Pteropine Orthoreovirus in an Angolan Soft-Furred Fruit Bat (Lissonycteris angolensis) in Uganda Dramatically Expands the Global Distribution of an Emerging Bat-Borne Respiratory Virus


Abstract
1. Introduction
2. Materials and Methods
2.1. Collection of Samples
2.2. Metagenomics
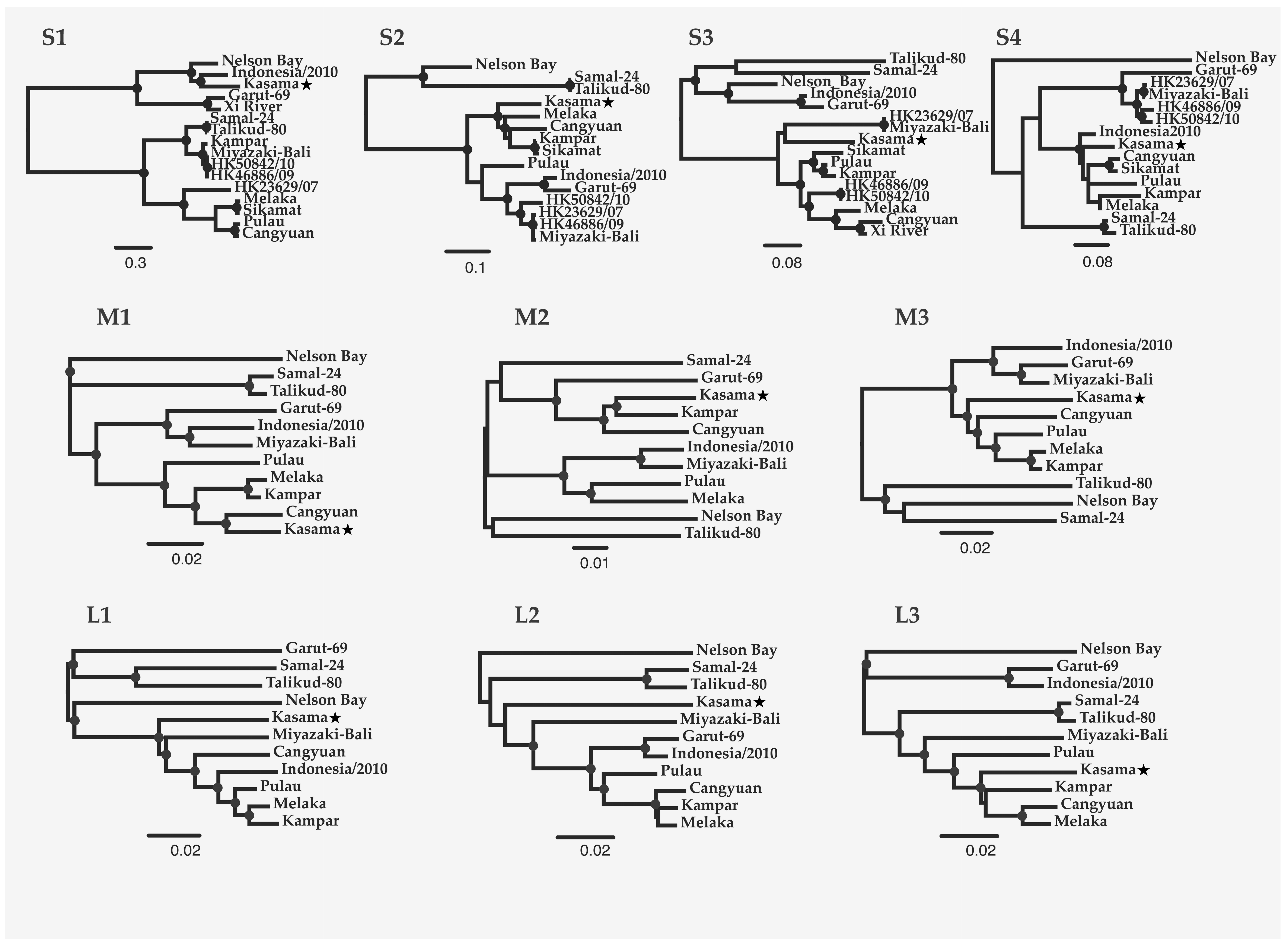
2.3. Phylogenetics of PRV16K Segments and Asia Pacific PRVs
2.4. Detection of Positive Selection on Cell-Attachment Protein (σC)
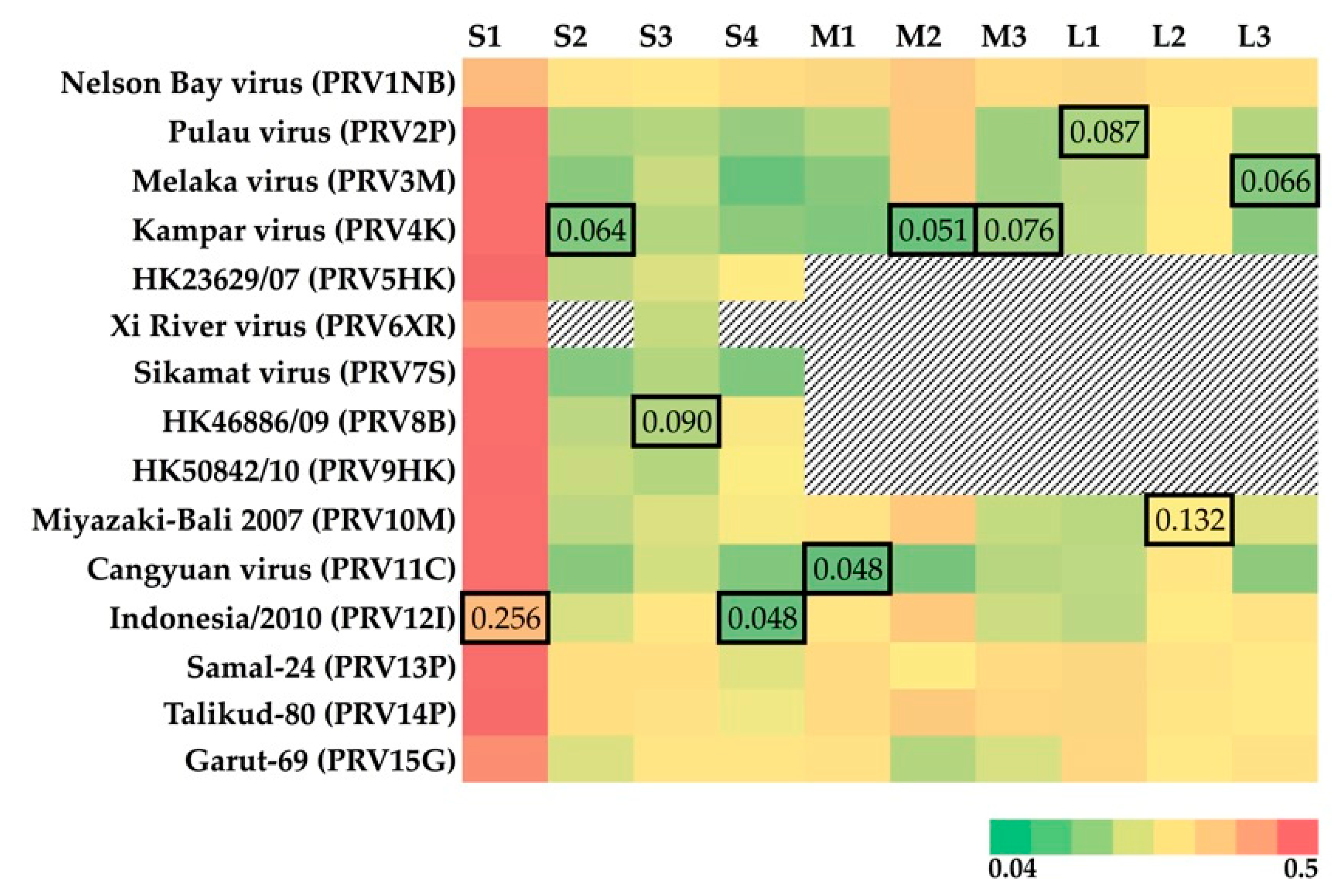
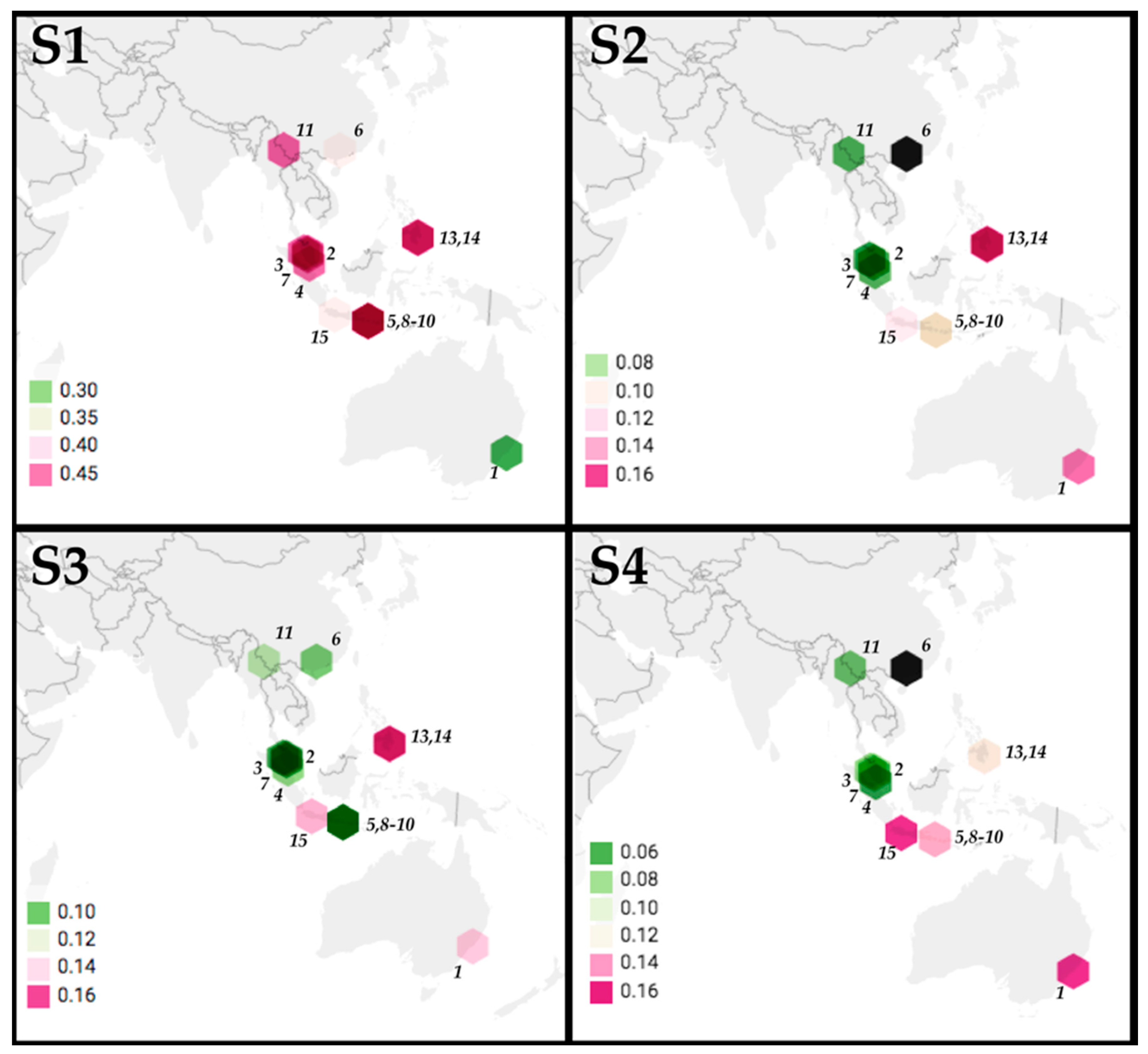
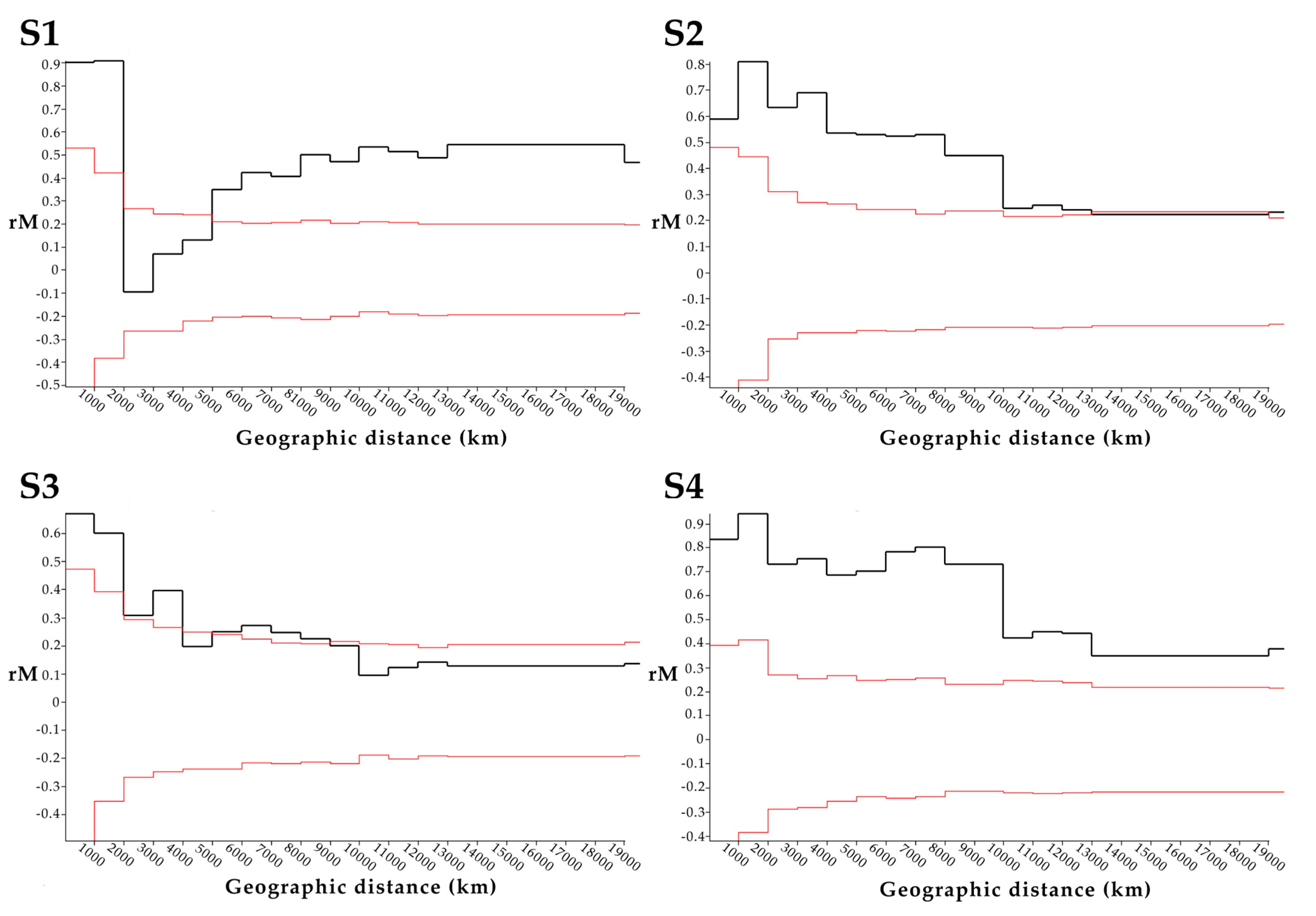
2.5. Correlation of Geographic Distance with Patristic Distance of Segments S1-4
3. Results
3.1. Sequencing and PRV Genome Assembly
3.2. Evolutionary Relationships among PRV Segments
3.3. Geographic Relationships among PRV Segments S1-4
3.4. Diversifying Selection on Cell-Attachment Protein (σC)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Voon, K.; Tan, Y.F.; Leong, P.P.; Teng, C.L.; Gunnasekaran, R.; Ujang, K.; Chua, K.B.; Wang, L.-F. Pteropine orthoreovirus infection among out-patients with acute upper respiratory tract infection in Malaysia. J. Med. Virol. 2015, 87, 2149–2153. [Google Scholar] [CrossRef] [PubMed]
- Uehara, A.; Tan, C.W.; Mani, S.; Chua, K.B.; Leo, Y.S.; Anderson, D.E.; Wang, L.-F. Serological evidence of human infection by bat orthoreovirus in Singapore. J. Med. Virol. 2018, 91, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.W.; Wittwer, K.; Lim, X.F.; Uehara, A.; Mani, S.; Wang, L.-F.; Anderson, D.E. Serological evidence and experimental infection of cynomolgus macaques with pteropine orthoreovirus reveal monkeys as potential hosts for transmission to humans. Emerg. Microbes Infect. 2019, 8, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.F.; Teng, C.L.; Chua, K.B.; Voon, K. Pteropine orthoreovirus: An important emerging virus causing infectious disease in the tropics? J. Infect. Dev. Ctries. 2017, 11, 215–219. [Google Scholar] [CrossRef]
- Day, J.M. The diversity of the orthoreoviruses: Molecular taxonomy and phylogentic divides. Infect. Genet. Evol. 2009, 9, 390–400. [Google Scholar] [CrossRef]
- Gard, G.P.; Marshall, I.D. Nelson Bay virus. Arch. Virol. 1973, 43, 34–42. [Google Scholar] [CrossRef]
- Taniguchi, S.; Maeda, K.; Horimoto, T.; Masangkay, J.S.; Puentespina, R.; Alvarez, J.; Eres, E.; Cosico, E.; Nagata, N.; Egawa, K.; et al. First isolation and characterization of pteropine orthoreoviruses in fruit bats in the Philippines. Arch. Virol. 2017, 162, 1529–1539. [Google Scholar] [CrossRef]
- Hu, T.-S.; Qiu, W.; He, B.; Zhang, Y.; Yu, J.; Liang, X.; Zhang, W.; Chen, G.; Zhang, Y.; Wang, Y.; et al. Characterization of a novel orthoreovirus isolated from fruit bat, China. BMC Microbiol. 2014, 14, 293. [Google Scholar] [CrossRef]
- Lorusso, A.; Teodori, L.; Leone, A.; Marcacci, M.; Mangone, I.; Orsini, M.; Dondona, A.C.; Cammà, C.; Monaco, F.; Savini, G. A new member of the Pteropine Orthoreovirus species isolated from fruit bats imported to Italy. Infect. Genet. Evol. 2015, 30, 55–58. [Google Scholar] [CrossRef]
- Pritchard, L.I.; Chua, K.B.; Cummins, D.; Hyatt, A.; Crameri, G.; Eaton, B.T.; Wang, L.-F. Pulau virus; A new member of the Nelson Bay orthoreovirus species isolated from fruit bats in Malaysia. Arch. Virol. 2005, 151, 229–239. [Google Scholar] [CrossRef]
- Wong, A.H.; Cheng, P.K.; Lai, M.Y.; Leung, P.C.; Wong, K.K.; Lee, W.; Lim, W.W. Virulence Potential of Fusogenic Orthoreoviruses. Emerg. Infect. Dis. 2012, 18, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Lau, C.S.; Lai, A.; Ho, E.; Leung, P.; Chan, F.; Wong, A.; Lim, W. A novel reovirus isolated from a patient with acute respiratory disease. J. Clin. Virol. 2009, 45, 79–80. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Yoshikawa, T.; Kobayashi, T.; Fukushi, S.; Tani, H.; Taniguchi, S.; Fukuma, A.; Yang, M.; Sugamata, M.; Shimojima, M.; et al. Rapid whole genome sequencing of Miyazaki-Bali/2007 Pteropine orthoreovirus by modified rolling circular amplification with adaptor ligation—Next generation sequencing. Sci. Rep. 2015, 5, 16517. [Google Scholar] [CrossRef] [PubMed]
- Reoviridae—DsRNA Viruses—DsRNA Viruses. 2011. Available online: https://talk.ictvonline.org/ictv-reports/ictv_9th_report/dsrna-viruses-2011/w/dsrna_viruses/188/reoviridae (accessed on 22 April 2020).
- Shmulevitz, M.; Duncan, R. A new class of fusion-associated small transmembrane (FAST) proteins encoded by the non-enveloped fusogenic reoviruses. EMBO J. 2000, 19, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Murphy, F.A.; Mirkovic, R.R. Characterization of a Novel Syncytium-Inducing Baboon Reovirus. Virology 1995, 212, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Lu, Z.; Fan, Y.; Meng, K.; Jiang, Y.; Zhu, Y.; Wang, S.; Gu, W.; Zou, X.; Tu, C. Xi River virus, a new bat reovirus isolated in southern China. Arch. Virol. 2010, 155, 1295–1299. [Google Scholar] [CrossRef]
- Chua, K.B.; Crameri, G.; Hyatt, A.; Yu, M.; Tompang, M.R.; Rosli, J.; McEachern, J.; Crameri, S.; Kumarasamy, V.; Eaton, B.T.; et al. A previously unknown reovirus of bat origin is associated with an acute respiratory disease in humans. Proc. Natl. Acad. Sci. USA 2007, 104, 11424–11429. [Google Scholar] [CrossRef]
- Chua, K.B.; Voon, K.; Crameri, G.; Tan, H.S.; Rosli, J.; McEachern, J.A.; Suluraju, S.; Yu, M.; Wang, L.-F. Identification and Characterization of a New Orthoreovirus from Patients with Acute Respiratory Infections. PLoS ONE 2008, 3, e3803. [Google Scholar] [CrossRef]
- Chua, K.B.; Voon, K.; Yu, M.; Keniscope, C.; Rasid, K.A.; Wang, L.-F. Investigation of a Potential Zoonotic Transmission of Orthoreovirus Associated with Acute Influenza-Like Illness in an Adult Patient. PLoS ONE 2011, 6, e25434. [Google Scholar] [CrossRef]
- Singh, H.; Shimojima, M.; Ngoc, T.C.; Huy, N.V.Q.; Chuong, T.X.; le van, A.; Saijo, M.; Yang, M.; Sugamata, M. Serological evidence of human infection with Pteropine orthoreovirus in Central Vietnam. J. Med Virol. 2015, 87, 2145–2148. [Google Scholar] [CrossRef]
- Wamala, J.F.; Lukwago, L.; Malimbo, M.; Nguku, P.; Yoti, Z.; Musenero, M.; Amone, J.; Mbabazi, W.; Nanyunja, M.; Zaramba, S.; et al. Ebola Hemorrhagic Fever Associated with Novel Virus Strain, Uganda, 2007–2008. Emerg. Infect. Dis. 2010, 16, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Voon, K.; Chua, K.B.; Yu, M.; Crameri, G.; Barr, J.; Malik, Y.; Wang, L.-F. Evolutionary relationship of the L- and M-class genome segments of bat-borne fusogenic orthoreoviruses in Malaysia and Australia. J. Gen. Virol. 2011, 92, 2930–2936. [Google Scholar] [CrossRef] [PubMed]
- Bennett, A.J.; Sibley, S.D.; Lauck, M.; Weny, G.; Hyeroba, D.; Tumukunde, A.; Friedrich, T.C.; O’Connor, D.H.; Johnson, C.A.; Rothman, J.M.; et al. Naturally Circulating Hepatitis A Virus in Olive Baboons, Uganda. Emerg. Infect. Dis. 2016, 22, 1308–1310. [Google Scholar] [CrossRef] [PubMed]
- Toohey-Kurth, K.; Sibley, S.; Goldberg, T.L. Metagenomic assessment of adventitious viruses in commercial bovine sera. Biologicals 2017, 47, 64–68. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Schiff, L.A.; Nibert, M.L.; Tyler, K.L. Orthoreoviruses and their replication. In Fields Virology; Lippincott/Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 1853–1915. [Google Scholar]
- Wang, L.-F.; Anderson, D.E. Viruses in bats and potential spillover to animals and humans. Curr. Opin. Virol. 2019, 34, 79–89. [Google Scholar] [CrossRef]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Löytynoja, A. Phylogeny-aware alignment with PRANK. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2013; Volume 1079, pp. 155–170. [Google Scholar]
- Computational Biology Unit—Ka/Ks Calculation Tool. Available online: http://services.cbu.uib.no/tools/kaks (accessed on 20 May 2020).
- Fourment, M.; Gibbs, M.J. PATRISTIC: A program for calculating patristic distances and graphically comparing the components of genetic change. BMC Evol. Biol. 2006, 6, 1. [Google Scholar] [CrossRef]
- Bailey, K.; Sokal, R.R.; Rohlf, F. Biometry: The Principles and Practice of Statistics in Biological Research. J. Am. Stat. Assoc. 1982, 77, 946. [Google Scholar] [CrossRef]
- XLSTAT Statistical and Data Analysis Solution; Addinsoft: Long Island, NY, USA, 2019.
- Remm, K.; Kelviste, T. An online calculator for spatial data and its applications. Comput. Ecol. Softw. 2014, 4, 22–34. [Google Scholar]
- Kawagishi, T.; Kanai, Y.; Tani, H.; Shimojima, M.; Saijo, M.; Matsuura, Y.; Kobayashi, T. Reverse Genetics for Fusogenic Bat-Borne Orthoreovirus Associated with Acute Respiratory Tract Infections in Humans: Role of Outer Capsid Protein σC in Viral Replication and Pathogenesis. PLoS Pathog. 2016, 12, e1005455. [Google Scholar] [CrossRef]
- Plowright, R.; Eby, P.; Hudson, P.J.; Smith, I.L.; Westcott, D.; Bryden, W.L.; Middleton, D.; Reid, P.A.; McFarlane, R.; Martin, G.; et al. Ecological dynamics of emerging bat virus spillover. Proc. R. Soc. Biol. Sci. 2015, 282, 20142124. [Google Scholar] [CrossRef] [PubMed]
- Almeida, F.C.; Giannini, N.P.; Simmons, N.B. The evolutionary history of the African fruit bats (Chiroptera: Pteropodidae). Acta Chiropterol. 2016, 18, 73–90. [Google Scholar] [CrossRef]
- African Chiroptera Report 2019. Available online: https://www.academia.edu/41472741/AFRICAN_CHIROPTERA_REPORT_2019 (accessed on 1 June 2020).
- Wright, S. Isolation by Distance. Genetics 1943, 28, 114–138. [Google Scholar] [PubMed]
- Tao, Y.; Shi, M.; Chommanard, C.; Queen, K.; Zhang, J.; Markotter, W.; Kuzmin, I.V.; Holmes, E.C.; Tong, S. Surveillance of Bat Coronaviruses in Kenya Identifies Relatives of Human Coronaviruses NL63 and 229E and Their Recombination History. J. Virol. 2017, 91, e01953-16. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.E.; Reeder, D.M. Mammal Species of the World: A Taxonomic and Geographic Reference; JHU Press: Baltimore, MD, USA, 2005; ISBN 978-0-8018-8221-0. [Google Scholar]
- Willoughby, A.R.; Phelps, K.L.; Olival, K.J. PREDICT Consortium A Comparative Analysis of Viral Richness and Viral Sharing in Cave-Roosting Bats. Diversity 2017, 9, 35. [Google Scholar] [CrossRef]
- Kosoltanapiwat, N.; Reamtong, O.; Okabayashi, T.; Ampawong, S.; Rungruengkitkun, A.; Thiangtrongjit, T.; Thippornchai, N.; Leaungwutiwong, P.; Mahittikorn, A.; Mori, H.; et al. Mass spectrometry-based identification and whole-genome characterisation of the first pteropine orthoreovirus isolated from monkey faeces in Thailand. BMC Microbiol. 2018, 18, 135. [Google Scholar] [CrossRef]
- Wickramasinghe, R.; Meanger, J.; Enriquez, C.E.; Wilcox, G.E. Avian Reovirus Proteins Associated with Neutralization of Virus Infectivity. Virology 1993, 194, 688–696. [Google Scholar] [CrossRef]
- Payne, S. Family Reoviridae. In Viruses; Elsevier BV: Amsterdam, The Netherlands, 2017; pp. 219–226. [Google Scholar]
- Spinner, M.L.; di Giovanni, G.D. Detection and Identification of Mammalian Reoviruses in Surface Water by Combined Cell Culture and Reverse Transcription-PCR. Appl. Environ. Microbiol. 2001, 67, 3016–3020. [Google Scholar] [CrossRef]
- Lim, M.C.Y.; Wang, Y.-F.; Huang, S.-W.; Yang, J.-Y.; Wang, J.-R. High Incidence of Mammalian Orthoreovirus Identified by Environmental Surveillance in Taiwan. PLoS ONE 2015, 10, e0142745. [Google Scholar] [CrossRef] [PubMed]
- McMorrow, M.L.; Emukule, G.O.; Njuguna, H.N.; Bigogo, G.; Montgomery, J.M.; Nyawanda, B.; Audi, A.; Breiman, R.F.; Katz, M.A.; Cosmas, L.; et al. The Unrecognized Burden of Influenza in Young Kenyan Children, 2008–2012. PLoS ONE 2015, 10, e0138272. [Google Scholar] [CrossRef]
- Nair, H.; Brooks, W.A.; Katz, M.; Roca, A.; Berkley, J.A.; Madhi, S.A.; Simmerman, J.M.; Gordon, A.; Sato, M.; Howie, S.; et al. Global burden of respiratory infections due to seasonal influenza in young children: A systematic review and meta-analysis. Lancet 2011, 378, 1917–1930. [Google Scholar] [CrossRef]
- Katz, M.A.; Schoub, B.D.; Heraud, J.-M.; Breiman, R.F.; Njenga, M.K.; Widdowson, M.-A. Influenza in Africa: Uncovering the Epidemiology of a Long-Overlooked Disease. J. Infect. Dis. 2012, 206, S1–S4. [Google Scholar] [CrossRef] [PubMed]
- Schoub, B. Surveillance and management of influenza on the African continent. Expert Rev. Respir. Med. 2010, 4, 167–169. [Google Scholar] [CrossRef]
- Razuri, H.; Romero, C.; Tinoco, Y.; Guezala, M.C.; Ortiz, E.; Silva, M.; Reaves, E.; Williams, M.; Laguna-Torres, V.A.; Halsey, E.S.; et al. Population-based active surveillance cohort studies for influenza: Lessons from Peru. Bull. World Heal. Organ. 2012, 90, 318–320. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | Common Name | Year | Host | Country of Origin |
|---|---|---|---|---|
| PRV1NB | Nelson Bay virus | 1968 | Bat (Pteropus policephalus) | Australia |
| PRV2P | Pulau virus | 1999 | Bat (Pteropus hypomelanus) | Malaysia |
| PRV3M | Melaka virus | 2006 | Human | Malaysia |
| PRV4K | Kampar virus | 2006 | Human | Malaysia |
| PRV5HK | HK23629/07 | 2007 | Human | Indonesia |
| PRV6XR | Xi River virus | 2006 | Bat (Rousettus leschenaultii) | China |
| PRV7S | Sikamat virus | 2010 | Human | Malaysia |
| PRV8B | HK46886/09 | 2009 | Human | Indonesia |
| PRV9HK | HK50842/10 | 2010 | Human | Indonesia |
| PRV10M | Miyazaki-Bali 2007 | 2007 | Human | Indonesia |
| PRV11C | Cangyuan virus | 2012 | Bat (Rousettus leschenaultii) | China |
| PRV12I | Indonesia/2010 | 2010 | Bat (Pteropus vampyrus) | Indonesia |
| PRV13P | Samal-24 | 2013 | Bat (Eonycteris spelaea) | Philippines |
| PRV14P | Talikud-80 | 2013 | Bat (Rousettus amplexicaudatus) | Philippines |
| PRV15G | Garut-69 | 2017 | Bat (Pteropus vampyrus) | Indonesia |
| PRV16K* | Kasama virus | 2017 | Bat (Lissonycteris angolensis ruwenzorii) | Uganda |
| PRV Genome Segment | Virus Protein(s) | Mantel’s r (rM) | p-Value (Two-Tailed) [99% CI] |
|---|---|---|---|
| S1 | P10 (FAST), P17 (unknown), σC (cell attachment protein) | 0.462 | <0.0001 * |
| S2 | σ1 (major inner-capsid protein) | 0.229 | 0.031 [0.027–0.036] * |
| S3 | σNS (nonstructural replication protein) | 0.132 | 0.179 [0.169–0.189] |
| S4 | σ2 (major outer-capsid protein) | 0.373 | <0.0001 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bennett, A.J.; Goldberg, T.L. Pteropine Orthoreovirus in an Angolan Soft-Furred Fruit Bat (Lissonycteris angolensis) in Uganda Dramatically Expands the Global Distribution of an Emerging Bat-Borne Respiratory Virus. Viruses 2020, 12, 740. https://doi.org/10.3390/v12070740
Bennett AJ, Goldberg TL. Pteropine Orthoreovirus in an Angolan Soft-Furred Fruit Bat (Lissonycteris angolensis) in Uganda Dramatically Expands the Global Distribution of an Emerging Bat-Borne Respiratory Virus. Viruses. 2020; 12(7):740. https://doi.org/10.3390/v12070740
Chicago/Turabian StyleBennett, Andrew J., and Tony L. Goldberg. 2020. "Pteropine Orthoreovirus in an Angolan Soft-Furred Fruit Bat (Lissonycteris angolensis) in Uganda Dramatically Expands the Global Distribution of an Emerging Bat-Borne Respiratory Virus" Viruses 12, no. 7: 740. https://doi.org/10.3390/v12070740
APA StyleBennett, A. J., & Goldberg, T. L. (2020). Pteropine Orthoreovirus in an Angolan Soft-Furred Fruit Bat (Lissonycteris angolensis) in Uganda Dramatically Expands the Global Distribution of an Emerging Bat-Borne Respiratory Virus. Viruses, 12(7), 740. https://doi.org/10.3390/v12070740