Multi-Laboratory Comparison of Next-Generation to Sanger-Based Sequencing for HIV-1 Drug Resistance Genotyping

 , , , , , , , , ,
, , , , , , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Specimens
2.2. Sequencing Methods
2.3. Sequence Comparison
3. Results
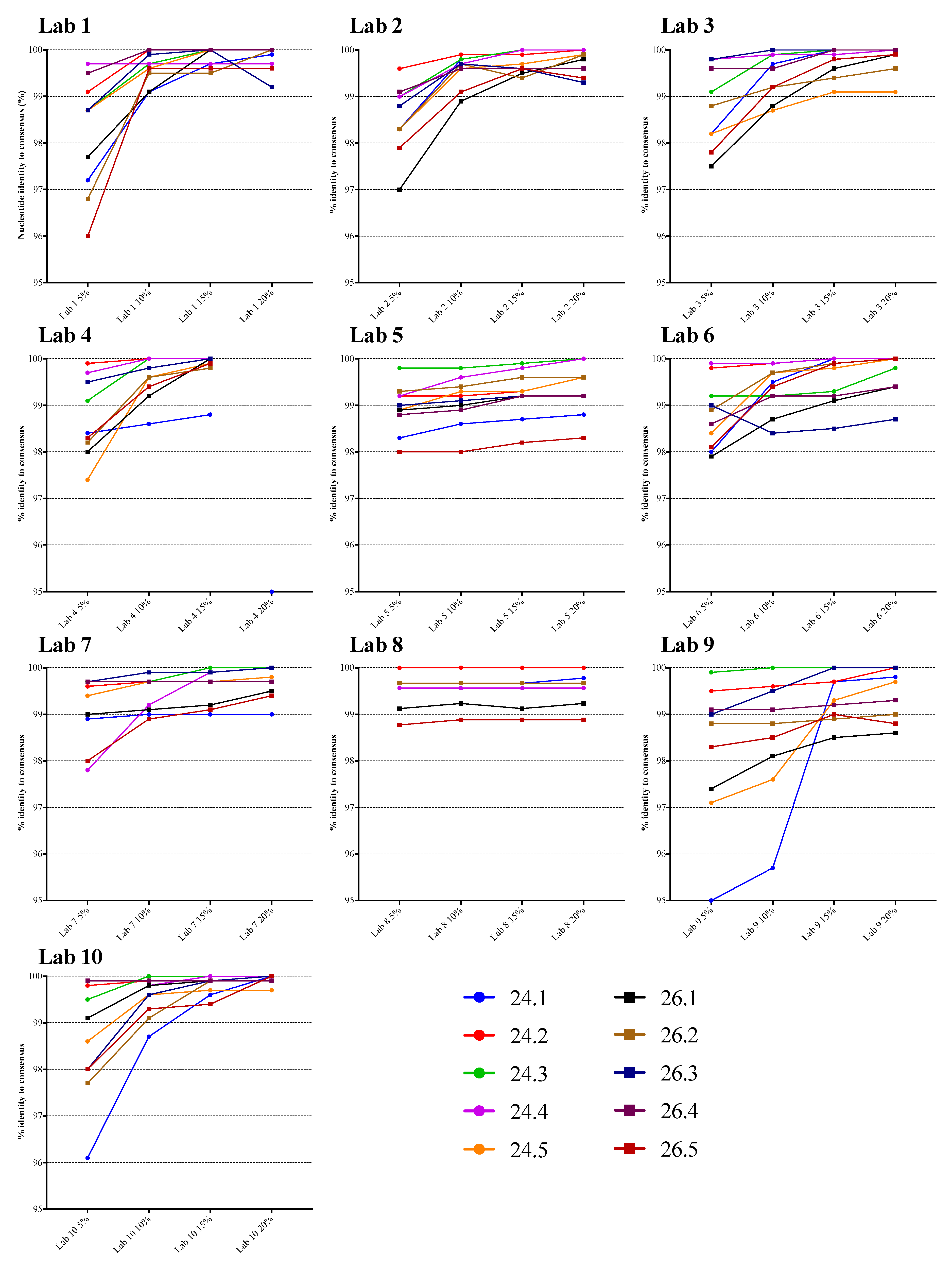
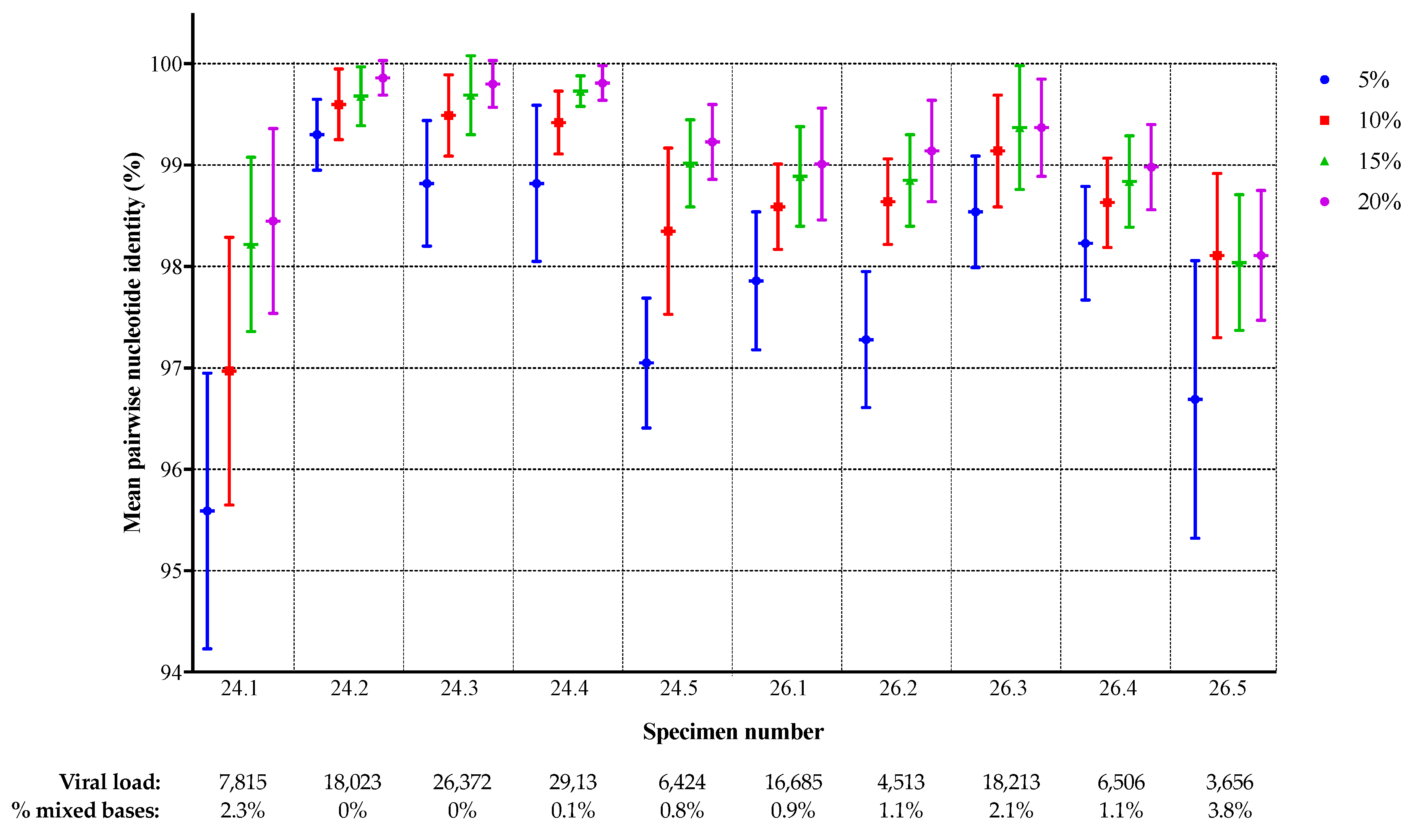
3.1. Comparison of NGS Sequences to VQA Sanger Consensus
3.2. Comparison of NGS Sequences Between Laboratories
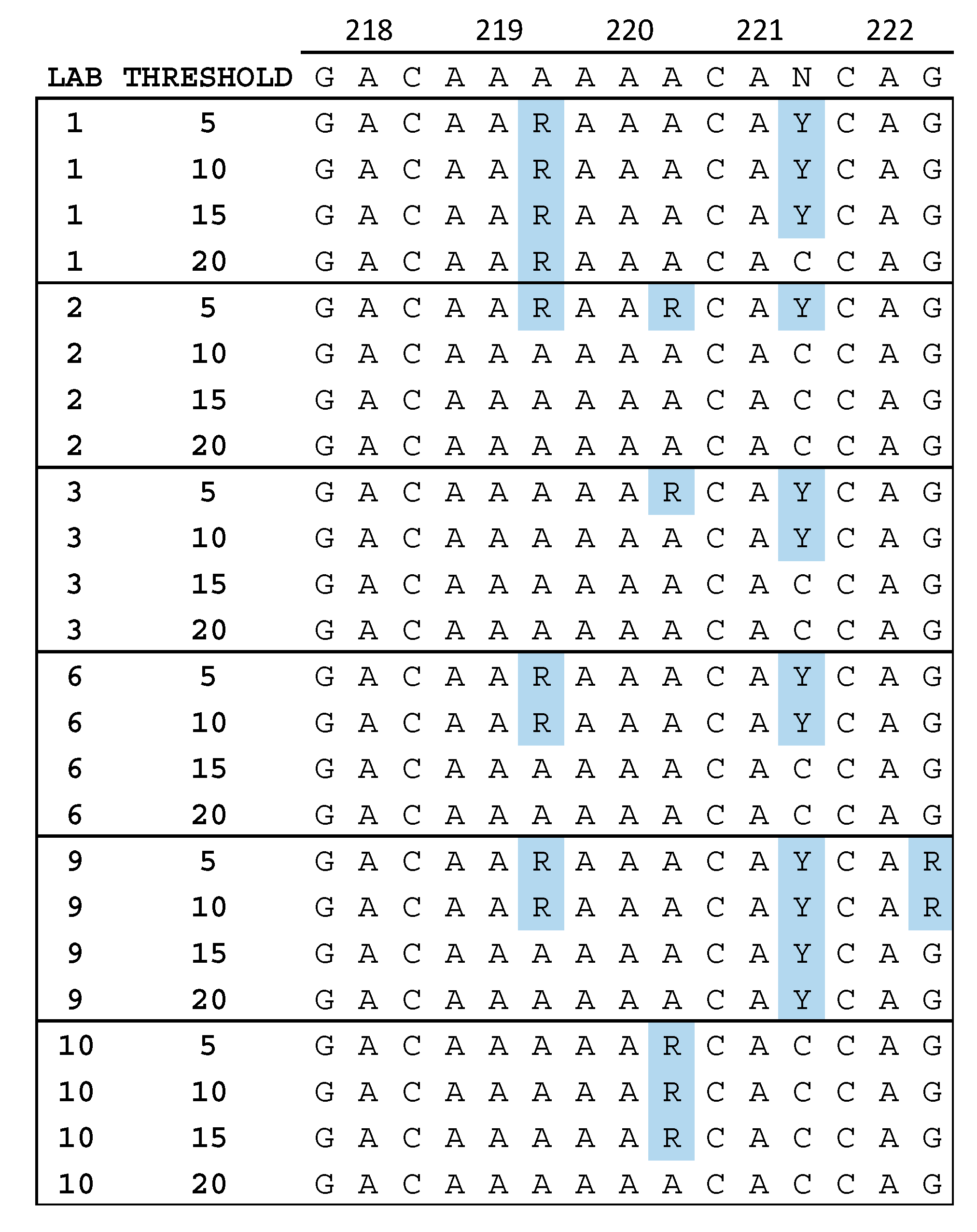
3.3. Quality Assurance Anomalies
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Laboratory ID | RNA Extraction Method (Specimen Volume) | RT-PCR Amplification Strategy | Negative Control | % of Extracted RNA Used | Coverage (PR, RT aa) | Minimum Read Depth a | Minimum Variant Count b | Analysis Pipeline |
|---|---|---|---|---|---|---|---|---|
| 1 | QIAamp Viral RNA Mini kit (0.14 mL) | RT with primerID, then nested PCR | Water | 50% | PR 1–99, RT 34–122 and 152–236 | Varies | NA c | TCS pipeline in house |
| 2 | ViroSeq RNA extraction kit (0.5 mL) | RT then nested PCR | Water | 10% | PR 1–99, RT 1–440 | 1000 | 1000 | CLC Genomics Workbench and In-house |
| 3 | QIAamp Viral RNA Mini kit (1 mL) | One-step RT-PCR then nested PCR | Water | 10% | PR 6–99, RT 1–251 | 1000 | 50 | HyDRA [35] |
| 4 | MagnaPure LC (0.5 mL) | One-step RT-PCR, then nested PCR | Water | 30% | PR 1–99, RT 1–250 | 330 | NA c | Geneious |
| 5 | QIAamp Viral RNA Mini kit (0.14 mL) | One-step RT-PCR then nested PCR | Water | 10% | PR 1–99 RT 1–300 | 100 | 5 | Trim Galore!, HydDRA [35] |
| 6 | NucliSENS easyMAG (0.4 mL) | One-step RT-PCR, then nested PCR | Water | ~9% | PR 1–99, RT 1–250 | 100 | 5 | HyDRA [35] |
| 7 | QIAamp UltraSens Virus kit (0.5 mL) | Primary RT-PCR, then nested PCR | Fetal bovine serum | 16.7% | PR 5–99, RT 1–320 | 1000 | NA c | In-house [36] |
| 8 | QIAamp Viral RNA Mini kit (0.14 mL) | One-step RT-PCR then nested PCR | Water | 25 % | PR 1–99, RT 1–440 | 1000 | 10 | PASeq.org [35] |
| 9 | EZ1 Advance XL (variable) d | RT then nested PCR | Water | 16.7% | PR 1–99, RT 1–240 | 1000 | 10 e | Hivmmer [9] |
| 10 | NucliSENS easyMAG (0.5 mL) | RT then nested PCR | Water | 6.7% | PR 1–99, RT 1–400 or 1–240 | 100 | NA c | MiCall [35] |
| 5% | 10% | 15% | 20% | |
|---|---|---|---|---|
| Number | 94 | 94 | 94 | 85 |
| Minimum | 95.0 | 95.7 | 98.2 | 98.3 |
| Median | 98.9 | 99.6 | 99.7 | 99.9 |
| Mean | 98.7 | 99.4 | 99.6 | 99.7 |
| Std. Deviation | 0.95 | 0.63 | 0.41 | 0.40 |
| Lower 95% CI of mean | 98.5 | 99.2 | 99.5 | 99.6 |
| Upper 95% CI of mean | 98.9 | 99.5 | 99.7 | 99.8 |
| Comparison | N | Rand Eff Model Mean (SEM) | Rand Eff Model p-value Test = 0 | Mean Diff | Median Diff | SD Diff | Min Diff | Max Diff | Paired t p value | Sign Test p value | Sign Rank p value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 10% vs 5% | 94 | 0.0062 (0.0016) | 0.004 | 0.0065 | 0.0044 | 0.0071 | −0.0056 | 0.0359 | 0 | 0 | 0 |
| 15% vs 5% | 94 | 0.0087 (0.0020) | 0.002 | 0.009 | 0.0056 | 0.0094 | −0.0052 | 0.0466 | 0 | 0 | 0 |
| 15% vs 10% | 94 | 0.0025 (0.00076) | 0.01 | 0.0025 | 0.0011 | 0.0048 | −0.0028 | 0.04 | 0.0000022 | 0 | 0 |
| 20% vs 5% | 85 | 0.0094 (0.0022) | 0.003 | 0.0097 | 0.0055 | 0.01 | −0.0034 | 0.0477 | 0 | 0 | 0 |
| 20% vs 10% | 85 | 0.0033 (0.00091) | 0.007 | 0.0034 | 0.0022 | 0.0055 | −0.0063 | 0.0411 | 0.0000002 | 0 | 0 |
| 20% vs 15% | 85 | 0.00084 (0.0002) | 0.003 | 0.0008 | 0 | 0.0017 | −0.0075 | 0.0056 | 0.0000265 | 0 | 0.0000012 |
References
- Parikh, U.M.; McCormick, K.; van Zyl, G.; Mellors, J.W. Future technologies for monitoring HIV drug resistance and cure. Curr. Opin. HIV AIDS 2017, 12, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Casadella, M.; Paredes, R. Deep sequencing for HIV-1 clinical management. Virus Res. 2017, 239, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Van Laethem, K.; Theys, K.; Vandamme, A.M. HIV-1 genotypic drug resistance testing: digging deep, reaching wide? Curr. Opin. Virol. 2015, 14, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Barzon, L.; Lavezzo, E.; Costanzi, G.; Franchin, E.; Toppo, S.; Palu, G. Next-generation sequencing technologies in diagnostic virology. J. Clin. Virol. 2013, 58, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Z.; Paredes, R.; Ribaudo, H.J.; Svarovskaia, E.S.; Metzner, K.J.; Kozal, M.J.; Hullsiek, K.H.; Balduin, M.; Jakobsen, M.R.; Geretti, A.M.; et al. Low-frequency HIV-1 drug resistance mutations and risk of NNRTI-based antiretroviral treatment failure: a systematic review and pooled analysis. JAMA 2011, 305, 1327–1335. [Google Scholar] [CrossRef]
- Paredes, R.; Lalama, C.M.; Ribaudo, H.J.; Schackman, B.R.; Shikuma, C.; Giguel, F.; Meyer, W.A., 3rd; Johnson, V.A.; Fiscus, S.A.; D’Aquila, R.T.; et al. Pre-existing minority drug-resistant HIV-1 variants, adherence, and risk of antiretroviral treatment failure. J. Infect. Dis. 2010, 201, 662–671. [Google Scholar] [CrossRef]
- Ji, H.; Enns, E.; Brumme, C.J.; Parkin, N.; Howison, M.; Lee, E.R.; Capina, R.; Marinier, E.; Avila-Rios, S.; Sandstrom, P.; et al. Bioinformatic data processing pipelines in support of next-generation sequencing-based HIV drug resistance testing: the Winnipeg Consensus. J. Int. AIDS Soc. 2018, 21, e25193. [Google Scholar] [CrossRef]
- Taylor, T.; Lee, E.R.; Nykoluk, M.; Enns, E.; Liang, B.; Capina, R.; Gauthier, M.K.; Domselaar, G.V.; Sandstrom, P.; Brooks, J.; et al. A MiSeq-HyDRA platform for enhanced HIV drug resistance genotyping and surveillance. Sci Rep. 2019, 9, 8970. [Google Scholar] [CrossRef]
- Howison, M.; Coetzer, M.; Kantor, R. Measurement error and variant-calling in deep Illumina sequencing of HIV. Bioinformatics 2019, 35, 2029–2035. [Google Scholar] [CrossRef]
- Raymond, S.; Nicot, F.; Carcenac, R.; Lefebvre, C.; Jeanne, N.; Saune, K.; Delobel, P.; Izopet, J. HIV-1 genotypic resistance testing using the Vela automated next-generation sequencing platform. J. Antimicrob. Chemother. 2018, 73, 1152–1157. [Google Scholar] [CrossRef]
- Weber, J.; Volkova, I.; Sahoo, M.K.; Tzou, P.L.; Shafer, R.W.; Pinsky, B.A. Prospective evaluation of the vela diagnostics next-generation sequencing platform for HIV-1 genotypic resistance testing. J. Mol. Diagn. 2019, 21, 961–970. [Google Scholar] [CrossRef] [PubMed]
- May, S.; Adamska, E.; Tang, J. Evaluation of Vela Diagnostics HIV-1 genotyping assay on an automated next generation sequencing platform. J. Clin. Virol. 2020, 127, 104376. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Diaz, A.; McCormick, A.; Booth, C.; Gonzalez, D.; Sayada, C.; Haque, T.; Johnson, M.; Webster, D. Analysis of transmitted HIV-1 drug resistance using 454 ultra-deep-sequencing and the DeepChek®-HIV system. J. Int. AIDS Soc. 2014, 17 (4 Suppl 3), 19752. [Google Scholar] [CrossRef]
- Li, J.Z.; Kuritzkes, D.R. Clinical implications of HIV-1 minority variants. Clin. Infect. Dis. 2013, 56, 1667–1674. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, J.Z.; Paredes, R.; Ribaudo, H.J.; Kozal, M.J.; Svarovskaia, E.S.; Johnson, J.A.; Geretti, A.M.; Metzner, K.J.; Jakobsen, M.R.; Hullsiek, K.H.; et al. Impact of minority nonnucleoside reverse transcriptase inhibitor resistance mutations on resistance genotype after virologic failure. J. Infect. Dis. 2013, 207, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Z.; Paredes, R.; Ribaudo, H.J.; Svarovskaia, E.S.; Kozal, M.J.; Hullsiek, K.H.; Miller, M.D.; Bangsberg, D.R.; Kuritzkes, D.R. Relationship between minority nonnucleoside reverse transcriptase inhibitor resistance mutations, adherence, and the risk of virologic failure. AIDS 2012, 26, 185–192. [Google Scholar] [CrossRef]
- Inzaule, S.C.; Hamers, R.L.; Noguera-Julian, M.; Casadella, M.; Parera, M.; Kityo, C.; Steegen, K.; Naniche, D.; Clotet, B.; Rinke de Wit, T.F.; et al. Clinically relevant thresholds for ultrasensitive HIV drug resistance testing: a multi-country nested case-control study. Lancet HIV 2018, 5, e638–e646. [Google Scholar] [CrossRef]
- Mbunkah, H.A.; Bertagnolio, S.; Hamers, R.L.; Hunt, G.; Inzaule, S.; Rinke De Wit, T.F.; Paredes, R.; Parkin, N.T.; Jordan, M.R.; Metzner, K.J.; et al. Low-abundance drug-resistant HIV-1 variants in antiretroviral drug-naive individuals: A systematic review of detection methods, prevalence, and clinical impact. J. Infect. Dis. 2020, 221, 1584–1597. [Google Scholar] [CrossRef]
- Avila-Rios, S.; Garcia-Morales, C.; Matias-Florentino, M.; Romero-Mora, K.A.; Tapia-Trejo, D.; Quiroz-Morales, V.S.; Reyes-Gopar, H.; Ji, H.; Sandstrom, P.; Casillas-Rodriguez, J.; et al. Pretreatment HIV-drug resistance in Mexico and its impact on the effectiveness of first-line antiretroviral therapy: a nationally representative 2015 WHO survey. Lancet HIV 2016, 3, e579–e591. [Google Scholar] [CrossRef]
- World Health Organization. WHO HIV Drug Resistance Report 2012; World Health Organization: Geneva, Switzerland, 2012. [Google Scholar]
- World Health Organization. WHO HIV Drug Resistance Report 2017; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- World Health Organization. WHO HIV Drug Resistance Report 2019; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Parkin, N.; de Mendoza, C.; Schuurman, R.; Jennings, C.; Bremer, J.; Jordan, M.R.; Bertagnolio, S. Evaluation of in-house genotyping assay performance using dried blood spot specimens in the Global World Health Organization laboratory network. Clin. Infect. Dis. 2012, 54 (Suppl 4), S273–S279. [Google Scholar] [CrossRef]
- Parkin, N.; Bremer, J.; Bertagnolio, S. Genotyping external quality assurance in the World Health Organization HIV drug resistance laboratory network during 2007–2010. Clin. Infect. Dis. 2012, 54 (Suppl 4), S266–S272. [Google Scholar] [CrossRef]
- World Health Organization. WHO/HIVResNet HIV Drug Resistance Laboratory Operational Framework; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Huang, D.D.; Bremer, J.W.; Brambilla, D.J.; Palumbo, P.E.; Aldrovandi, G.; Eshleman, S.; Brown, C.; Fiscus, S.; Frenkel, L.; Hamdan, H.; et al. Model for assessment of proficiency of human immunodeficiency virus type 1 sequencing-based genotypic antiretroviral assays. J. Clin. Microbiol. 2005, 43, 3963–3970. [Google Scholar] [CrossRef] [PubMed]
- Jabara, C.B.; Jones, C.D.; Roach, J.; Anderson, J.A.; Swanstrom, R. Accurate sampling and deep sequencing of the HIV-1 protease gene using a Primer ID. Proc. Natl. Acad. Sci. USA 2011, 108, 20166–20171. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Jones, C.; Mieczkowski, P.; Swanstrom, R. Primer ID validates template sampling depth and greatly reduces the error rate of next-generation sequencing of HIV-1 genomic RNA populations. J. Virol. 2015, 89, 8540–8555. [Google Scholar] [CrossRef] [PubMed]
- Tzou, P.L.; Kosakovsky Pond, S.L.; Avila-Rios, S.; Holmes, S.P.; Kantor, R.; Shafer, R.W. Analysis of unusual and signature APOBEC-mutations in HIV-1 pol next-generation sequences. PLoS ONE 2020, 15, e0225352. [Google Scholar] [CrossRef] [PubMed]
- Church, J.D.; Jones, D.; Flys, T.; Hoover, D.; Marlowe, N.; Chen, S.; Shi, C.; Eshleman, J.R.; Guay, L.A.; Jackson, J.B.; et al. Sensitivity of the ViroSeq HIV-1 genotyping system for detection of the K103N resistance mutation in HIV-1 subtypes A, C, and D. J. Mol. Diagn. 2006, 8, 430–432; [Google Scholar] [CrossRef] [PubMed]
- Leitner, T.; Halapi, E.; Scarlatti, G.; Rossi, P.; Albert, J.; Fenyo, E.M.; Uhlen, M. Analysis of heterogeneous viral populations by direct DNA sequencing. Biotechniques 1993, 15, 120–127. [Google Scholar]
- Halvas, E.K.; Aldrovandi, G.M.; Balfe, P.; Beck, I.A.; Boltz, V.F.; Coffin, J.M.; Frenkel, L.M.; Hazelwood, J.D.; Johnson, V.A.; Kearney, M.; et al. Blinded, multicenter comparison of methods to detect a drug-resistant mutant of human immunodeficiency virus type 1 at low frequency. J. Clin. Microbiol. 2006, 44, 2612–2614. [Google Scholar] [CrossRef]
- Larder, B.A.; Kohli, A.; Kellam, P.; Kemp, S.D.; Kronick, M.; Henfrey, R.D. Quantitative detection of HIV-1 drug resistance mutations by automated DNA sequencing. Nature 1993, 365, 671–673. [Google Scholar] [CrossRef]
- Lee, E.R.; Parkin, N.; Jennings, C.; Brumme, C.J.; Enns, E.; Casadella, M.; Howison, M.; Coetzer, M.; Avila-Rios, S.; Capina, R.; et al. Performance comparison of next generation sequencing analysis pipelines for HIV-1 drug resistance testing. Sci. Rep. 2020, 10, 1634. [Google Scholar] [CrossRef]
- Noguera-Julian, M.; Edgil, D.; Harrigan, P.R.; Sandstrom, P.; Godfrey, C.; Paredes, R. Next-Generation Human Immunodeficiency Virus sequencing for patient management and drug resistance surveillance. J. Infect. Dis. 2017, 216 (suppl_9), S829–s833. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, E.; Chan, Y.T.; Aghaizu, A.; Bibby, D.F.; Murphy, G.; Tosswill, J.; Harris, R.J.; Myers, R.; Field, N.; Delpech, V.; et al. Enhanced surveillance of HIV-1 drug resistance in recently infected MSM in the UK. J. Antimicrob. Chemother. 2017, 72, 227–234. [Google Scholar] [CrossRef] [PubMed]




| Specimen | Viral Load a | Subtype b | PR DRMs c | RT DRMs c | % Mixed Bases in SS Consensus d | Number of Amplification Failures |
|---|---|---|---|---|---|---|
| 24.1 | 7815 | B | None | T215C | 2.3% | 0 |
| 24.2 | 18,023 | F | K20R, M36I | None | 0.0% | 0 |
| 24.3 | 26,372 | C | M36I | M41L, V75T, V90I, V106M, V179D | 0.0% | 0 |
| 24.4 | 29,139 | C | M36I | M41L, K103N, M184V, T215Y | 0.1% | 1 |
| 24.5 | 6424 | B | L10I, L33F, M46L, I54V, A71I/T, V82A, L90M | M41L, E44D, A62V, D67N, L74V, L100I, K103N, H208Y, L210W, T215Y H221Y | 0.8% | 1 |
| 26.1 | 16,685 | C | M36I, T74S | D67N, K70R, V90I, M184V | 0.9% | 0 |
| 26.2 e | 4513 | B | L10I, L33F, M46L, I54V, A71I/T, V82A, L90M | M41L, E44D, A62V, D67N, L74V, L100I, K103N, H208Y, L210W, T215Y, H221Y | 1.1% | 1 |
| 26.3 | 18,213 | C | K20R, M36I | A62V, K65R, D67N, V75A/I/T, K101Q, K103N, V106M, E138A, M184V | 2.1% | 1 |
| 26.4 | 6506 | D | M36I | None | 1.1% | 2 |
| 26.5 | 3656 | B | none | V90I, K103N | 3.8% | 0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parkin, N.T.; Avila-Rios, S.; Bibby, D.F.; Brumme, C.J.; Eshleman, S.H.; Harrigan, P.R.; Howison, M.; Hunt, G.; Ji, H.; Kantor, R.; et al. Multi-Laboratory Comparison of Next-Generation to Sanger-Based Sequencing for HIV-1 Drug Resistance Genotyping. Viruses 2020, 12, 694. https://doi.org/10.3390/v12070694
Parkin NT, Avila-Rios S, Bibby DF, Brumme CJ, Eshleman SH, Harrigan PR, Howison M, Hunt G, Ji H, Kantor R, et al. Multi-Laboratory Comparison of Next-Generation to Sanger-Based Sequencing for HIV-1 Drug Resistance Genotyping. Viruses. 2020; 12(7):694. https://doi.org/10.3390/v12070694
Chicago/Turabian StyleParkin, Neil T., Santiago Avila-Rios, David F. Bibby, Chanson J. Brumme, Susan H. Eshleman, P. Richard Harrigan, Mark Howison, Gillian Hunt, Hezhao Ji, Rami Kantor, and et al. 2020. "Multi-Laboratory Comparison of Next-Generation to Sanger-Based Sequencing for HIV-1 Drug Resistance Genotyping" Viruses 12, no. 7: 694. https://doi.org/10.3390/v12070694
APA StyleParkin, N. T., Avila-Rios, S., Bibby, D. F., Brumme, C. J., Eshleman, S. H., Harrigan, P. R., Howison, M., Hunt, G., Ji, H., Kantor, R., Ledwaba, J., Lee, E. R., Matías-Florentino, M., Mbisa, J. L., Noguera-Julian, M., Paredes, R., Rivera-Amill, V., Swanstrom, R., Zaccaro, D. J., ... Jennings, C. (2020). Multi-Laboratory Comparison of Next-Generation to Sanger-Based Sequencing for HIV-1 Drug Resistance Genotyping. Viruses, 12(7), 694. https://doi.org/10.3390/v12070694