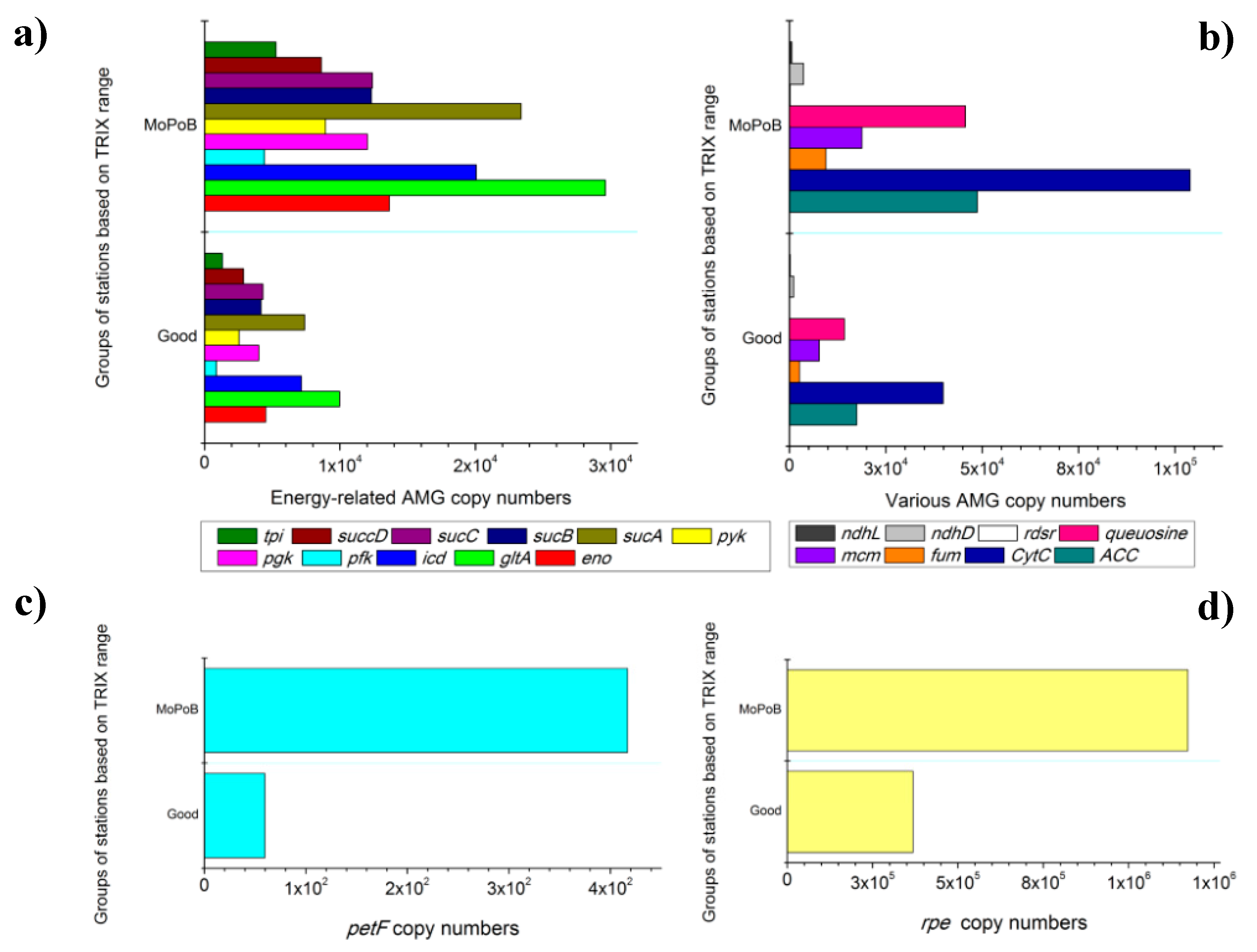
3.2. AMG Diversity Differs Based on Ecological Quality (TRIX Range)
3.3. Ecosystem Function Based on Viral and Bacterial Community Composition versus Virome Content
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nogales, B.; Lanfranconi, M.P.; Piña-Villalonga, J.M.; Bosch, R. Anthropogenic perturbations in marine microbial communities. FEMS Microbiol. Rev. 2011, 35, 275–298. [Google Scholar] [CrossRef]
- Halpern, B.S.; Frazier, M.; Potapenko, J.; Casey, K.S.; Koenig, K.; Longo, C.; Lowndes, J.S.; Rockwood, R.C.; Selig, E.R.; Selkoe, K.A.; et al. Spatial and temporal changes in cumulative human impacts on the world’s ocean. Nat. Commun. 2015, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Quero, G.M.; Cassin, D.; Botter, M.; Perini, L.; Luna, G.M. Patterns of benthic bacterial diversity in coastal areas contaminated by heavy metals, polycyclic aromatic hydrocarbons (PAHs) and polychlorinated biphenyls (PCBs). Front. Microbiol. 2015, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Borja, A. Testing the efficiency of a bacterial community-based index (microgAMBI) to assess distinct impact sources in six locations around the world. Ecol. Indic. 2018, 85, 594–602. [Google Scholar] [CrossRef]
- Sun, M.Y.; Dafforn, K.A.; Brown, M.V.; Johnston, E.L. Bacterial communities are sensitive indicators of contaminant stress. Mar. Pollut. Bull. 2012, 64, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Ager, D.; Evans, S.; Li, H.; Lilley, A.K.; Gast, C.J. Van Der Anthropogenic disturbance affects the structure of bacterial communities. Environ. Microbiol. 2010, 12, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Falkowski, P.G.; Fenchel, T.; Delong, E.F. The microbial engines that drive earth’s biogeochemical cycles. Science (80-.) 2008, 320, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Warwick-Dugdale, J.; Buchholz, H.H.; Allen, M.J.; Temperton, B. Host-hijacking and planktonic piracy: How phages command the microbial high seas. Virol. J. 2019, 16. [Google Scholar] [CrossRef]
- Cantalupo, P.G.; Calgua, B.; Zhao, G.; Hundesa, A.; Wier, A.D.; Katz, J.P.; Grabe, M.; Hendrix, R.W.; Girones, R.; Wang, D.; et al. Raw Sewage Harbors Diverse Viral Populations. MBio 2011, 2, 1–11. [Google Scholar] [CrossRef]
- Ng, T.F.F.; Marine, R.; Wang, C.; Simmonds, P.; Kapusinszky, B.; Bodhidatta, L.; Oderinde, B.S.; Wommack, K.E.; Delwart, E.; Phyloge-, N. High Variety of Known and New RNA and DNA Viruses of Diverse Origins in Untreated Sewage. J. Virol. 2012, 86, 12161–12175. [Google Scholar] [CrossRef]
- Suttle, C. A Marine viruses--major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Keshri, J.; Sriram, A.; Ram, P.; Colombet, J.; Perriere, F. Differential impact of lytic viruses on the taxonomical resolution of freshwater bacterioplankton community structure. Water Res. 2017, 124, 129–138. [Google Scholar] [PubMed]
- Koskella, B.; Brockhurst, M.A. Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Ecol. 2014, 38, 916–931. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Sullivan, M.B. Rising to the challenge: Accelerated pace of discovery transforms marine virology. Nat. Rev. Microbiol. 2015, 13, 147–159. [Google Scholar] [CrossRef]
- Hurwitz, B.L.; U’Ren, J.M. Viral metabolic reprogramming in marine ecosystems. Curr. Opin. Microbiol. 2016, 31, 161–168. [Google Scholar] [CrossRef]
- Roitman, S.; Hornung, E.; Flores-Uribe, J.; Sharon, I.; Feussner, I.; Beja, O.; Biology, F.; Israel, T. Cyanophage-encoded lipid desaturases: Oceanic distribution, diversity and function. ISME J. 2018, 12, 343–355. [Google Scholar] [CrossRef]
- Micheli, F.; Levin, N.; Giakoumi, S.; Katsanevakis, S.; Abdulla, A.; Coll, M.; Fraschetti, S.; Kark, S.; Koutsoubas, D.; Mackelworth, P.; et al. Setting Priorities for Regional Conservation Planning in the Mediterranean Sea. PLoS ONE 2013, 8, e59038. [Google Scholar] [CrossRef]
- Stock, A.; Crowder, L.B.; Halpern, B.S.; Micheli, F. Uncertainty analysis and robust areas of high and low modeled human impact on the global oceans. Conserv. Biol. 2018, 32, 1368–1379. [Google Scholar] [CrossRef]
- Piante, C.; Ody, D. Blue Growth in the Mediterranean Sea: The Challenge of Good Environmental Status; MedTrends Project; WWF-France: Le Pre-Saint-Gervais, France, 2015. [Google Scholar]
- Simboura, N.; Pavlidou, A.; Bald, J.; Tsapakis, M.; Pagou, K.; Zeri, C.; Androni, A.; Panayotidis, P. Response of ecological indices to nutrient and chemical contaminant stress factors in Eastern Mediterranean coastal waters. Ecol. Indic. 2016, 70, 89–105. [Google Scholar] [CrossRef]
- Simboura, N.; Maragou, P.; Paximadis, G.; Kapiris, K.; Papadopoulos, V.; Sakellariou, D.; Pavlidou, A.; Hatzianestis, I.; Salomidi, M.; Arvanitidis, C.; et al. Ch. 9. Greece. In World Seas: An Environmental Evaluation Volume I: Europe, The Americas and West Africa, 2nd ed.; Sheppard, C., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 227–256. [Google Scholar]
- Borja, A.; Prins, T.C.; Simboura, N.; Andersen, J.H.; Berg, T.; Marques, J.; Neto, J.M.; Papadopoulou, N.; Reker, J.; Teixeira, H. Tales from a thousand and one ways to integrate marine ecosystem components when assessing the environmenta l status. Front. Mar. Sci. 2014, 1. [Google Scholar] [CrossRef]
- Primpas, I.; Karydis, M. Scaling the trophic index (TRIX) in oligotrophic marine environments. Environ. Monit. Assess. 2011, 178, 257–269. [Google Scholar] [CrossRef] [PubMed]
- John, S.G.; Mendez, C.B.; Deng, L.; Poulos, B.; Kauffman, A.K.M.; Kern, S.; Brum, J.; Polz, M.F.; Boyle, E.A.; Sullivan, M.B. A simple and efficient method for concentration of ocean viruses by chemical flocculation. Environ. Microbiol. Rep. 2011, 3, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Holm-Hansen, O.; Lorenzen, C.; Holmes, R.; Strickland, J. Fluorometric determination of chlorophyll. CIES J. Mar. Sci. 1965, 30, 3–15. [Google Scholar] [CrossRef]
- Brussaard, C.P.D. Optimization of Procedures for Counting Viruses by Flow Cytometry. Appl. Environ. Microbiol. 2004, 70, 1506–1513. [Google Scholar] [CrossRef]
- Marie, D.; Partensky, F.; Jacquet, S.; Vaulot, D. Enumeration and cell cycle analysis of natural populations of marine picoplankton by flow cytometry using the nucleic acid stain SYBR Green I. Appl. Environ. Microbiol. 1997, 63, 186–193. [Google Scholar] [CrossRef]
- Tsiola, A.; Pitta, P.; Giannakourou, A.; Bourdin, G.; Marro, S.; Maugendre, L.; Pedrotti, M.L.; Gazeau, F. Ocean acidification and viral replication cycles: Frequency of lytically infected and lysogenic cells during a mesocosm experiment in the NW Mediterranean Sea. Estuar. Coast. Shelf Sci. 2017, 186, 139–151. [Google Scholar] [CrossRef]
- Winnepenninckx, B.T.; Backeljau, T.; Wachter, R. De Extraction of high molecular weight DNA from molluscs. Trends Genet. 1993, 9, 407. [Google Scholar]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Apprill, A.; Mcnally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef]
- Tsiola, A.; Toncelli, C.; Fodelianakis, S.; Michoud, G.; Bucheli, T.D.; Gavriilidou, A.; Kagiorgi, M.; Kalantzi, I.; Knauer, K.; Kotoulas, G.; et al. Low-dose addition of silver nanoparticles stresses marine plankton communities. Environ. Sci. Nano 2018, 5, 1965–1980. [Google Scholar] [CrossRef]
- Bolduc, B.; Youens-Clark, K.; Roux, S.; Hurwitz, B.L.; Sullivan, M.B. iVirus: Facilitating new insights in viral ecology with software and community data sets imbedded in a cyberinfrastructure. ISME J. 2017, 11, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.A.; Vaughn, M.; McKay, S.; Lyons, E.; Stapleton, A.E.; Gessler, D.; Matasci, N.; Wang, L.; Hanlon, M.; Lenards, A.; et al. The iPlant Collaborative: Cyberinfrastructure for Plant Biology. Front. Plant. Sci. 2011, 2, 1–16. [Google Scholar] [CrossRef]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A novel k -mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 2017, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Gregory, A.C.; Zayed, A.A.; Sunagawa, S.; Wincker, P.; Sullivan, M.B.; Ferland, J.; Kandels, S.; Liu, Y.; Marec, C.; Vik, D. Marine DNA Viral Macro- and Microdiversity from Pole to Pole Article Marine DNA Viral Macro- and Microdiversity from Pole to Pole. Cell 2019, 177, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, B.; Jang, H.B.; Doulcier, G.; You, Z.; Roux, S.; Sullivan, M.B. vConTACT: An iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ 2017, 5, e3243. [Google Scholar] [CrossRef] [PubMed]
- Clarke, K.R.; Warwick, R.M. Change in Marine Communities: An Approach to Statistical Analysis and Interpretation; Primer-E Ltd.: Plymouth, UK, 1994. [Google Scholar]
- Pielou, E. An Introduction to Mathematical Ecology; Wiley: Interscience, NY, USA, 1971. [Google Scholar]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef]
- Clarke, K.R.; Ainsworth, M. A method of linking multivaritate community structure to environmental variable. Mar. Ecol. Prog. Ser. 1993, 92, 205–219. [Google Scholar] [CrossRef]
- Anderson, M.J.; Gorley, R.N.; Clarke, K.R. PERMANOVA + for PRIMER: Guide to Software and Statistical Methods; PRIMER-E: Plymouth, UK, 2008. [Google Scholar]
- Somerfield, P.J. Identification of the Bray-Curtis similarity index: Comment on Yoshioka (2008). Mar. Ecol. Prog. Ser. 2008, 372, 303–306. [Google Scholar] [CrossRef]
- Ruiz-Perez, C.A.; Tsementzi, D.; Hatt, J.K.; Sullivan, M.B.; Konstantinidis, K.T. Prevalence of viral photosynthesis genes along a freshwater to saltwater transect in Southeast USA. Environ. Microbiol. Rep. Rep. 2019, 11, 672–689. [Google Scholar] [CrossRef] [PubMed]
- Yooseph, S.; Sutton, G.; Rusch, D.B.; Halpern, A.L.; Williamson, S.J.; Remington, K.; Eisen, J.A.; Heidelberg, K.B.; Manning, G.; Li, W.; et al. The Sorcerer II global ocean sampling expedition: Expanding the universe of protein families. PLoS Biol. 2007, 5, 0432–0466. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.J.; Rusch, D.B.; Yooseph, S.; Halpern, A.L.; Heidelberg, K.B.; Glass, J.I.; Pfannkoch, C.A.; Fadrosh, D.; Miller, C.S.; Sutton, G.; et al. The Corcerer II global ocean sampling expedition: Metagenomic characterization of viruses within aquatic microbial samples. PLoS ONE 2008, 3, e1456. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.B.; Larsen, A.; Thyrhaug, R.; Bratbak, G.; Sandaa, R. A Response of marine viral populations to a nutrient induced phytoplankton bloom at different pCO2 levels. Biogeosciences 2008, 5, 523–533. [Google Scholar] [CrossRef]
- Weynberg, K.D.; Allen, M.J.; Ashelford, K.; Scanlan, D.J.; Wilson, W.H. From small hosts come big viruses: The complete genome of a second Ostreococcus tauri virus, OtV-1. Environ. Microbiol. 2009, 11, 2821–2839. [Google Scholar] [CrossRef]
- Roux, S.; Krupovic, M.; Daly, R.A.; Borges, A.L.; Nayfach, S.; Schulz, F.; Sharrar, A.; Carnevali, P.B.M.; Cheng, J.; Ivanova, N.N.; et al. Cryptic inoviruses revealed as pervasive in bacteria and archaea across Earth’s biomes. Nat. Microbiol. 2019, 4, 1895–1906. [Google Scholar] [CrossRef]
- McMinn, B.R.; Ashbolt, N.J.; Korajkic, A. Bacteriophages as indicators of fecal pollution and enteric virus removal. Lett. Appl. Microbiol. 2018, 65, 11–26. [Google Scholar] [CrossRef]
- Brum, J.R.; Ignacio-espinoza, J.C.; Roux, S.; Doulcier, G.; Acinas, S.G.; Alberti, A.; Chaffron, S.; Cruaud, C.; de Vargas, C.; Gasol, J.M.; et al. Patterns and ecological drivers of ocean viral communities. Science (80-.) 2015, 348, 1261498. [Google Scholar] [CrossRef]
- Pitta, P.; Tsapakis, M.; Apostolaki, E.; Tsagaraki, T.; Holmer, M.; Karakassis, I. ‘Ghost nutrients’ from fish farms are transferred up the food web by phytoplankton grazers. Mar. Ecol. Prog. Ser. 2009, 374, 1–6. [Google Scholar] [CrossRef]
- Tsagaraki, T.M.; Pitta, P.; Frangoulis, C.; Petihakis, G.; Karakassis, I. Plankton response to nutrient enrichment is maximized at intermediate distances from fish farms. Mar. Ecol. Prog. Ser. 2013, 493, 31–42. [Google Scholar] [CrossRef][Green Version]
- Coutinho, F.H.; Silveira, C.B.; Gregoracci, G.B.; Thompson, C.C.; Edwards, R.A.; Brussaard, C.P.D.; Dutilh, B.E.; Thompson, F.L. Marine viruses discovered via metagenomics shed light on viral strategies throughout the oceans. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dann, L.M.; Rosales, S.; Mckerral, J.; Paterson, J.S.; Smith, R.J.; Jeffries, T.C.; Oliver, R.L.; Mitchell, J.G. Marine and giant viruses as indicators of a marine microbial community in a riverine system. Microbiologyopen 2016, 5, 1071–1084. [Google Scholar] [CrossRef] [PubMed]
- Vellend, M.; Lajoie, G.; Bourret, A.; Murria, C.; Kembel, S.W.; Garant, D. Drawing ecological inferences from coincident patterns of population- and community-level biodiversity. Mol. Ecol. 2014, 23, 2890–2901. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.Z.; Mccrow, J.P.; Ininbergs, K.; Dupont, C.L.; Badger, J.H.; Hoffman, J.M.; Ekman, M.; Allen, A.E.; Bergman, B.; Craig, J. The Baltic Sea Virome: Diversity and transcriptional activity of DNA and RNA viruses. mSystems 2017, 2, 1–16. [Google Scholar]
- Castelán-Sánchez, H.G.; Lopéz-Rosas, I.; García-Suastegui, W.A.; Peralta, R.; Dobson, A.D.W.; Batista-García, R.A.; Dávila-ramos, S. Extremophile deep-sea viral communities from hydrothermal vents: Structural and functional analysis. Mar. Genomics 2019, 46, 16–28. [Google Scholar] [CrossRef]
- Thompson, L.R.; Zeng, Q.; Kelly, L.; Huang, K.H.; Singer, A.U.; Stubbe, J.; Chisholm, S.W. Phage auxiliary metabolic genes and the redirection of cyanobacterial host carbon metabolism. Proc. Natl. Acad. Sci. USA 2011, 108, E757–E764. [Google Scholar] [CrossRef]
- Hurwitz, B.L.; Hallam, S.J.; Sullivan, M.B. Metabolic reprogramming by viruses in the sunlit and dark ocean. Genome Biol. 2013, 14, R123. [Google Scholar] [CrossRef]
- Puxty, R.J.; Millard, A.D.; Evans, D.J.; Scanlan, D.J. Shedding new light on viral photosynthesis. Photosynth. Res. 2015, 126, 71–97. [Google Scholar] [CrossRef]
- Sullivan, M.B.; Huang, K.H.; Ignacio-Espinoza, J.C.; Berlin, A.M.; Kelly, L.; Weigele, P.R.; DeFrancesco, A.S.; Kern, S.E.; Thompson, L.R.; Young, S.; et al. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ. Microbiol. 2010, 12, 3035–3056. [Google Scholar] [CrossRef]
- Gao, E.; Huang, Y.; Ning, D. Metabolic Genes within Cyanophage Genomes: Implications for diversity and evolution. Genes 2016, 7. [Google Scholar] [CrossRef]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 2016, 537, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.; Saez, J.M.; Davila Costa, J.S.; Colin, V.L.; Fuentes, M.S.; Cuozzo, S.A.; Benimeli, C.S.; Polti, M.A.; Amoroso, M.J. Actinobacteria: Current research and perspectives for bioremediation of pesticides and heavy metals. Chemosphere 2017, 166, 41–62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Tsementzi, D.; Hatt, J.K.; Bivins, A.; Khelurkar, N.; Brown, J. Intensive allochthonous inputs along the Ganges River and their effect on microbial community composition and dynamics. Environ. Microbiol. 2019, 21, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.G.; Luo, Y.R.; Huang, L.F. Dynamics of size-fractionated bacterial communities during the coastal dispersal of treated municipal effluents. Appl. Microbiol. Biotechnol. 2016, 100, 5839–5848. [Google Scholar] [CrossRef]
- Bellas, C.M.; Anesio, A.M.; Barker, G. Analysis of virus genomes from glacial environments reveals novel virus groups with unusual host interactions. Front. Microbiol. 2015, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hampson, D.J.; Ahmed, N. Spirochaetes as intestinal pathogens: Lessons from a Brachyspira genome. Gut Pathog. 2009, 3, 1–3. [Google Scholar] [CrossRef]
- Eggers, C.H.; Caimano, M.J.; Malizia, R.A.; Kariu, T.; Cusack, B.; Desrosiers, D.C.; Hazlett, K.R.O.; Claiborne, A.; Pal, U.; Radolf, J.D. The coenzyme A disulphide reductase of Borrelia burgdorferi is important for rapid growth throughout the enzootic cycle and essential for infection of the mammalian host. Mol. Microbiol. 2011, 82, 679–697. [Google Scholar] [CrossRef]
- Jumas-Bilak, E.; Marchandin, H. The Phylum Synergistetes. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 931–954. [Google Scholar]
- Nõges, P.; Argillier, C.; Borja, Á.; Mikel, J.; Kode, V.; Pletterbauer, F.; Sagouis, A.; Birk, S. Quantified biotic and abiotic responses to multiple stress in freshwater, marine and ground waters. Sci. Total Environ. 2016, 540, 43–52. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Station ID | T | C | S | Dens | DO | DO Dev% | SiO4 | PO4 | NO3 | NO2 | NH4 | TN | TP | PP | POC | Chl α | Surface-Waters March TRIX | Surface-Waters Annual TRIX | Depth-Integrated Annual TRIX |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Heraklion | 17.49 | 49.91 | 38.94 | 28.80 | 5.30 | 0.14 | 0.99 | 2.85 | 3.26 | 0.48 | 5.34 | 0.00 | 0.00 | 0.06 | 1.74 | 0.24 | 1.19 (H) | 2.22 (GOOD) | 1.75 (G) |
| Kefalonia | 14.18 | 45.33 | 38.01 | 28.47 | 5.78 | 1.75 | 1.56 | 2.25 | 9.67 | 1.56 | 3.29 | 7.73 | 0.23 | 0.09 | 11.58 | 0.94 | 2.69 (G) | 2.62 (GOOD) | 2.87 (M) |
| South Patraikos | 14.76 | 46.08 | 38.15 | 28.46 | 5.88 | 4.87 | 4.87 | 13.25 | 350.11 | 8.65 | 15.96 | 8.00 | 0.80 | 0.09 | 14.00 | 0.43 | 4.60 (P) | 2.12 (GOOD) | 2.45 (G) |
| West Patraikos | 14.18 | 45.78 | 38.44 | 28.81 | 5.88 | 3.75 | 4.97 | 8.30 | 57.47 | 7.87 | 3.29 | 6.38 | 0.54 | 0.06 | 10.83 | 0.33 | 3.63 (M) | 2.58 (GOOD) | 3.02 (M) |
| Messolonghi | 14.05 | 45.09 | 37.91 | 28.43 | 5.79 | 1.50 | 3.69 | 2.25 | 48.79 | 7.91 | 3.11 | 6.40 | 0.20 | 0.04 | 7.02 | 0.33 | 2.77 (G) | 2.21 (GOOD) | 2.06 (G) |
| Messiniakos | 16.37 | 48.07 | 38.39 | 28.28 | 5.45 | 0.35 | 1.49 | 3.69 | 13.21 | 3.17 | 10.77 | 4.49 | 0.10 | 0.01 | 5.52 | 0.21 | 1.97 (G) | 2.19 (GOOD) | 2..16 (G) |
| Saronikos Epidavros | 14.73 | 46.50 | 38.56 | 28.78 | 5.98 | 6.73 | 0.56 | 2.80 | 0.74 | 2.35 | 0.92 | 6.09 | 0.16 | 0.05 | 11.39 | 0.40 | 2.48 (G) | 2.14 (GOOD) | 5.32 (B) |
| Arachthos | 14.78 | 36.31 | 29.23 | 21.57 | 6.65 | 11.74 | 21.62 | 4.19 | 6.70 | 2.25 | 0.92 | 10.53 | 0.45 | 0.15 | 34.65 | 0.12 | 2.73 (G) | 3.73 (MODERATE) | 4.47 (P) |
| Igoumenitsa | 13.81 | 44.22 | 37.32 | 28.02 | 5.73 | 0.27 | 3.02 | 1.71 | 49.48 | 2.67 | 8.28 | 8.75 | 0.30 | 0.12 | 15.44 | 1.10 | 2.5 (G) | 3.83 (MODERATE) | 3.82 (M) |
| Kalamas | 15.51 | 46.24 | 37.56 | 27.83 | 5.61 | 4.99 | 4.24 | 1.64 | 39.18 | 5.29 | 2.87 | 5.41 | 0.17 | 0.04 | 8.99 | 0.52 | 2.88 (M) | 3.25 (MODERATE) | 3.45 (M) |
| Louros | 15.23 | 33.83 | 26.72 | 19.55 | 6.79 | 12.80 | 23.28 | 7.26 | 45.20 | 3.36 | 0.92 | 16.40 | 0.62 | 0.18 | 53.56 | 0.06 | 3.30 (M) | 4.46 (POOR) | 4.39 (P) |
| South Amvrakikos | 14.47 | 34.73 | 28.03 | 20.71 | 6.68 | 10.82 | 24.87 | 3.17 | 1.12 | 2.07 | 0.92 | 10.87 | 0.45 | 0.13 | 50.73 | 0.17 | 2.41 (G) | 3.15 (MODERATE) | 5.77 (B) |
| Saronikos Elefsina | 12.72 | 43.44 | 37.66 | 28.51 | 5.98 | 2.00 | 1.79 | 5.56 | 281.17 | 7.54 | 16.79 | 16.93 | 0.29 | 0.07 | 17.28 | 0.64 | 4.03 (P) | 4.22 (POOR) | 3.67 (M) |
| Saronikos Psittalia | 14.33 | 46.13 | 38.61 | 28.90 | 5.51 | 2.38 | 1.34 | 11.61 | 44.21 | 5.34 | 11.52 | 6.85 | 0.32 | 0.05 | 9.31 | 0.64 | 3.78 (M) | 3.52 (MODERATE) | 3.62 (M) |
| Sample ID | Lytic Contigs | % Assigned Viral | AB | HB | VLP | TB | VBR | %LDNA V | %MDNA V | %HDNA V | %LDNA B | %HDNA B | S-W Bacteria | S-W Viruses | Total AMGs | S-W AMGs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Heraklion | 638 | 8.0% | 3 × 104 | 9 × 105 | 9 × 106 | 9 × 105 | 10.69 | 72.8% | 23.4% | 3.7% | 44.6% | 55.4% | 5.92 | 1.67 | 1,493,947 | 5.11 |
| Kefalonia | 700 | 25.1% | 3 × 104 | 9 × 105 | 2 × 107 | 1 × 106 | 16.79 | 78.9% | 17.1% | 4.0% | 58.2% | 41.8% | 4.82 | 1.87 | 636,782 | 5.15 |
| South Patraikos | 185 | 9.9% | 2 × 104 | 5 × 105 | 9 × 106 | 5 × 105 | 17.65 | 62.8% | 29.5% | 7.7% | 52.7% | 47.3% | 6.08 | 2.07 | 456,142 | 5.06 |
| West Patraikos | 261 | 9.1% | 1 × 104 | 4 × 105 | 8 × 106 | 4 × 105 | 17.07 | 63.5% | 27.4% | 9.1% | 60.4% | 39.6% | 5.82 | 1.92 | 395,112 | 5.04 |
| Messolonghi | 116 | 17.5% | 1 × 104 | 4 × 105 | 1 × 107 | 4 × 105 | 32.26 | 75.2% | 18.9% | 5.9% | 62.0% | 38.0% | 6.03 | 1.95 | 164,837 | 5.12 |
| Messiniakos | 521 | 31.5% | 5 × 104 | 2 × 106 | 1 × 107 | 2 × 106 | 5.04 | 82.0% | 15.6% | 2.4% | 68.3% | 31.7% | 4.76 | 1.67 | ||
| Saronikos Epidavros | 125 | 28.1% | 2 × 104 | 8 × 105 | 1 × 107 | 9 × 105 | 16.10 | 68.7% | 26.9% | 4.4% | 54.8% | 45.2% | 4.56 | 1.49 | 325,742 | 5.16 |
| Arachthos | 805 | 16.8% | 1 × 105 | 4 × 105 | 6 × 107 | 5 × 105 | 111.33 | 67.1% | 26.9% | 6.0% | 35.0% | 65.0% | 4.82 | 1.83 | 1,319,746 | 5.21 |
| Igoumenitsa | 1196 | 16.0% | 1 × 104 | 2 × 106 | 2 × 107 | 2 × 106 | 12.06 | 76.9% | 16.7% | 6.4% | 63.1% | 36.9% | 4.34 | 1.98 | 1,387,594 | 5.07 |
| Kalamas | 1009 | 15.3% | 5 × 104 | 1 × 106 | 1 × 107 | 1 × 106 | 13.55 | 78.1% | 18.5% | 3.4% | 55.3% | 44.7% | 5.04 | 1.79 | 545,599 | 5.21 |
| Louros | 736 | 17.7% | 2 × 105 | 7 × 105 | 8 × 107 | 9 × 105 | 84.19 | 78.2% | 18.1% | 3.7% | 36.2% | 63.8% | 5.13 | 1.76 | 4,532,574 | 5.21 |
| South Amvrakikos | 425 | 9.2% | 1 × 105 | 3 × 105 | 6 × 107 | 5 × 105 | 131.32 | 75.4% | 20.4% | 4.3% | 36.2% | 63.8% | 4.78 | 1.80 | 3,361,543 | 5.06 |
| Saronikos Elefsina | 1387 | 38.1% | 2 × 102 | 8 × 105 | 6 × 107 | 8 × 105 | 75.73 | 79.4% | 18.4% | 2.2% | 30.6% | 69.4% | 4.55 | 1.81 | 1,530,439 | 5.15 |
| Saronikos Psittalia | 98 | 9.4% | 2 × 103 | 8 × 105 | 2 × 107 | 8 × 105 | 22.71 | 77.1% | 20.4% | 2.5% | 58.6% | 41.4% | 4.86 | 1.56 | 380,858 | 5.13 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsiola, A.; Michoud, G.; Fodelianakis, S.; Karakassis, I.; Kotoulas, G.; Pavlidou, A.; Pavloudi, C.; Pitta, P.; Simboura, N.; Daffonchio, D.; et al. Viral Metagenomic Content Reflects Seawater Ecological Quality in the Coastal Zone. Viruses 2020, 12, 806. https://doi.org/10.3390/v12080806
Tsiola A, Michoud G, Fodelianakis S, Karakassis I, Kotoulas G, Pavlidou A, Pavloudi C, Pitta P, Simboura N, Daffonchio D, et al. Viral Metagenomic Content Reflects Seawater Ecological Quality in the Coastal Zone. Viruses. 2020; 12(8):806. https://doi.org/10.3390/v12080806
Chicago/Turabian StyleTsiola, Anastasia, Grégoire Michoud, Stilianos Fodelianakis, Ioannis Karakassis, Georgios Kotoulas, Alexandra Pavlidou, Christina Pavloudi, Paraskevi Pitta, Nomiki Simboura, Daniele Daffonchio, and et al. 2020. "Viral Metagenomic Content Reflects Seawater Ecological Quality in the Coastal Zone" Viruses 12, no. 8: 806. https://doi.org/10.3390/v12080806
APA StyleTsiola, A., Michoud, G., Fodelianakis, S., Karakassis, I., Kotoulas, G., Pavlidou, A., Pavloudi, C., Pitta, P., Simboura, N., Daffonchio, D., & Tsapakis, M. (2020). Viral Metagenomic Content Reflects Seawater Ecological Quality in the Coastal Zone. Viruses, 12(8), 806. https://doi.org/10.3390/v12080806