Extracellular Vesicle Activation of Latent HIV-1 Is Driven by EV-Associated c-Src and Cellular SRC-1 via the PI3K/AKT/mTOR Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Infection and Treatment of PBMCs
2.3. EV Isolation and Ultracentrifugation
2.4. EV Characterization Using ZetaView
2.5. Nanoparticle Capture of EVs/Virions
2.6. Cell Transfection
2.7. Cell Lysis
2.8. Western Blot Analysis
2.9. Cytotoxicity Assay
2.10. RNA Isolation and RT-qPCR
2.11. Kinase Assay
2.12. ChIP Assay
2.13. Statistical Analysis
3. Results
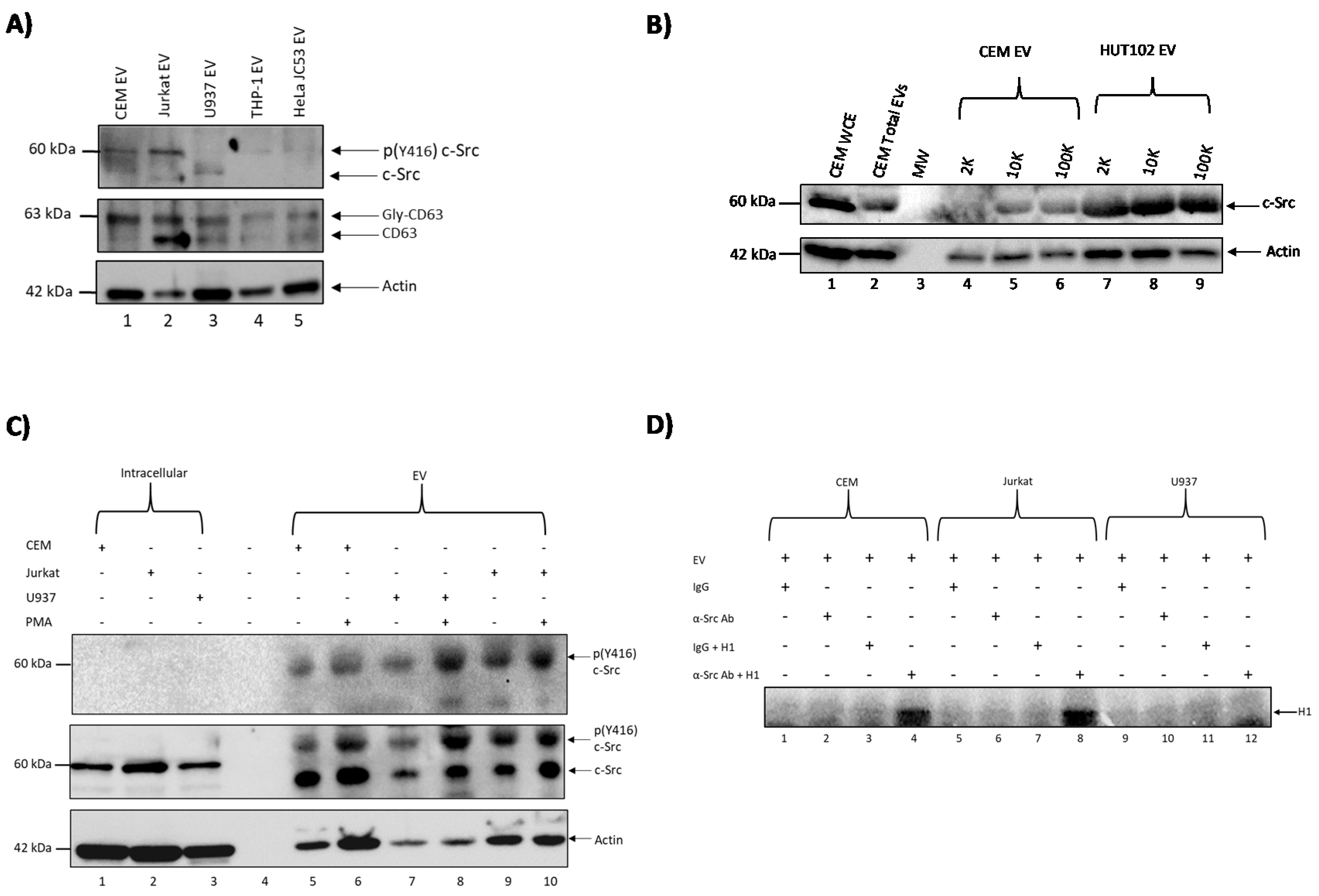
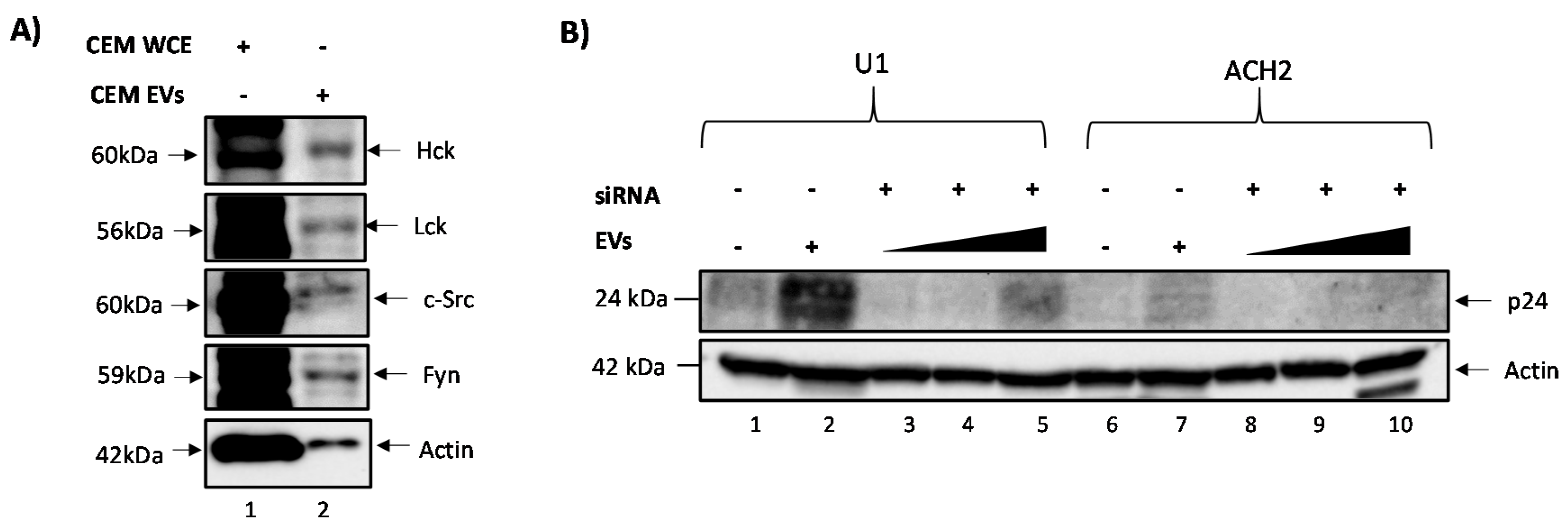
3.1. c-Src Is Present in Multiple Cell Lines and Different EV Populations
3.2. Elucidating the Activation Pathway of Latent HIV-1 by c-Src
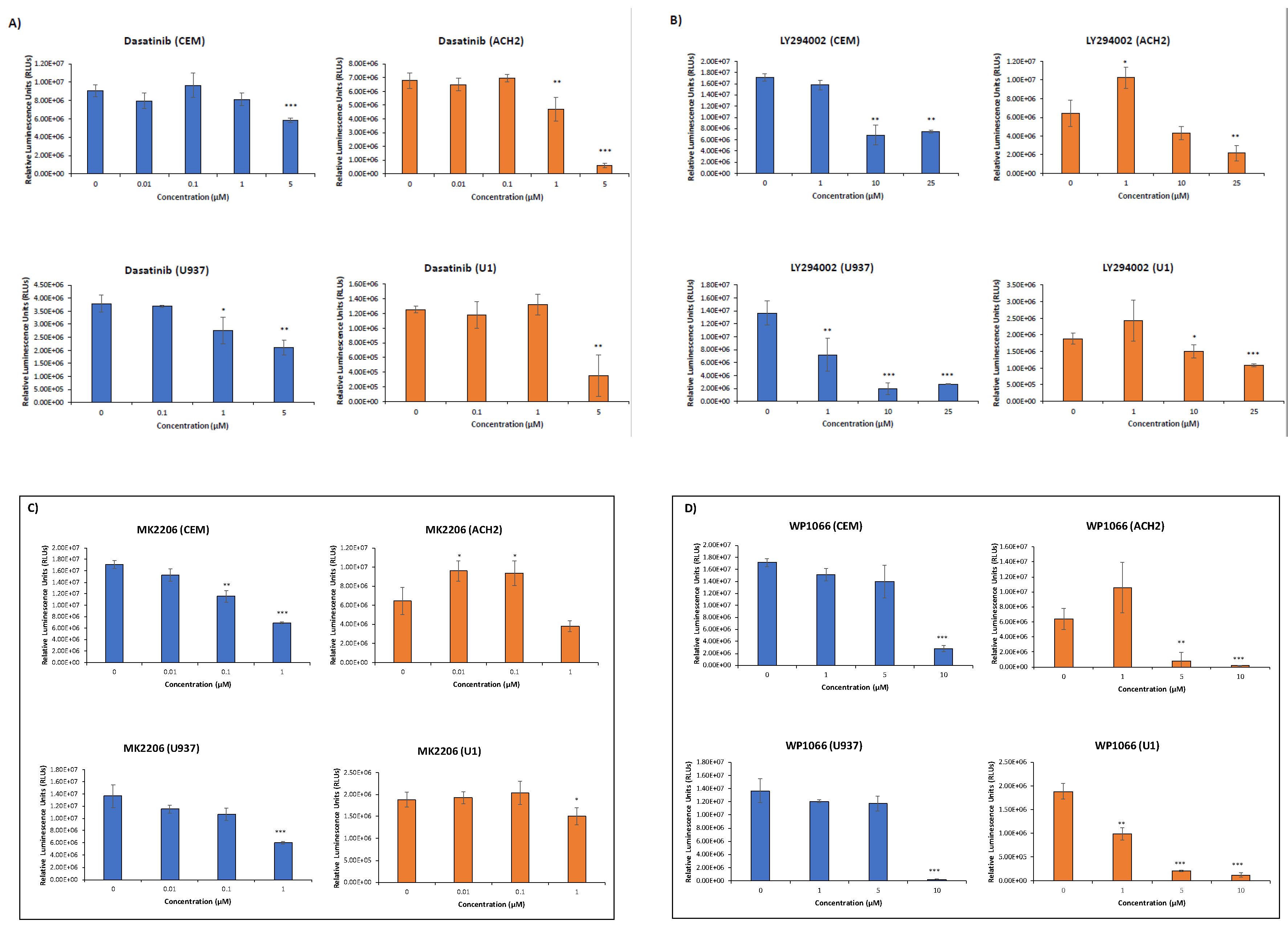
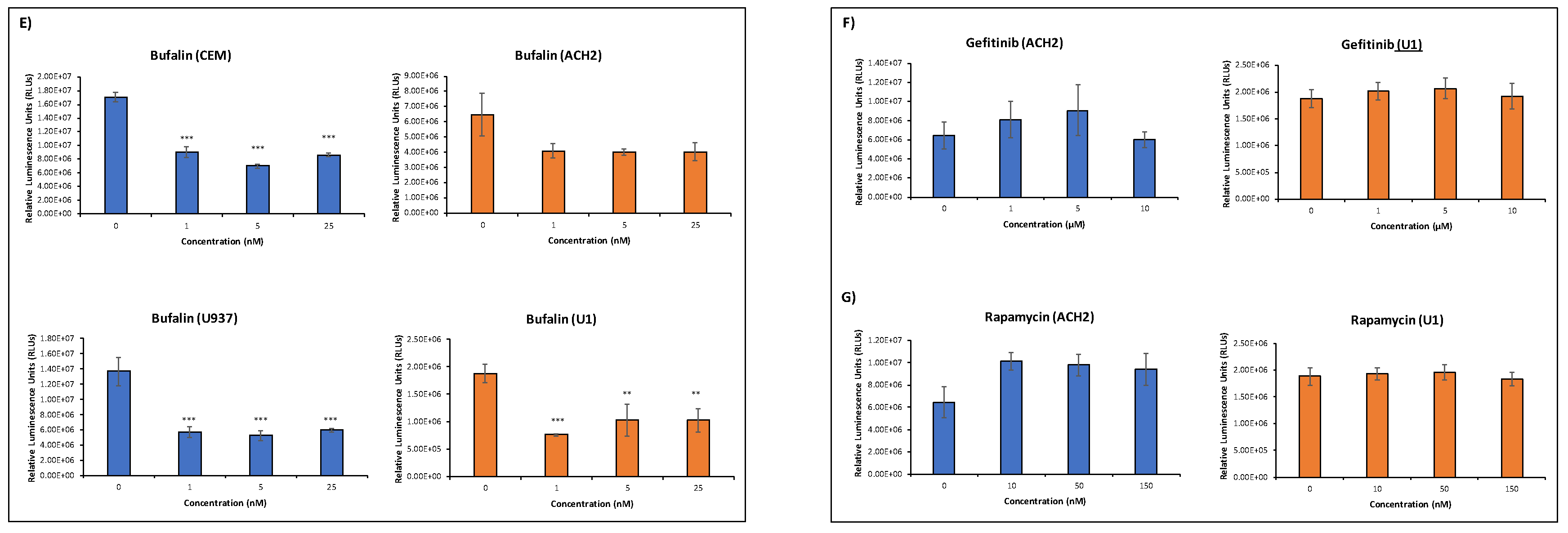
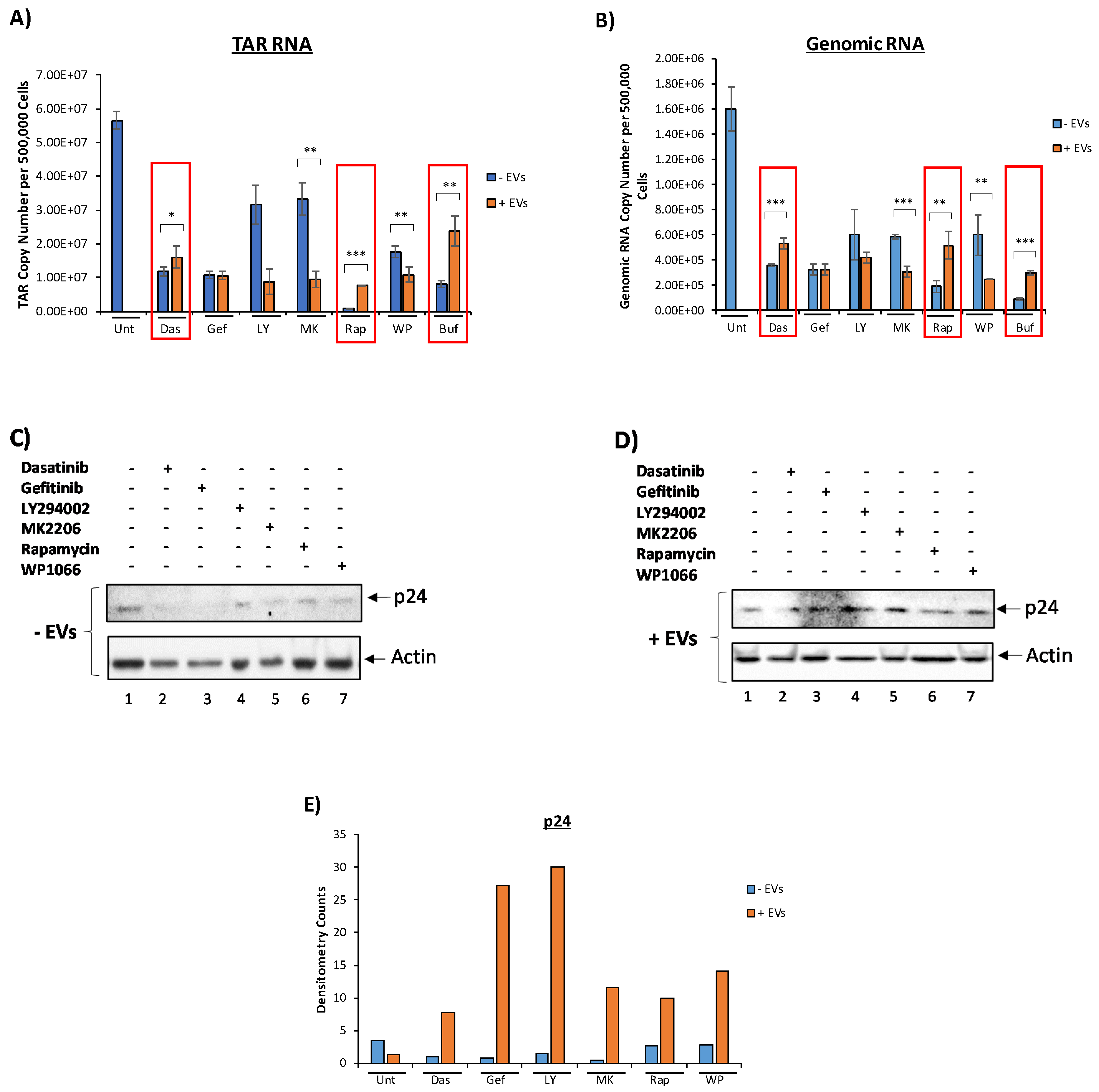
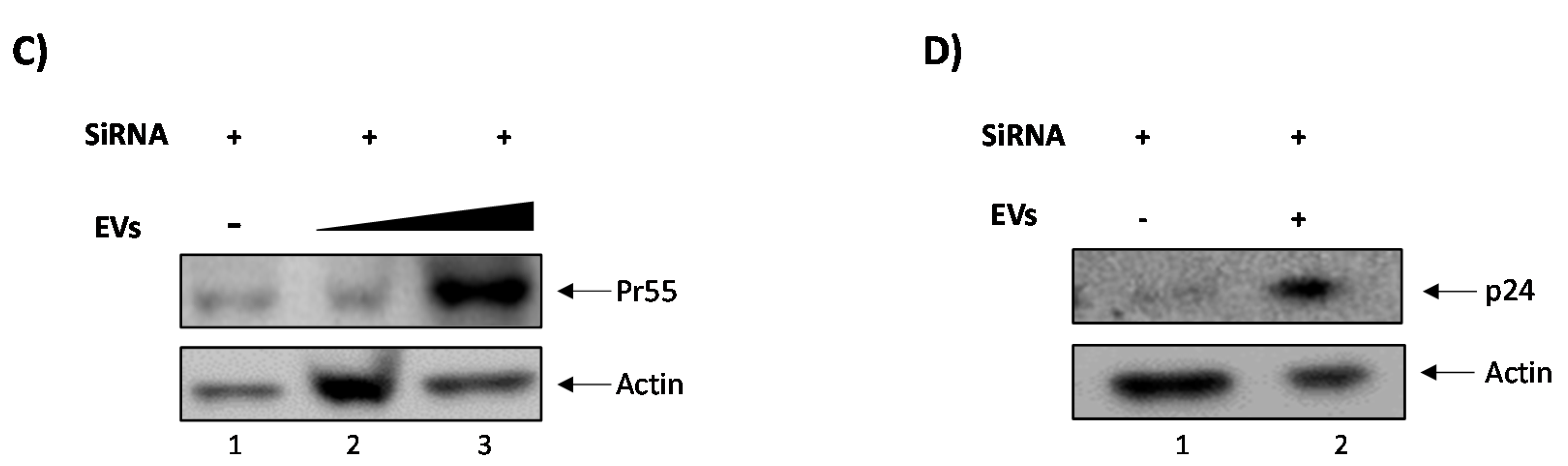
3.3. EVs Containing c-Src rescue HIV-1 Levels in Inhibitor-Treated Cells
3.4. Confirming EV-Associated c-Src Activates Latent HIV-1 in Infected Cells
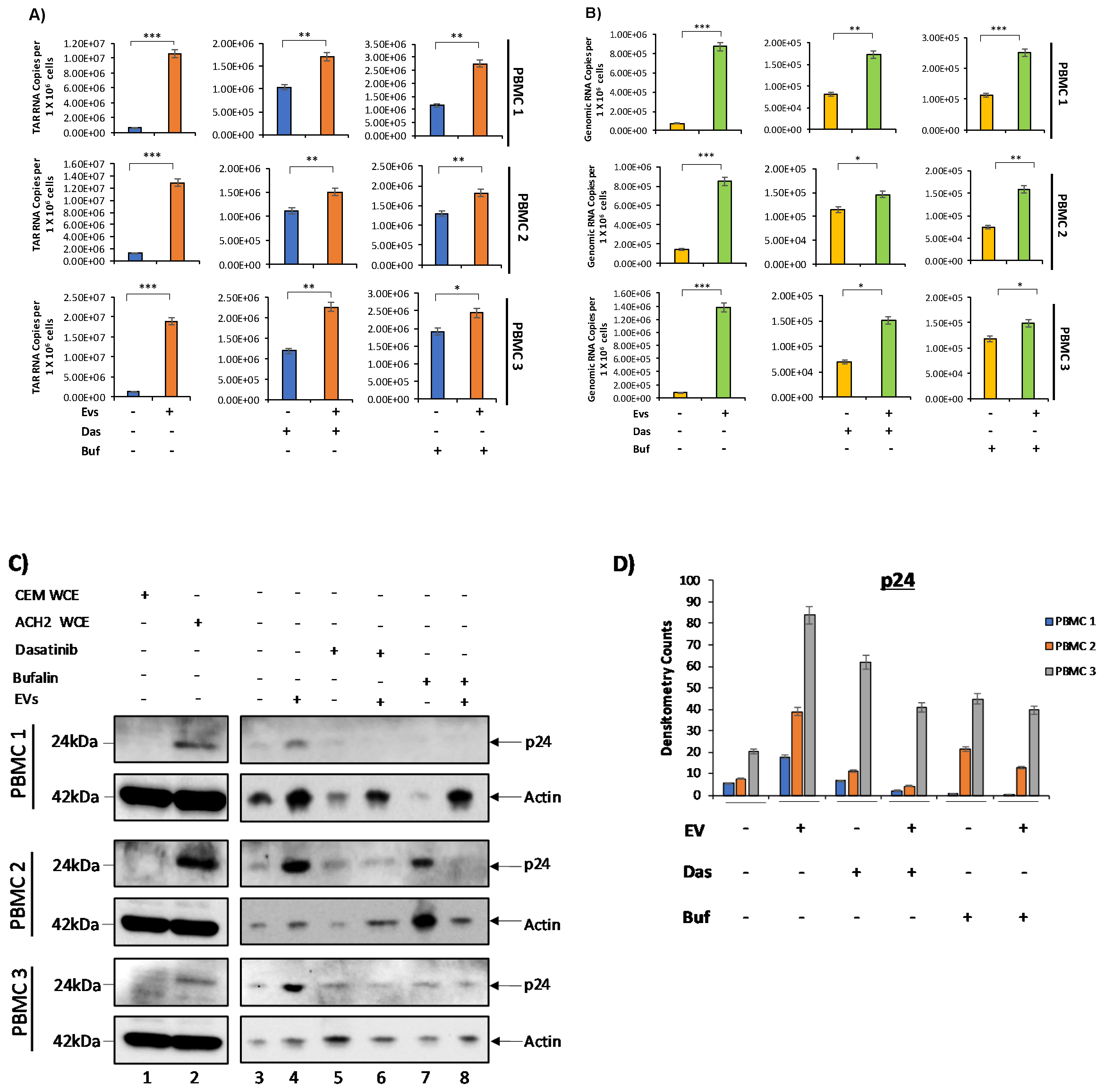
3.5. c-Src and SRC-1 Critical in HIV-1 Activation in Primary Cells
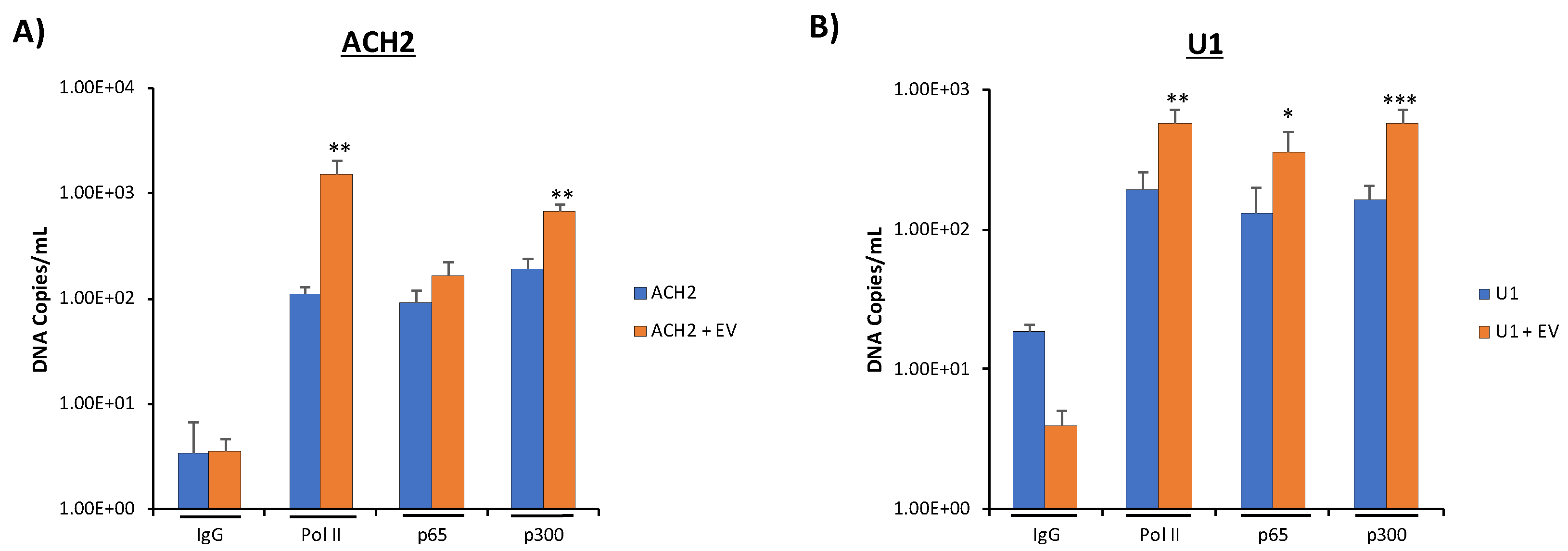
3.6. Increased Basal Transcription Is Driven by NF-κB/p300 Pathway
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ortblad, K.F.; Lozano, R.; Murray, C.J.L. The burden of HIV: Insights from the Global Burden of Disease Study 2010. AIDS 2013, 27, 2003–2017. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Abbas, W.; Herbein, G. HIV-1 latency in monocytes/macrophages. Viruses 2014, 6, 1837–1860. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.M. Latent HIV dynamics and implications for sustained viral suppression in the absence of antiretroviral therapy. J. Virus Erad. 2018, 4, 91–98. [Google Scholar]
- Barton, K.; Burch, B.; Soriano-Sarabia, N.; Margolis, D. Prospects for treatment of latent HIV. Clin. Pharmacol. Ther. 2013, 93, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Eisele, E.; Siliciano, R.F. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity 2012, 37, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Pasquier, E.; Watkins, J.; Bourgarel-Rey, V.; Peyrot, V.; Esquieu, D.; Barbier, P.; de Mareuil, J.; Braguer, D.; Kaleebu, P.; et al. The Glutamine-rich Region of the HIV-1 Tat Protein Is Involved in T-cell Apoptosis. J. Biol. Chem. 2004, 279, 48197–48204. [Google Scholar] [CrossRef]
- Barclay, R.A.; Schwab, A.; DeMarino, C.; Akpamagbo, Y.; Lepene, B.; Kassaye, S.; Iordanskiy, S.; Kashanchi, F. Exosomes from uninfected cells activate transcription of latent HIV-1. J. Biol. Chem. 2017, 292, 11682–11701. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Mathew, A.; Mason, A.B.; Teng, K. Exosome formation during maturation of mammalian and avian reticulocytes: Evidence that exosome release is a major route for externalization of obsolete membrane proteins. J. Cell. Physiol. 1991, 147, 27–36. [Google Scholar] [CrossRef]
- Gu, Y.; Li, M.; Wang, T.; Liang, Y.; Zhong, Z.; Wang, X.; Zhou, Q.; Chen, L.; Lang, Q.; He, Z.; et al. Lactation-Related MicroRNA Expression Profiles of Porcine Breast Milk Exosomes. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Rashed, M.H.; Bayraktar, E.; Helal, G.K.; Abd-Ellah, M.F.; Amero, P.; Chavez-Reyes, A.; Rodriguez-Aguayo, C. Exosomes: From Garbage Bins to Promising Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 538. [Google Scholar] [CrossRef]
- Admyre, C.; Johansson, S.M.; Qazi, K.R.; Filén, J.-J.; Lahesmaa, R.; Norman, M.; Neve, E.P.A.; Scheynius, A.; Gabrielsson, S. Exosomes with Immune Modulatory Features Are Present in Human Breast Milk. J. Immunol. 2007, 179, 1969–1978. [Google Scholar] [CrossRef]
- Batagov, A.O.; Kurochkin, I.V. Exosomes secreted by human cells transport largely mRNA fragments that are enriched in the 3′-untranslated regions. Biol. Direct 2013, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Pleet, M.L.; DeMarino, C.; Lepene, B.; Aman, M.J.; Kashanchi, F. The Role of Exosomal VP40 in Ebola Virus Disease. DNA Cell Biol. 2017, 36, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Sampey, G.C.; Meyering, S.S.; Zadeh, M.A.; Saifuddin, M.; Hakami, R.M.; Kashanchi, F. Exosomes and their role in CNS viral infections. J. Neurovirol. 2014, 20, 199–208. [Google Scholar] [CrossRef]
- Ahsan, N.A.; Sampey, G.C.; Lepene, B.; Akpamagbo, Y.; Barclay, R.A.; Iordanskiy, S.; Hakami, R.M.; Kashanchi, F. Presence of Viral RNA and Proteins in Exosomes from Cellular Clones Resistant to Rift Valley Fever Virus Infection. Front. Microbiol. 2016, 7, 139. [Google Scholar] [CrossRef]
- Fleming, A.; Sampey, G.; Chung, M.-C.; Bailey, C.; Van Hoek, M.L.; Kashanchi, F.; Hakami, R.M. The carrying pigeons of the cell: Exosomes and their role in infectious diseases caused by human pathogens. Pathog. Dis. 2014, 71, 109–120. [Google Scholar] [CrossRef]
- Alenquer, M.; Amorim, M.J. Exosome Biogenesis, Regulation, and Function in Viral Infection. Viruses 2015, 7, 5066–5083. [Google Scholar] [CrossRef]
- Kalani, A.; Tyagi, A.; Tyagi, N. Exosomes: Mediators of Neurodegeneration, Neuroprotection and Therapeutics. Mol. Neurobiol. 2014, 49, 590–600. [Google Scholar] [CrossRef]
- Narayanan, A.; Iordanskiy, S.; Das, R.; Van Duyne, R.; Santos, S.; Jaworski, E.; Guendel, I.; Sampey, G.; Dalby, E.; Iglesias-Ussel, M.; et al. Exosomes Derived from HIV-1-infected Cells Contain Trans-activation Response Element RNA. J. Biol. Chem. 2013, 288, 20014–20033. [Google Scholar] [CrossRef]
- DeMarino, C.; Pleet, M.L.; Cowen, M.; Barclay, R.A.; Akpamagbo, Y.; Erickson, J.; Ndembe, N.; Charurat, M.; Jumare, J.; Bwala, S.; et al. Antiretroviral Drugs Alter the Content of Extracellular Vesicles from HIV-1-Infected Cells. Sci. Rep. 2018, 8, 7653. [Google Scholar] [CrossRef] [PubMed]
- Akpamagbo, Y.A.; DeMarino, C.; Pleet, M.L.; Schwab, A.; Rodriguez, M.; Barclay, R.A.; Sampey, G.; Iordanskiy, S.; El-Hage, N.; Kashanchi, F. HIV-1 Transcription Inhibitors Increase the Synthesis of Viral Non-Coding RNA that Contribute to Latency. Curr. Pharm. Des. 2017, 23, 4133–4144. [Google Scholar] [CrossRef]
- Ung, T.H.; Madsen, H.J.; Hellwinkel, J.E.; Lencioni, A.M.; Graner, M.W. Exosome proteomics reveals transcriptional regulator proteins with potential to mediate downstream pathways. Cancer Sci. 2014, 105, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.M.; Brugge, J.S. Cellular Functions Regulated by Src Family Kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef] [PubMed]
- Irby, R.B.; Yeatman, T.J. Role of Src expression and activation in human cancer. Oncogene 2000, 19, 5636–5642. [Google Scholar] [CrossRef] [PubMed]
- Maa, M.C.; Leu, T.H.; McCarley, D.J.; Schatzman, R.C.; Parsons, S.J. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: Implications for the etiology of multiple human cancers. Proc. Natl. Acad. Sci. USA 1995, 92, 6981–6985. [Google Scholar] [CrossRef]
- Biscardi, J.S.; Ishizawar, R.C.; Silva, C.M.; Parsons, S.J. Tyrosine kinase signalling in breast cancer: Epidermal growth factor receptor and c-Src interactions in breast cancer. Breast Cancer Res. 2000, 2, 203–210. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef]
- LoPiccolo, J.; Blumenthal, G.M.; Bernstein, W.B.; Dennis, P.A. Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist. Updates 2008, 11, 32–50. [Google Scholar] [CrossRef]
- Heredia, A.; Le, N.; Gartenhaus, R.B.; Sausville, E.; Medina-Moreno, S.; Zapata, J.C.; Davis, C.; Gallo, R.C.; Redfield, R.R. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proc. Natl. Acad. Sci. USA 2015, 112, 9412–9417. [Google Scholar] [CrossRef]
- McCarthy, S.D.S.; Sakac, D.; Neschadim, A.; Branch, D.R. c-SRC protein tyrosine kinase regulates early HIV-1 infection post-entry. AIDS 2016, 30, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Gallay, P.; Swingler, S.; Aiken, C.; Trono, D. HIV-1 infection of nondividing cells: C-terminal tyrosine phosphorylation of the viral matrix protein is a key regulator. Cell 1995, 80, 379–388. [Google Scholar] [CrossRef][Green Version]
- Harmon, B.; Campbell, N.; Ratner, L. Role of Abl Kinase and the Wave2 Signaling Complex in HIV-1 Entry at a Post-Hemifusion Step. PLoS Pathog. 2010, 6, e1000956. [Google Scholar] [CrossRef]
- Readinger, J.A.; Schiralli, G.M.; Jiang, J.-K.; Thomas, C.J.; August, A.; Henderson, A.J.; Schwartzberg, P.L. Selective targeting of ITK blocks multiple steps of HIV replication. Proc. Natl. Acad. Sci. USA 2008, 105, 6684–6689. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Molecular Med. 2005, 9, 59–71. [Google Scholar] [CrossRef]
- Ersahin, T.; Tuncbag, N.; Cetin-Atalay, R. The PI3K/AKT/mTOR interactive pathway. Mol. BioSyst. 2015, 11, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Yao, S.; Hu, M.; Li, W.; Hao, T.; Zhou, F.; Zhu, X.; Lu, H.; Qin, D.; Yan, Q.; et al. HIV-1 Nef and KSHV oncogene K1 synergistically promote angiogenesis by inducing cellular miR-718 to regulate the PTEN/AKT/mTOR signaling pathway. Nucleic Acids Res. 2014, 42, 9862–9879. [Google Scholar] [CrossRef] [PubMed]
- Sampey, G.C.; Saifuddin, M.; Schwab, A.; Barclay, R.; Punya, S.; Chung, M.-C.; Hakami, R.M.; Zadeh, M.A.; Lepene, B.; Klase, Z.A.; et al. Exosomes from HIV-1-infected Cells Stimulate Production of Pro-inflammatory Cytokines through Trans-activating Response (TAR) RNA. J. Biol. Chem. 2016, 291, 1251–1266. [Google Scholar] [CrossRef]
- Gilbert, C.; Barat, C.; Cantin, R.; Tremblay, M.J. Involvement of Src and Syk Tyrosine Kinases in HIV-1 Transfer from Dendritic Cells to CD4+ T Lymphocytes. J. Immunol. 2007, 178, 2862–2871. [Google Scholar] [CrossRef]
- Trible, R.P.; Emert-Sedlak, L.; Smithgall, T.E. HIV-1 Nef Selectively Activates Src Family Kinases Hck, Lyn, and c-Src through Direct SH3 Domain Interaction. J. Biol. Chem. 2006, 281, 27029–27038. [Google Scholar] [CrossRef] [PubMed]
- Why the Need and How to Approach the Functional Diversity of Extracellular Vesicles. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5717434/ (accessed on 3 January 2020).
- Walker, V.G.; Ammer, A.; Cao, Z.; Clump, A.C.; Jiang, B.-H.; Kelley, L.C.; Weed, S.A.; Zot, H.; Flynn, D.C. PI3K activation is required for PMA-directed activation of cSrc by AFAP-110. Am. J. Physiol. Cell Physiol. 2007, 293, C119–C132. [Google Scholar] [CrossRef]
- Amata, I.; Maffei, M.; Pons, M. Phosphorylation of unique domains of Src family kinases. Front. Genet 2014, 5. [Google Scholar] [CrossRef]
- Xu, Y.Z.; Thuraisingam, T.; Kanagaratham, C.; Tao, S.; Radzioch, D. c-Src kinase is involved in the tyrosine phosphorylation and activity of SLC11A1 in differentiating macrophages. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Yu, G.; Glazer, R.I. Purification and characterization of p93fes- and p60src-related tyrosine protein kinase activities in differentiated HL-60 leukemia cells. J. Biol. Chem. 1987, 262, 17543–17548. [Google Scholar]
- Sato, K.; Sato, A.; Aoto, M.; Fukami, Y. c-Src phosphorylates epidermal growth factor receptor on tyrosine 845. Biochem. Biophys. Res. Commun. 1995, 215, 1078–1087. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Rodon, J.; Serra, V.; Tabernero, J. Picking the point of inhibition: A comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031. [Google Scholar] [CrossRef]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: An updated review. Ann. Med. 2014, 46, 372–383. [Google Scholar] [CrossRef]
- Freudlsperger, C.; Burnett, J.R.; Friedman, J.A.; Kannabiran, V.R.; Chen, Z.; Van Waes, C. EGFR-PI3K-AKT-mTOR signaling in head and neck squamous cell carcinomas: Attractive targets for molecular-oriented therapy. Expert Opin. Ther. Targets 2011, 15, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E.; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar] [CrossRef]
- Yuan, Z.-L.; Guan, Y.-J.; Wang, L.; Wei, W.; Kane, A.B.; Chin, Y.E. Central role of the threonine residue within the p+1 loop of receptor tyrosine kinase in STAT3 constitutive phosphorylation in metastatic cancer cells. Mol. Cell. Biol. 2004, 24, 9390–9400. [Google Scholar] [CrossRef] [PubMed]
- Stephanou, A.; Latchman, D.S. Opposing actions of STAT-1 and STAT-3. Growth Factors 2005, 23, 177–182. [Google Scholar] [CrossRef]
- Giraud, S.; Bienvenu, F.; Avril, S.; Gascan, H.; Heery, D.M.; Coqueret, O. Functional interaction of STAT3 transcription factor with the coactivator NcoA/SRC1a. J. Biol. Chem. 2002, 277, 8004–8011. [Google Scholar] [CrossRef]
- Walsh, C.A.; Qin, L.; Tien, J.C.-Y.; Young, L.S.; Xu, J. The function of steroid receptor coactivator-1 in normal tissues and cancer. Int. J. Biol. Sci. 2012, 8, 470–485. [Google Scholar] [CrossRef] [PubMed]
- Lindauer, M.; Hochhaus, A. Dasatinib. Recent Results Cancer Res. 2014, 201, 27–65. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, C.W. The Akt/PKB family of protein kinases: A review of small molecule inhibitors and progress towards target validation: A 2009 update. Curr. Top. Med. Chem. 2010, 10, 458–477. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lonard, D.M.; Yu, Y.; Chow, D.-C.; Palzkill, T.G.; Wang, J.; Qi, R.; Matzuk, A.J.; Song, X.; Madoux, F.; et al. Bufalin is a potent small-molecule inhibitor of the steroid receptor coactivators SRC-3 and SRC-1. Cancer Res. 2014, 74, 1506–1517. [Google Scholar] [CrossRef]
- Lu, K.; Chen, N.; Zhou, X.; Ge, X.; Feng, L.; Li, P.; Li, X.; Geng, L.; Wang, X. The STAT3 inhibitor WP1066 synergizes with vorinostat to induce apoptosis of mantle cell lymphoma cells. Biochem. Biophys. Res. Commun. 2015, 464, 292–298. [Google Scholar] [CrossRef]
- Nika, K.; Soldani, C.; Salek, M.; Paster, W.; Gray, A.; Etzensperger, R.; Fugger, L.; Polzella, P.; Cerundolo, V.; Dushek, O.; et al. Constitutively Active Lck Kinase in T Cells Drives Antigen Receptor Signal Transduction. Immunity 2010, 32, 766–777. [Google Scholar] [CrossRef]
- Masiello, D.; Gorospe, G.; Yang, A.S. The occurrence and management of fluid retention associated with TKI therapy in CML, with a focus on dasatinib. J. Hematol. Oncol. 2009, 2, 46. [Google Scholar] [CrossRef][Green Version]
- Archin, N.M.; Sung, J.M.; Garrido, C.; Soriano-Sarabia, N.; Margolis, D.M. Eradicating HIV-1 infection: Seeking to clear a persistent pathogen. Nat. Rev. Microbiol. 2014, 12, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Lusic, M.; Marcello, A.; Cereseto, A.; Giacca, M. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 2003, 22, 6550–6561. [Google Scholar] [CrossRef]
- Vo, N.; Goodman, R.H. CREB-binding protein and p300 in transcriptional regulation. J. Biol. Chem. 2001, 276, 13505–13508. [Google Scholar] [CrossRef]
- Mantelingu, K.; Reddy, B.A.A.; Swaminathan, V.; Kishore, A.H.; Siddappa, N.B.; Kumar, G.V.P.; Nagashankar, G.; Natesh, N.; Roy, S.; Sadhale, P.P.; et al. Specific inhibition of p300-HAT alters global gene expression and represses HIV replication. Chem. Biol. 2007, 14, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Tetel, M.J. Nuclear receptor coactivators: Essential players for steroid hormone action in the brain and in behaviour. J. Neuroendocrinol. 2009, 21, 229–237. [Google Scholar] [CrossRef]
- Schwab, A.; Meyering, S.S.; Lepene, B.; Iordanskiy, S.; Van Hoek, M.L.; Hakami, R.M.; Kashanchi, F. Extracellular vesicles from infected cells: Potential for direct pathogenesis. Front. Microbiol. 2015, 6, 1132. [Google Scholar] [CrossRef] [PubMed]
- Vlassov, A.V.; Magdaleno, S.; Setterquist, R.; Conrad, R. Exosomes: Current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochimica et Biophysica Acta (BBA)—General Subjects 2012, 1820, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Prieto, D.; Sotelo, N.; Seija, N.; Sernbo, S.; Abreu, C.; Durán, R.; Gil, M.; Sicco, E.; Irigoin, V.; Oliver, C.; et al. S100-A9 protein in exosomes from chronic lymphocytic leukemia cells promotes NF-κB activity during disease progression. Blood 2017, 130, 777–788. [Google Scholar] [CrossRef]
- Konadu, K.A.; Chu, J.; Huang, M.B.; Amancha, P.K.; Armstrong, W.; Powell, M.D.; Villinger, F.; Bond, V.C. Association of Cytokines With Exosomes in the Plasma of HIV-1-Seropositive Individuals. J. Infect. Dis. 2015, 211, 1712–1716. [Google Scholar] [CrossRef]
- Litterst, C.M.; Pfitzner, E. Transcriptional activation by STAT6 requires the direct interaction with NCoA-1. J. Biol. Chem. 2001, 276, 45713–45721. [Google Scholar] [CrossRef]
- Marzio, G.; Tyagi, M.; Gutierrez, M.I.; Giacca, M. HIV-1 tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc. Natl. Acad. Sci. USA 1998, 95, 13519–13524. [Google Scholar] [CrossRef] [PubMed]
- Benkirane, M.; Chun, R.F.; Xiao, H.; Ogryzko, V.V.; Howard, B.H.; Nakatani, Y.; Jeang, K.T. Activation of integrated provirus requires histone acetyltransferase. p300 and P/CAF are coactivators for HIV-1 Tat. J. Biol. Chem. 1998, 273, 24898–24905. [Google Scholar] [CrossRef]
- Hatano, H.; Jain, V.; Hunt, P.W.; Lee, T.-H.; Sinclair, E.; Do, T.D.; Hoh, R.; Martin, J.N.; McCune, J.M.; Hecht, F.; et al. Cell-based measures of viral persistence are associated with immune activation and programmed cell death protein 1 (PD-1)-expressing CD4+ T cells. J. Infect. Dis. 2013, 208, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Imamichi, H.; Dewar, R.L.; Adelsberger, J.W.; Rehm, C.A.; O’Doherty, U.; Paxinos, E.E.; Fauci, A.S.; Lane, H.C. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2016, 113, 8783–8788. [Google Scholar] [CrossRef]
- Maldarelli, F.; Kearney, M.; Palmer, S.; Stephens, R.; Mican, J.; Polis, M.A.; Davey, R.T.; Kovacs, J.; Shao, W.; Rock-Kress, D.; et al. HIV populations are large and accumulate high genetic diversity in a nonlinear fashion. J. Virol. 2013, 87, 10313–10323. [Google Scholar] [CrossRef] [PubMed]
- Boltz, V.F.; Zheng, Y.; Lockman, S.; Hong, F.; Halvas, E.K.; McIntyre, J.; Currier, J.S.; Chibowa, M.C.; Kanyama, C.; Nair, A.; et al. Role of low-frequency HIV-1 variants in failure of nevirapine-containing antiviral therapy in women previously exposed to single-dose nevirapine. Proc. Natl. Acad. Sci. USA 2011, 108, 9202–9207. [Google Scholar] [CrossRef]
- Kumar, A.M.; Borodowsky, I.; Fernandez, B.; Gonzalez, L.; Kumar, M. Human immunodeficiency virus type 1 RNA Levels in different regions of human brain: Quantification using real-time reverse transcriptase-polymerase chain reaction. J. Neurovirol. 2007, 13, 210–224. [Google Scholar] [CrossRef]
- Cihlar, T.; Fordyce, M. Current status and prospects of HIV treatment. Curr. Opin. Virol. 2016, 18, 50–56. [Google Scholar] [CrossRef]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef]
- Honda, S.; Sadatomi, D.; Yamamura, Y.; Nakashioya, K.; Tanimura, S.; Takeda, K. WP1066 suppresses macrophage cell death induced by inflammasome agonists independently of its inhibitory effect on STAT3. Cancer Sci. 2017, 108, 520–527. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barclay, R.A.; Mensah, G.A.; Cowen, M.; DeMarino, C.; Kim, Y.; Pinto, D.O.; Erickson, J.; Kashanchi, F. Extracellular Vesicle Activation of Latent HIV-1 Is Driven by EV-Associated c-Src and Cellular SRC-1 via the PI3K/AKT/mTOR Pathway. Viruses 2020, 12, 665. https://doi.org/10.3390/v12060665
Barclay RA, Mensah GA, Cowen M, DeMarino C, Kim Y, Pinto DO, Erickson J, Kashanchi F. Extracellular Vesicle Activation of Latent HIV-1 Is Driven by EV-Associated c-Src and Cellular SRC-1 via the PI3K/AKT/mTOR Pathway. Viruses. 2020; 12(6):665. https://doi.org/10.3390/v12060665
Chicago/Turabian StyleBarclay, Robert A., Gifty A. Mensah, Maria Cowen, Catherine DeMarino, Yuriy Kim, Daniel O. Pinto, James Erickson, and Fatah Kashanchi. 2020. "Extracellular Vesicle Activation of Latent HIV-1 Is Driven by EV-Associated c-Src and Cellular SRC-1 via the PI3K/AKT/mTOR Pathway" Viruses 12, no. 6: 665. https://doi.org/10.3390/v12060665
APA StyleBarclay, R. A., Mensah, G. A., Cowen, M., DeMarino, C., Kim, Y., Pinto, D. O., Erickson, J., & Kashanchi, F. (2020). Extracellular Vesicle Activation of Latent HIV-1 Is Driven by EV-Associated c-Src and Cellular SRC-1 via the PI3K/AKT/mTOR Pathway. Viruses, 12(6), 665. https://doi.org/10.3390/v12060665