The Interplay between Adeno-Associated Virus and Its Helper Viruses



Abstract
1. Introduction
1.1. Adeno-Associated Virus
1.2. AAV Biology
2. Helper Viruses and AAV
2.1. Herpesviruses
2.1.1. HSV-1 Biology
2.1.2. HSV-1 Helper Functions
2.2. Adenoviruses
2.2.1. Adenovirus Biology
2.2.2. AdV Helper Functions
2.3. Other Helper Viruses
2.3.1. Other Herpesviruses
2.3.2. Papillomaviruses
2.3.3. Bocaviruses
3. AAV-Mediated Inhibition of Helper Virus Replication
3.1. AAV Inhibits HSV-1 DNA Replication
3.2. AAV2 Inhibits AdV Replication
3.3. AAV Inhibits Other Helper Viruses
4. AAV Vectors for Gene Therapy
4.1. Production of AAV Gene Therapy Vectors
4.2. AAV Hybrid Vectors
5. Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Atchison, R.W.; Casto, B.C.; Hammon, W.M. Adenovirus-Associated Defective Virus Particles. Science 1965. [Google Scholar] [CrossRef]
- Hoggan, M.D.; Blacklow, N.R.; Rowe, W.P. Studies of small DNA viruses found in various adenovirus preparations: Physical, biological, and immunological characteristics. Proc. Natl. Acad. Sci. USA 1966, 55, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Asokan, A.; Schaffer, D.V.; Jude Samulski, R. The AAV Vector Toolkit: Poised at the Clinical Crossroads. Mol. Ther. 2012, 20, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.E.; Rolling, F.; Li, C.; Conrath, H.; Xiao, W.; Xiao, X.; Samulski, R.J. Cross-Packaging of a Single Adeno-Associated Virus (AAV) Type 2 Vector Genome into Multiple AAV Serotypes Enables Transduction with Broad Specificity. J. Virol. 2002, 76, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Calcedo, R.; Vandenberghe, L.H.; Gao, G.; Lin, J.; Wilson, J.M. Worldwide Epidemiology of Neutralizing Antibodies to Adeno-Associated Viruses. J. Infect. Dis. 2009, 199, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Huang, W.; Zhang, H.; Wang, Y.; Zhao, J.; Song, A.; Xie, H.; Zhao, C.; Gao, D.; Wang, Y. Neutralizing antibodies against AAV2, AAV5 and AAV8 in healthy and HIV-1-infected subjects in China: Implications for gene therapy using AAV vectors. Gene Ther. 2014, 21, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Louis Jeune, V.; Joergensen, J.A.; Hajjar, R.J.; Weber, T. Pre-existing Anti–Adeno-Associated Virus Antibodies as a Challenge in AAV Gene Therapy. Hum. Gene Ther. Methods 2013, 24, 59–67. [Google Scholar] [CrossRef]
- Atchison, R.W. The Role of Herpesvirus in Adeno-Associated Virus Replication in Vitro. Virology 1970, 42, 155–162. [Google Scholar] [CrossRef]
- Georg-Fries, B.; Biederlack, S.; Wolf, J.; Zur Hausen, H. Analysis of proteins, helper dependence, and seroepidemiology of a new human parvovirus. Virology 1984, 134, 64–71. [Google Scholar] [CrossRef]
- Ogston, P.; Raj, K.; Beard, P. Productive Replication of Adeno-Associated Virus Can Occur in Human Papillomavirus Type 16 (HPV-16) Episome-Containing Keratinocytes and Is Augmented by the HPV-16 E2 Protein. J. Virol. 2000, 74, 3494–3504. [Google Scholar] [CrossRef]
- Urabe, M.; Nakakura, T.; Xin, K.-Q.; Obara, Y.; Mizukami, H.; Kume, A.; Kotin, R.M.; Ozawa, K. Scalable Generation of High-Titer Recombinant Adeno-Associated Virus Type 5 in Insect Cells. J. Virol. 2006, 80, 1874–1885. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Deng, X.; Zou, W.; Engelhardt, J.F.; Yan, Z.; Qiu, J. Human Bocavirus 1 Is a Novel Helper for Adeno-associated Virus Replication. J. Virol. 2017, 91, e00710–e00717. [Google Scholar] [CrossRef] [PubMed]
- Heilbronn, R.; Schlehofer, J.R.; Yalkinoglu, A.O.; Zur Hausen, H. Selective DNA-amplification induced by carcinogens (initiators): Evidence for a role of proteases and DNA polymerase alpha. Int. J. Cancer 1985, 36, 85–91. [Google Scholar] [CrossRef]
- Schlehofer, J.R.; Heilbronn, R.; Georg-Fries, B.; zur Hausen, H. Inhibition of initiator-induced SV40 gene amplification in SV40-transformed Chinese hamster cells by infection with a defective parvovirus. Int. J. Cancer 1983, 32, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Yakobson, B.; Koch, T.; Winocour, E. Replication of adeno-associated virus in synchronized cells without the addition of a helper virus. J. Virol. 1987, 61, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Yalkinoglu, A.Ö.; Heilbronn, R.; Bürkle, A.; Schlehofer, J.R.; zur Hausen, H. DNA amplification of adeno-associated virus as a response to cellular genotoxic stress. Cancer Res. 1988, 48, 3123–3129. [Google Scholar] [PubMed]
- Yue, Y.; Duan, D. Double strand interaction is the predominant pathway for intermolecular recombination of adeno-associated viral genomes. Virology 2003, 313, 1–7. [Google Scholar] [CrossRef][Green Version]
- Sun, X.; Lu, Y.; Bish, L.T.; Calcedo, R.; Wilson, J.M.; Gao, G. Molecular Analysis of Vector Genome Structures After Liver Transduction by Conventional and Self-Complementary Adeno-Associated Viral Serotype Vectors in Murine and Nonhuman Primate Models. Hum. Gene Ther. 2010, 21, 750–761. [Google Scholar] [CrossRef]
- Samulski, R.J.; Muzyczka, N. AAV-Mediated Gene Therapy for Research and Therapeutic Purposes. Annu. Rev. Virol. 2014, 1, 427–451. [Google Scholar] [CrossRef]
- Rose, J.A.; Maizel, J.V.; Inman, J.K.; Shatkin, A.J. Structural Proteins of Adenovirus-Associated Viruses. J. Virol. 1971, 8, 766–770. [Google Scholar] [CrossRef]
- Xie, Q.; Bu, W.; Bhatia, S.; Hare, J.; Somasundaram, T.; Azzi, A.; Chapman, M.S. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 10405–10410. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, S.; Kleinschmidt, J.A.; Böttcher, B. Electron cryo-microscopy and image reconstruction of adeno-associated virus type 2 empty capsids. Embo Rep. 2001, 2, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Lusby, E.W.; Berns, K.I. Nucleotide sequence and organization of the adeno-associated virus 2 genome. J. Virol. 1983, 45, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Laughlin, C.A.; Westphal, H.; Carter, B.J. Spliced adenovirus-associated virus RNA. Proc. Natl. Acad. Sci. USA 1979, 76, 5567–5571. [Google Scholar] [CrossRef]
- Green, M.R.; Roeder, R.G. Transcripts of the adeno-associated virus genome: Mapping of the major RNAs. J. Virol. 1980, 36, 79–92. [Google Scholar] [CrossRef]
- Green, M.R.; Roeder, R.G. Definition of a novel promoter for the major adenovirus-associated virus mRNA. Cell 1980, 22, 231–242. [Google Scholar] [CrossRef]
- Lusby, E.W.; Berns, K.I. Mapping of the 5’ termini of two adeno-associated virus 2 RNAs in the left half of the genome. J. Virol. 1982, 41, 518–526. [Google Scholar] [CrossRef]
- Mendelson, E.; Trempe, J.P.; Carter, B.J. Identification of the trans-acting Rep proteins of adeno-associated virus by antibodies to a synthetic oligopeptide. J. Virol. 1986, 60, 823–832. [Google Scholar] [CrossRef]
- Trempe, J.P.; Carter, B.J. Alternate mRNA splicing is required for synthesis of adeno-associated virus VP1 capsid protein. J. Virol. 1988, 62, 3356–3363. [Google Scholar] [CrossRef]
- Sonntag, F.; Schmidt, K.; Kleinschmidt, J.A. A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc. Natl. Acad. Sci. USA 2010, 107, 10220–10225. [Google Scholar] [CrossRef]
- Lusby, E.W.; Fife, K.H.; Berns, K.I. Nucleotide Sequence of the Inverted Terminal Repetition in Adeno-Associated Virus DNA. J. Virol. 1980, 34, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Samulski, R.J.; Berns, K.I.; Tan, M.; Muzyczka, N. Cloning of adeno-associated virus into pBR322: Rescue of intact virus from the recombinant plasmid in human cells. Proc. Natl. Acad. Sci. USA 1982, 79, 2077–2081. [Google Scholar] [CrossRef] [PubMed]
- Samulski, R.J.; Srivastava, A.; Berns, K.I.; Muzyczka, N. Rescue of adeno-associated virus from recombinant plasmids: Gene correction within the terminal repeats of AAV. Cell 1983, 33, 135–143. [Google Scholar] [CrossRef]
- Senapathy, P.; Tratschin, J.-D.; Carter, B.J. Replication of Adeno-associated virus DNA. J. Mol. Biol. 1984. [Google Scholar] [CrossRef]
- Berns, K.I. The Unusual Properties of the AAV Inverted Terminal Repeat. Hum. Gene Ther. 2020. [Google Scholar] [CrossRef]
- Berns, K.I.; Adler, S. Separation of Two Types of Adeno-Associated Virus Particles Containing Complementary Polynucleotide Chains. J. Virol. 1972, 9, 394–396. [Google Scholar] [CrossRef]
- Samulski, R.J.; Chang, L.S.; Shenk, T. A recombinant plasmid from which an infectious adeno-associated virus genome can be excised in vitro and its use to study viral replication. J. Virol. 1987, 61, 3096–3101. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zeng, X.; Fan, Z.; Li, C.; McCown, T.; Samulski, R.J.; Xiao, X. Adeno-associated Virus of a Single-polarity DNA Genome Is Capable of Transduction In Vivo. Mol. Ther. 2008, 16, 494–499. [Google Scholar] [CrossRef]
- Nonnenmacher, M.; Weber, T. Intracellular transport of recombinant adeno-associated virus vectors. Gene Ther. 2012, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Summerford, C.; Bartlett, J.S.; Samulski, R.J. αVβ5 integrin: A co-receptor for adeno-associated virus type 2 infection. Nat. Med. 1999, 5, 78–82. [Google Scholar] [CrossRef]
- Qiu, J.; Brown, K.E. Integrin alphaVbeta5 is not involved in adeno-associated virus type 2 (AAV2) infection. Virology 1999, 264, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Akache, B.; Grimm, D.; Shen, X.; Fuess, S.; Yant, S.R.; Glazer, D.S.; Park, J.; Kay, M.A. A Two-hybrid Screen Identifies Cathepsins B and L as Uncoating Factors for Adeno-associated Virus 2 and 8. Mol. Ther. 2007, 15, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Wallen, A.J.; Barker, G.A.; Fein, D.E.; Jing, H.; Diamond, S.L. Enhancers of Adeno-associated Virus AAV2 Transduction via High Throughput siRNA Screening. Mol. Ther. 2011, 19, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Pillay, S.; Meyer, N.L.; Puschnik, A.S.; Davulcu, O.; Diep, J.; Ishikawa, Y.; Jae, L.T.; Wosen, J.E.; Nagamine, C.M.; Chapman, M.S.; et al. An essential receptor for adeno-associated virus infection. Nature 2016, 530, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Jensen, R.L.; Schnepp, B.C.; Connell, M.J.; Shell, R.; Sferra, T.J.; Bartlett, J.S.; Clark, K.R.; Johnson, P.R. Molecular Characterization of Adeno-Associated Viruses Infecting Children. J. Virol. 2005, 79, 14781–14792. [Google Scholar] [CrossRef]
- Dudek, A.M.; Zabaleta, N.; Zinn, E.; Pillay, S.; Zengel, J.; Porter, C.; Franceschini, J.S.; Estelien, R.; Carette, J.E.; Zhou, G.L.; et al. GPR108 Is a Highly Conserved AAV Entry Factor. Mol. Ther. 2020, 28, 367–381. [Google Scholar] [CrossRef]
- Bartlett, J.S.; Wilcher, R.; Samulski, R.J. Infectious Entry Pathway of Adeno-Associated Virus and Adeno-Associated Virus Vectors. J. Virol. 2000, 74, 2777–2785. [Google Scholar] [CrossRef]
- Nonnenmacher, M.; Weber, T. Adeno-Associated Virus 2 Infection Requires Endocytosis through the CLIC/GEEC Pathway. Cell Host Microbe. 2011, 10, 563–576. [Google Scholar] [CrossRef]
- Nonnenmacher, M.E.; Cintrat, J.-C.; Gillet, D.; Weber, T. Syntaxin 5-Dependent Retrograde Transport to the trans -Golgi Network Is Required for Adeno-Associated Virus Transduction. J. Virol. 2015, 89, 1673–1687. [Google Scholar] [CrossRef]
- Sonntag, F.; Bleker, S.; Leuchs, B.; Fischer, R.; Kleinschmidt, J.A. Adeno-Associated Virus Type 2 Capsids with Externalized VP1/VP2 Trafficking Domains Are Generated prior to Passage through the Cytoplasm and Are Maintained until Uncoating Occurs in the Nucleus. J. Virol. 2006, 80, 11040–11054. [Google Scholar] [CrossRef]
- Zádori, Z.; Szelei, J.; Lacoste, M.-C.; Li, Y.; Gariépy, S.; Raymond, P.; Allaire, M.; Nabi, I.R.; Tijssen, P. A Viral Phospholipase A2 Is Required for Parvovirus Infectivity. Dev. Cell 2001, 1, 291–302. [Google Scholar] [CrossRef]
- Grieger, J.C.; Snowdy, S.; Samulski, R.J. Separate Basic Region Motifs within the Adeno-Associated Virus Capsid Proteins Are Essential for Infectivity and Assembly. J. Virol. 2006, 80, 5199–5210. [Google Scholar] [CrossRef]
- Girod, A.; Wobus, C.E.; Zádori, Z.; Ried, M.; Leike, K.; Tijssen, P.; Kleinschmidt, J.A.; Hallek, M. The VP1 capsid protein of adeno-associated virus type 2 is carrying a phospholipase A2 domain required for virus infectivity. J. Gen. Virol. 2002, 83, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Stahnke, S.; Lux, K.; Uhrig, S.; Kreppel, F.; Hösel, M.; Coutelle, O.; Ogris, M.; Hallek, M.; Büning, H. Intrinsic phospholipase A2 activity of adeno-associated virus is involved in endosomal escape of incoming particles. Virology 2011, 409, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.S.; Samulski, R.J. Enhancement of Adeno-Associated Virus Infection by Mobilizing Capsids into and Out of the Nucleolus. J. Virol. 2009, 83, 2632–2644. [Google Scholar] [CrossRef]
- Thomas, C.E.; Storm, T.A.; Huang, Z.; Kay, M.A. Rapid Uncoating of Vector Genomes Is the Key to Efficient Liver Transduction with Pseudotyped Adeno-Associated Virus Vectors. J. Virol. 2004, 78, 3110–3122. [Google Scholar] [CrossRef]
- Rossi, A.; Dupaty, L.; Aillot, L.; Zhang, L.; Gallien, C.; Hallek, M.; Odenthal, M.; Adriouch, S.; Salvetti, A.; Büning, H. Vector uncoating limits adeno-associated viral vector-mediated transduction of human dendritic cells and vector immunogenicity. Sci. Rep. 2019, 9, 3631. [Google Scholar] [CrossRef]
- Cheung, A.K.; Hoggan, M.D.; Hauswirth, W.W.; Berns, K.I. Integration of the adeno-associated virus genome into cellular DNA in latently infected human Detroit 6 cells. J. Virol. 1980, 33, 739–748. [Google Scholar] [CrossRef]
- Dyall, J.; Szabo, P.; Berns, K.I. Adeno-associated virus (AAV) site-specific integration: Formation of AAV-AAVS1 junctions in an in vitro system. Proc. Natl. Acad. Sci. USA 1999, 96, 12849–12854. [Google Scholar] [CrossRef]
- Huser, D.; Weger, S.; Heilbronn, R. Kinetics and Frequency of Adeno-Associated Virus Site-Specific Integration into Human Chromosome 19 Monitored by Quantitative Real-Time PCR. J. Virol. 2002, 76, 7554–7559. [Google Scholar] [CrossRef]
- McCarty, D.M.; Young, S.M.; Samulski, R.J. Integration of Adeno-Associated Virus (AAV) and Recombinant AAV Vectors. Annu. Rev. Genet. 2004, 38, 819–845. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.J.; McCarty, D.M.; Muzyczka, N. The adeno-associated virus (AAV) Rep protein acts as both a repressor and an activator to regulate AAV transcription during a productive infection. J. Virol. 1997, 71, 1079–1088. [Google Scholar] [CrossRef] [PubMed]
- Nash, K.; Chen, W.; McDonald, W.F.; Zhou, X.; Muzyczka, N. Purification of Host Cell Enzymes Involved in Adeno-Associated Virus DNA Replication. J. Virol. 2007, 81, 5777–5787. [Google Scholar] [CrossRef] [PubMed]
- Nash, K.; Chen, W.; Muzyczka, N. Complete In Vitro Reconstitution of Adeno-Associated Virus DNA Replication Requires the Minichromosome Maintenance Complex Proteins. J. Virol. 2008, 82, 1458–1464. [Google Scholar] [CrossRef]
- Ni, T.-H.; McDonald, W.F.; Zolotukhin, I.; Melendy, T.; Waga, S.; Stillman, B.; Muzyczka, N. Cellular Proteins Required for Adeno-Associated Virus DNA Replication in the Absence of Adenovirus Coinfection. J. Virol. 1998, 72, 2777–2787. [Google Scholar] [CrossRef]
- Fields, B.N.; Knipe, D.M.; Howley, P.M. Fields’ Virology; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Im, D.-S.; Muzyczka, N. The AAV Origin Binding Protein Rep68 Is an ATP-Dependent Site-Specific Endonuclease with DNA Helicase Activity. Cell 1990, 447–457. [Google Scholar] [CrossRef]
- Maurer, A.C.; Pacouret, S.; Cepeda Diaz, A.K.; Blake, J.; Andres-Mateos, E.; Vandenberghe, L.H. The Assembly-Activating Protein Promotes Stability and Interactions between AAV’s Viral Proteins to Nucleate Capsid Assembly. Cell Rep. 2018, 23, 1817–1830. [Google Scholar] [CrossRef]
- Bleker, S.; Sonntag, F.; Kleinschmidt, J.A. Mutational Analysis of Narrow Pores at the Fivefold Symmetry Axes of Adeno-Associated Virus Type 2 Capsids Reveals a Dual Role in Genome Packaging and Activation of Phospholipase A2 Activity. J. Virol. 2005, 79, 2528–2540. [Google Scholar] [CrossRef] [PubMed]
- King, J.A.; Dubielzig, R.; Grimm, D.; Kleinschmidt, J.A. DNA helicase-mediated packaging of adeno-associated virus type 2 genomes into preformed capsids. Embo J. 2001, 20, 3282–3291. [Google Scholar] [CrossRef] [PubMed]
- Nony, P.; Chadeuf, G.; Tessier, J.; Moullier, P.; Salvetti, A. Evidence for Packaging of rep-cap Sequences into Adeno-Associated Virus (AAV) Type 2 Capsids in the Absence of Inverted Terminal Repeats: A Model for Generation of rep-Positive AAV Particles. J. Virol. 2003, 77, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Buller, R.M.L.; Janik, J.E.; Sebring, E.D.; Rose, J.A. Herpes Simplex Virus Types 1 and 2 Completely Help Adenovirus-Associated Virus Replication. J. Virol. 1981, 40, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Blacklow, N.R. Brief Communication: Potentiation of an Adenovirus-Associated Virus by Herpes Simplex Virus Type-2-Transformed Cells2. Jnci J. Natl. Cancer Inst. 1975, 54, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Thomson, B.J.; Weindler, F.W.; Gray, D.; Schwaab, V.; Heilbronn, R. Human Herpesvirus 6 (HHV-6) Is a Helper Virus for Adeno-Associated Virus Type 2 (AAV-2) and the AAV-2 rep Gene Homologue in HHV-6 Can Mediate AAV-2 DNA Replication and Regulate Gene Expression. Virology 1994, 204, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Urabe, M.; Ding, C.; Kotin, R.M. Insect Cells as a Factory to Produce Adeno-Associated Virus Type 2 Vectors. Hum. Gene Ther. 2002, 13, 1935–1943. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, K.; Desai, P.; Winkler, D.C.; Heymann, J.B.; Belnap, D.M.; Baumeister, W.; Steven, A.C. Three-Dimensional Structure of Herpes Simplex Virus from Cryo-Electron Tomography. Science 2003, 302, 1396–1398. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.J.; Brockman, M.A.; McNamee, E.E.; Knipe, D.M. Herpes simplex virus. Front. Biosci. 2002, 7, d752–d764. [Google Scholar] [CrossRef]
- Zhou, Z.H.; Dougherty, M.; Jakana, J.; He, J.; Rixon, F.J.; Chiu, W. Seeing the Herpesvirus Capsid at 8.5 A. Science 2000, 288, 877–880. [Google Scholar] [CrossRef]
- Bauer, D.W.; Huffman, J.B.; Homa, F.L.; Evilevitch, A. Herpes Virus Genome, The Pressure Is On. J. Am. Chem. Soc. 2013, 135, 11216–11221. [Google Scholar] [CrossRef]
- Hayward, G.S.; Jacob, R.J.; Wadsworth, S.C.; Roizman, B. Anatomy of herpes simplex virus DNA: Evidence for four populations of molecules that differ in the relative orientations of their long and short components. Proc. Natl. Acad. Sci. USA 1975, 72, 4243–4247. [Google Scholar] [CrossRef]
- Mocarski, E.S.; Roizman, B. Structure and Role of the Herpes Simplex Virus DNA Termini in Inversion, Circularization and Generation of Virion DNA. Cell 1982, 31. [Google Scholar] [CrossRef]
- Honess, R.W.; Roizman, B. Regulation of herpesvirus macromolecular synthesis I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 1974, 14, 8–19. [Google Scholar] [CrossRef]
- Spruance, S.L.; Kriesel, J.D.; Evans, T.G.; McKeough, M.B. Susceptibility to herpes labialis following multiple experimental exposures to ultraviolet radiation. Antivir. Res. 1995, 28, 57–67. [Google Scholar] [CrossRef]
- Smith, M.G.; Lennette, E.H.; Reames, H.R. Isolation of the virus of herpes simplex and the demonstration of intranuclear inclusions in a case of acute encephalitis. Am. J. Pathol. 1941, 15. [Google Scholar]
- Whitley, R.J.; Kimberlin, D.W. Herpes simplex: Encephalitis children and adolescents. Semin. Pediatric Infect. Dis. 2005, 16, 17–23. [Google Scholar] [CrossRef]
- Farooq, A.V.; Shukla, D. Herpes Simplex Epithelial and Stromal Keratitis: An Epidemiologic Update. Surv. Ophthalmol. 2012, 57, 448–462. [Google Scholar] [CrossRef]
- Akhtar, J.; Shukla, D. Viral entry mechanisms: Cellular and viral mediators of herpes simplex virus entry: Cellular and viral mediators of HSV entry. Febs. J. 2009, 276, 7228–7236. [Google Scholar] [CrossRef] [PubMed]
- Campadelli-Fiume, G.; Menotti, L.; Avitabile, E.; Gianni, T. Viral and cellular contributions to herpes simplex virus entry into the cell. Curr. Opin. Virol. 2012, 2, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Herold, B.C.; WuDunn, D.; Soltys, N.; Spear, P.G. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 1991, 65, 1090–1098. [Google Scholar] [CrossRef]
- Herold, B.C.; Visalli, R.J.; Susmarski, N.; Brandt, C.R.; Spear, P.G. Glycoprotein C-independent binding of herpes simplex virus to cells requires cell surface heparan sulphate and glycoprotein B. J. Gen. Virol. 1994, 75, 1211–1222. [Google Scholar] [CrossRef]
- Geraghty, R.J. Entry of Alphaherpesviruses Mediated by Poliovirus Receptor-Related Protein 1 and Poliovirus Receptor. Science 1998, 280, 1618–1620. [Google Scholar] [CrossRef]
- Spear, P.G.; Eisenberg, R.J.; Cohen, G.H. Three Classes of Cell Surface Receptors for Alphaherpesvirus Entry. Virology 2000, 275, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, B.; Grünewald, K. Herpesvirus membrane fusion – a team effort. Curr. Opin. Struct. Biol. 2020, 62, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Sodeik, B.; Ebersold, M.W.; Helenius, A. Microtubule-mediated Transport of Incoming Herpes Simplex Virus 1 Capsids to the Nucleus. J. Cell Biol. 1997, 136, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Radtke, K.; Kieneke, D.; Wolfstein, A.; Michael, K.; Steffen, W.; Scholz, T.; Karger, A.; Sodeik, B. Plus- and Minus-End Directed Microtubule Motors Bind Simultaneously to Herpes Simplex Virus Capsids Using Different Inner Tegument Structures. PloS Pathog. 2010, 6, e1000991. [Google Scholar] [CrossRef]
- Campbell, M.E.M.; Palfreyman, J.W.; Preston, C.M. Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate early transcription. J. Mol. Biol. 1984, 180, 1–19. [Google Scholar] [CrossRef]
- Stern, S.; Herr, W. The herpes simplex virus trans-activator VP16 recognizes the Oct-1 homeo domain: Evidence for a homeo domain recognition subdomain. Genes Dev. 1991. [Google Scholar] [CrossRef]
- Stringer, K.F.; Ingles, C.J.; Greenblatt, J. Direct and selective binding of an acidic transcriptional activation domain to the TATA-box factor TFIID. Nature 1990, 345, 783–786. [Google Scholar] [CrossRef]
- Miranda-Saksena, M.; Armati, P.; Boadle, R.A.; Holland, D.J.; Cunningham, A.L. Anterograde Transport of Herpes Simplex Virus Type 1 in Cultured, Dissociated Human and Rat Dorsal Root Ganglion Neurons. J. Virol. 2000, 74, 1827–1839. [Google Scholar] [CrossRef]
- Quinlan, M.P.; Chen, L.B.; Knipe, D.M. The lntranuclear Location of a Herpes Simplex Virus DNA-Binding Protein Is Determined by the Status of Viral DNA Replication. Cell 1984, 12. [Google Scholar]
- Glauser, D.L.; Strasser, R.; Laimbacher, A.S.; Saydam, O.; Clement, N.; Linden, R.M.; Ackermann, M.; Fraefel, C. Live Covisualization of Competing Adeno-Associated Virus and Herpes Simplex Virus Type 1 DNA Replication: Molecular Mechanisms of Interaction. J. Virol. 2007, 81, 4732–4743. [Google Scholar] [CrossRef]
- Weller, S.K.; Coen, D.M. Herpes Simplex Viruses: Mechanisms of DNA Replication. Cold Spring Harb. Perspect. Biol. 2012, 4, a013011-a013011. [Google Scholar] [CrossRef] [PubMed]
- Stow, N.D. Localization of an origin of DNA replication within the TRS/IRS repeated region of the herpes simplex virus type 1 genome. Embo J. 1982, 1, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Weller, S.K.; Spadaro, A.; Schaffer, J.E.; Murray, A.W.; Maxam, A.M.; Schaffer, P.A. Cloning, sequencing, and functional analysis of oriL, a herpes simplex virus type 1 origin of DNA synthesis. Mol. Cell. Biol. 1985, 5, 930–942. [Google Scholar] [CrossRef]
- Bruckner, R.C.; Crute, J.J.; Dodson, M.S.; Lehman, I.R. The Herpes Simplex Virus 1 Origin Binding Protein: A DNA Helicase. J. Biol. Chem. 1991, 266, 2669–2674. [Google Scholar] [PubMed]
- Boehmer, P.E.; Dodson, M.S.; Lehman, I.R. The Herpes Simplex Virus Type-1 Origin Binding Protein DNA Helicase Activity. J. Biol. Chem. 1993. [Google Scholar]
- Boehmer, P.E.; Lehman, I.R. Physical interaction between the herpes simplex virus 1 origin-binding protein and single-stranded DNA-binding protein ICP8. Proc. Natl. Acad. Sci. USA 1993, 90, 8444–8448. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Knipe, D.M. An immunoassay for the study of DNA-binding activities of herpes simplex virus protein ICP8. J. Virol. 1985, 54, 731–738. [Google Scholar] [CrossRef] [PubMed]
- McLean, G.W.; Abbotts, A.P.; Parry, M.E.; Marsden, H.S.; Stow, N.D. The herpes simplex virus type 1 origin-binding protein interacts specifically with the viral UL8 protein. J. Gen. Virol. 1994, 75, 2699–2706. [Google Scholar] [CrossRef]
- Monahan, S.J.; Grinstead, L.A.; Olivieri, W.; Parris, D.S. Interaction between the Herpes Simplex Virus Type 1 Origin-Binding and DNA Polymerase Accessory Proteins. Virology 1998, 241, 122–130. [Google Scholar] [CrossRef]
- Crute, J.J.; Mocarski, E.S.; Lehman, I.R. A DNA helicase induced by herpes simplex virus type 1. Nucleic Acids Res. 1988, 16, 6585–6596. [Google Scholar] [CrossRef]
- Crute, J.J.; Tsurumi, T.; Zhu, L.A.; Weller, S.K.; Olivo, P.D.; Challberg, M.D.; Mocarski, E.S.; Lehman, I.R. Herpes simplex virus 1 helicase-primase: A complex of three herpes-encoded gene products. Proc. Natl. Acad. Sci. USA 1989, 86, 2186–2189. [Google Scholar] [CrossRef] [PubMed]
- Poffenberger, K.L.; Roizman, B. A noninverting genome of a viable herpes simplex virus 1: Presence of head-to-tail linkages in packaged genomes and requirements for circularization after infection. J. Virol. 1985, 53, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Vlazny, D.A.; Frenkel, N. Replication of herpes simplex virus DNA: Localization of replication recognition signals within defective virus genomes. Proc. Natl. Acad. Sci. USA 1981, 78, 742–746. [Google Scholar] [CrossRef] [PubMed]
- Reuven, N.B.; Willcox, S.; Griffith, J.D.; Weller, S.K. Catalysis of Strand Exchange by the HSV-1 UL12 and ICP8 Proteins: Potent ICP8 Recombinase Activity is Revealed upon Resection of dsDNA Substrate by Nuclease. J. Mol. Biol. 2004, 342, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, A.J.; Mohni, K.N.; Kan, Y.; Hendrickson, E.A.; Stark, J.M.; Weller, S.K. The HSV-1 Exonuclease, UL12, Stimulates Recombination by a Single Strand Annealing Mechanism. PloS Pathog. 2012, 8, e1002862. [Google Scholar] [CrossRef] [PubMed]
- Weerasooriya, S.; DiScipio, K.A.; Darwish, A.S.; Bai, P.; Weller, S.K. Herpes simplex virus 1 ICP8 mutant lacking annealing activity is deficient for viral DNA replication. Proc. Natl. Acad. Sci. USA 2019, 116, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Newcomb, W.W.; Trus, B.L.; Booy, F.; Steven, A.; Wall, J.S.; Brown, J.C. Structure of Herpes Simplex Virus Capsid Molecular Composition of the Pentons and the Triplexes. J. Mol. Biol. 1993, 232, 499–511. [Google Scholar] [CrossRef]
- Homa, F.L.; Brown, J.C. Capsid assembly and DNA packaging in herpes simplex virus. Rev. Med. Virol. 1997, 7, 107–122. [Google Scholar] [CrossRef]
- Newcomb, W.W.; Homa, F.L.; Thomsen, D.R.; Trus, B.L.; Cheng, N.; Steven, A.; Booy, F.; Brown, J.C. Assembly of the Herpes Simplex Virus Procapsid from Purified Components and Identification of Small Complexes Containing the Major Capsid and Scaffolding Proteins. J. Virol. 1999, 73, 4239–4250. [Google Scholar] [CrossRef]
- Newcomb, W.W.; Juhas, R.M.; Thomsen, D.R.; Homa, F.L.; Burch, A.D.; Weller, S.K.; Brown, J.C. The UL6 Gene Product Forms the Portal for Entry of DNA into the Herpes Simplex Virus Capsid. J. Virol. 2001, 75, 10923–10932. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Jih, J.; Dai, X.; Bi, G.-Q.; Zhou, Z.H. Cryo-EM structures of herpes simplex virus type 1 portal vertex and packaged genome. Nature 2019, 570, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Mettenleiter, T.C.; Müller, F.; Granzow, H.; Klupp, B.G. The way out: What we know and do not know about herpesvirus nuclear egress: Herpesvirus nuclear egress. Cell. Microbiol. 2013, 15, 170–178. [Google Scholar] [CrossRef]
- Campadelli-Fiume, G.; Roizman, B.; Wild, P.; Mettenleiter, T.C.; Minson, T. The Egress of Herpesviruses from Cells: The Unanswered Questions. J. Virol. 2006, 80, 6716–6719. [Google Scholar] [CrossRef] [PubMed]
- Stackpole, C.W. Herpes-Type Virus of the Frog Renal Adenocarcinoma. J. Virol. 1969, 4, 75–93. [Google Scholar] [CrossRef]
- Arii, J.; Watanabe, M.; Maeda, F.; Tokai-Nishizumi, N.; Chihara, T.; Miura, M.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. ESCRT-III mediates budding across the inner nuclear membrane and regulates its integrity. Nat. Commun. 2018, 9, 3379. [Google Scholar] [CrossRef] [PubMed]
- Skepper, J.N.; Whiteley, A.; Browne, H.; Minson, A. Herpes Simplex Virus Nucleocapsids Mature to Progeny Virions by an Envelopment Deenvelopment Reenvelopment Pathway. J. Virol. 2001, 75, 5697–5702. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Baines, J.D. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 2011, 9, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; Engels, M.; Senn, C.; Tobler, K.; Ziegler, U.; Schraner, E.M.; Loepfe, E.; Ackermann, M.; Mueller, M.; Walther, P. Impairment of Nuclear Pores in Bovine Herpesvirus 1-Infected MDBK Cells. J. Virol. 2005, 79, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Leuzinger, H.; Ziegler, U.; Schraner, E.M.; Fraefel, C.; Glauser, D.L.; Heid, I.; Ackermann, M.; Mueller, M.; Wild, P. Herpes Simplex Virus 1 Envelopment Follows Two Diverse Pathways. J. Virol. 2005, 79, 13047–13059. [Google Scholar] [CrossRef]
- Pante, N.; Kann, M. Nuclear Pore Complex Is Able to Transport Macromolecules with Diameters of ϳ39 nm. Mol. Biol. Cell 2002, 13, 10. [Google Scholar] [CrossRef]
- Weindler, F.W.; Heilbronn, R. A subset of herpes simplex virus replication genes provides helper functions for productive adeno-associated virus replication. J. Virol. 1991, 65, 2476–2483. [Google Scholar] [CrossRef] [PubMed]
- Heilbronn, R. ssDNA-dependent colocalization of adeno-associated virus Rep and herpes simplex virus ICP8 in nuclear replication domains. Nucleic Acids Res. 2003, 31, 6206–6213. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nicolas, A.; Alazard-Dany, N.; Biollay, C.; Arata, L.; Jolinon, N.; Kuhn, L.; Ferro, M.; Weller, S.K.; Epstein, A.L.; Salvetti, A.; et al. Identification of Rep-Associated Factors in Herpes Simplex Virus Type 1-Induced Adeno-Associated Virus Type 2 Replication Compartments. J. Virol. 2010, 84, 8871–8887. [Google Scholar] [CrossRef]
- Slanina, H.; Weger, S.; Stow, N.D.; Kuhrs, A.; Heilbronn, R. Role of the Herpes Simplex Virus Helicase-Primase Complex during Adeno-Associated Virus DNA Replication. J. Virol. 2006, 80, 5241–5250. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Im, D.-S.; Muzyczka, N. Partial Purification of Adeno-Associated Virus Rep78, Rep52, and Rep4O and Their Biochemical Characterization. J. Virol. 1992, 66, 10. [Google Scholar] [CrossRef]
- Beaton, A.; Palumbo, P.; Berns, K.I. Expression from the adeno-associated virus p5 and p19 promoters is negatively regulated in trans by the rep protein. J. Virol. 1989, 63, 4450–4454. [Google Scholar] [CrossRef] [PubMed]
- Kyostio, S.R.M.; Owens, R.A.; Weitzman, T.D.; Chejanovsky, N.; Carterii, B.J. Analysis of Adeno-Associated Virus (AAV) Wild-Type and Mutant Rep Proteins for Their Abilities To Negatively Regulate AAV P5 and Plg mRNA Levels. J. Virol. 1994, 68, 11. [Google Scholar] [CrossRef]
- Alazard-Dany, N.; Nicolas, A.; Ploquin, A.; Strasser, R.; Greco, A.; Epstein, A.L.; Fraefel, C.; Salvetti, A. Definition of Herpes Simplex Virus Type 1 Helper Activities for Adeno-Associated Virus Early Replication Events. PloS Pathog. 2009, 5, e1000340. [Google Scholar] [CrossRef]
- Everett, R.D. A detailed mutational analysis of Vmw110, a trans-acting transcriptional activator encoded by herpes simplex virus type 1. Embo J. 1987, 6, 2069–2076. [Google Scholar] [CrossRef]
- Quinlan, M.P.; Knipe, D.M. Stimulation of expression of a herpes simplex virus DNA-binding protein by two viral functions. Mol. Cell. Biol. 1985, 5, 957–963. [Google Scholar] [CrossRef]
- Boutell, C.; Sadis, S.; Everett, R.D. Herpes Simplex Virus Type 1 Immediate-Early Protein ICP0 and Its Isolated RING Finger Domain Act as Ubiquitin E3 Ligases In Vitro. J. Virol. 2002, 76, 841–850. [Google Scholar] [CrossRef]
- Hagglund, R.; Van Sant, C.; Lopez, P.; Roizman, B. Herpes simplex virus 1-infected cell protein 0 contains two E3 ubiquitin ligase sites specific for different E2 ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. USA 2002, 99, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, M.-C.; Epstein, A.L.; Toublanc, E.; Moullier, P.; Salvetti, A. Herpes Simplex Virus Type 1 ICP0 Protein Mediates Activation of Adeno-Associated Virus Type 2 rep Gene Expression from a Latent Integrated Form. J. Virol. 2004, 78, 10977–10986. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, M.-C.; Chadeuf, G.; Orr, A.; Salvetti, A.; Everett, R.D. Impact of the Interaction between Herpes Simplex Virus Type 1 Regulatory Protein ICP0 and Ubiquitin-Specific Protease USP7 on Activation of Adeno-Associated Virus Type 2 rep Gene Expression. J. Virol. 2006, 80, 3650–3654. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Handa, H.; Carter, B.J. Adeno-associated Virus DNA Replication Complexes in Herpes Simplex Virus or Adenovirus-infected Cells. J. Biol. Chem. 1979, 254, 6603–6610. [Google Scholar] [PubMed]
- Mao, J.C.H.; Robishaw, E.E. Mode of inhibition of Herpes simplex virus DNA polymerase by phosphonoacetate. Biochemistry 1975, 14, 5475–5479. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.; Falkenberg, M.; Elias, P.; Weitzman, M.; Linden, R.M. Rep-Dependent Initiation of Adeno-Associated Virus Type 2 DNA Replication by a Herpes Simplex Virus Type 1 Replication Complex in a Reconstituted System. J. Virol. 2001, 75, 10250–10258. [Google Scholar] [CrossRef]
- Reuven, N.B.; Staire, A.E.; Myers, R.S.; Weller, S.K. The Herpes Simplex Virus Type 1 Alkaline Nuclease and Single-Stranded DNA Binding Protein Mediate Strand Exchange In Vitro. J. Virol. 2003, 77, 7425–7433. [Google Scholar] [CrossRef]
- Stutika, C.; Hüser, D.; Weger, S.; Rutz, N.; Heßler, M.; Heilbronn, R. Definition of herpes simplex virus helper functions for the replication of adeno-associated virus type 5. J. Gen. Virol. 2015, 96, 840–850. [Google Scholar] [CrossRef]
- Bantel-Schaal, U.; Zur Hausen, H. Characterization of the DNA of a defective human parvovirus isolated from a genital site. Virology 1984, 134, 52–63. [Google Scholar] [CrossRef]
- Gao, G.; Vandenberghe, L.H.; Alvira, M.R.; Lu, Y.; Calcedo, R.; Zhou, X.; Wilson, J.M. Clades of Adeno-Associated Viruses Are Widely Disseminated in Human Tissues. J. Virol. 2004, 78, 6381–6388. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.; Clement, N.; Linden, R.M. cis Effects in Adeno-Associated Virus Type 2 Replication. J. Virol. 2007, 81, 9976–9989. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weitzman, M.D.; Linden, R.M. Adeno-associated virus biology. Methods Mol. Biol. 2011, 807, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Handa, H.; Shiroki, K.; Shimojo, H. Helper factor(s) for growth of adeno-associated virus in cells transformed by adenovirus 12. Proc. Natl. Acad. Sci. USA 1977, 74, 4508–4510. [Google Scholar] [CrossRef]
- Khanal, S.; Ghimire, P.; Dhamoon, A.S. The Repertoire of Adenovirus in Human Disease: The Innocuous to the Deadly. Biomedicines 2018, 6, 30. [Google Scholar] [CrossRef]
- Doerfler, W.; Böhm, P. Adenoviruses: Model. and Vectors in Virus-Host Interactions; Doerfler, W., Böhm, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2003. [Google Scholar]
- Sussenbach, J.S.; van der Vliet, P.C. The mechanism of adenovirus DNA replication and the characterization of replication proteins. Curr Top. Microbiol. Immunol. 1984, 109, 53–73. [Google Scholar] [CrossRef]
- Hoeben, R.C.; Uil, T.G. Adenovirus DNA replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a013003. [Google Scholar] [CrossRef]
- Rochette-Egly, C.; Fromental, C.; Chambon, P. General repression of enhanson activity by the adenovirus-2 E1A proteins. Genes Dev. 1990, 4, 137–150. [Google Scholar] [CrossRef]
- Bayley, S.T.; Mymryk, J.S. Adenovirus e1a proteins and transformation (review). Int. J. Oncol. 1994, 5, 425–444. [Google Scholar] [CrossRef]
- Edwards, A.S.; Scott, J.D. A-kinase anchoring proteins: Protein kinase A and beyond. Curr. Opin. Cell Biol. 2000, 12, 217–221. [Google Scholar] [CrossRef]
- Hagiwara, M.; Brindle, P.; Harootunian, A.; Armstrong, R.; Rivier, J.; Vale, W.; Tsien, R.; Montminy, M.R. Coupling of hormonal stimulation and transcription via the cyclic AMP-responsive factor CREB is rate limited by nuclear entry of protein kinase A. Mol. Cell Biol. 1993, 13, 4852–4859. [Google Scholar] [CrossRef] [PubMed]
- Chrivia, J.C.; Kwok, R.P.; Lamb, N.; Hagiwara, M.; Montminy, M.R.; Goodman, R.H. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 1993, 365, 855–859. [Google Scholar] [CrossRef] [PubMed]
- Fax, P.; Lehmkuhler, O.; Kuhn, C.; Esche, H.; Brockmann, D. E1A12S-mediated activation of the adenovirus type 12 E2 promoter depends on the histone acetyltransferase activity of p300/CBP. J. Biol. Chem. 2000, 275, 40554–40560. [Google Scholar] [CrossRef] [PubMed]
- Fax, P.; Lipinski, K.S.; Esche, H.; Brockmann, D. cAMP-independent activation of the adenovirus type 12 E2 promoter correlates with the recruitment of CREB-1/ATF-1, E1A(12S), and CBP to the E2-CRE. J. Biol. Chem. 2000, 275, 8911–8920. [Google Scholar] [CrossRef]
- Ahn, S.; Olive, M.; Aggarwal, S.; Krylov, D.; Ginty, D.D.; Vinson, C. A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol. Cell Biol. 1998, 18, 967–977. [Google Scholar] [CrossRef]
- Van der Vliet PC, H.R. Adenovirus. In DNA Replication and Human Disease; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2006. [Google Scholar]
- Han, J.; Sabbatini, P.; Perez, D.; Rao, L.; Modha, D.; White, E. The E1B 19K protein blocks apoptosis by interacting with and inhibiting the p53-inducible and death-promoting Bax protein. Genes Dev. 1996, 10, 461–477. [Google Scholar] [CrossRef]
- Tarakanova, V.L.; Wold, W.S. Adenovirus E1A and E1B-19K proteins protect human hepatoma cells from transforming growth factor beta1-induced apoptosis. Virus Res. 2010, 147, 67–76. [Google Scholar] [CrossRef]
- Cuconati, A.; Degenhardt, K.; Sundararajan, R.; Anschel, A.; White, E. Bak and Bax function to limit adenovirus replication through apoptosis induction. J. Virol. 2002, 76, 4547–4558. [Google Scholar] [CrossRef]
- Piya, S.; White, E.J.; Klein, S.R.; Jiang, H.; McDonnell, T.J.; Gomez-Manzano, C.; Fueyo, J. The E1B19K oncoprotein complexes with Beclin 1 to regulate autophagy in adenovirus-infected cells. PLoS ONE 2011, 6, e29467. [Google Scholar] [CrossRef]
- White, E. Regulation of the cell cycle and apoptosis by the oncogenes of adenovirus. Oncogene 2001, 20, 7836–7846. [Google Scholar] [CrossRef]
- Querido, E.; Marcellus, R.C.; Lai, A.; Charbonneau, R.; Teodoro, J.G.; Ketner, G.; Branton, P.E. Regulation of p53 levels by the E1B 55-kilodalton protein and E4orf6 in adenovirus-infected cells. J. Virol. 1997, 71, 3788–3798. [Google Scholar] [CrossRef] [PubMed]
- Steegenga, W.T.; Riteco, N.; Jochemsen, A.G.; Fallaux, F.J.; Bos, J.L. The large E1B protein together with the E4orf6 protein target p53 for active degradation in adenovirus infected cells. Oncogene 1998, 16, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Grand, R.J. Adenovirus E1B 55-kilodalton protein: Multiple roles in viral infection and cell transformation. J. Virol. 2009, 83, 4000–4012. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, P.; Ip, W.H.; Dobner, T.; Gonzalez, R.A. The biology of the adenovirus E1B 55K protein. Febs. Lett. 2019, 593, 3504–3517. [Google Scholar] [CrossRef]
- Weitzman, M.D. Functions of the adenovirus E4 proteins and their impact on viral vectors. Front. Biosci. 2005, 10, 1106–1117. [Google Scholar] [CrossRef]
- Falgout, B.; Ketner, G. Adenovirus early region 4 is required for efficient virus particle assembly. J. Virol. 1987, 61, 3759–3768. [Google Scholar] [CrossRef]
- Bridge, E.; Ketner, G. Redundant control of adenovirus late gene expression by early region 4. J. Virol. 1989, 63, 631–638. [Google Scholar] [CrossRef]
- Sarnow, P.; Hearing, P.; Anderson, C.W.; Halbert, D.N.; Shenk, T.; Levine, A.J. Adenovirus early region 1B 58,000-dalton tumor antigen is physically associated with an early region 4 25,000-dalton protein in productively infected cells. J. Virol. 1984, 49, 692–700. [Google Scholar] [CrossRef]
- Weiden, M.D.; Ginsberg, H.S. Deletion of the E4 region of the genome produces adenovirus DNA concatemers. Proc. Natl. Acad. Sci. USA 1994, 91, 153–157. [Google Scholar] [CrossRef]
- Stracker, T.H.; Carson, C.T.; Weitzman, M.D. Adenovirus oncoproteins inactivate the Mre11–Rad50–NBS1 DNA repair complex. Nature 2002, 418, 348–352. [Google Scholar] [CrossRef]
- Vachon, V.K.; Conn, G.L. Adenovirus VA RNA: An essential pro-viral non-coding RNA. Virus Res. 2016, 212, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Mathews, M.B.; Shenk, T. Adenovirus virus-associated RNA and translation control. J. Virol. 1991, 65, 5657–5662. [Google Scholar] [CrossRef] [PubMed]
- Kitajewski, J.; Schneider, R.J.; Safer, B.; Munemitsu, S.M.; Samuel, C.E.; Thimmappaya, B.; Shenk, T. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell 1986, 45, 195–200. [Google Scholar] [CrossRef]
- O’Malley, R.P.; Mariano, T.M.; Siekierka, J.; Mathews, M.B. A mechanism for the control of protein synthesis by adenovirus VA RNAI. Cell 1986, 44, 391–400. [Google Scholar] [CrossRef]
- Ward, P.; Berns, K.I. In vitro replication of adeno-associated virus DNA: Enhancement by extracts from adenovirus-infected HeLa cells. J Virol. 1996, 70, 4495–4501. [Google Scholar] [CrossRef]
- Nayak, R.; Pintel, D.J. Positive and negative effects of adenovirus type 5 helper functions on adeno-associated virus type 5 (AAV5) protein accumulation govern AAV5 virus production. J. Virol. 2007, 81, 2205–2212. [Google Scholar] [CrossRef]
- Laughlin, C.A.; Jones, N.; Carter, B.J. Effect of deletions in adenovirus early region 1 genes upon replication of adeno-associated virus. J. Virol. 1982, 41, 868–876. [Google Scholar] [CrossRef]
- Tratschin, J.D.; Miller, I.L.; Carter, B.J. Genetic analysis of adeno-associated virus: Properties of deletion mutants constructed in vitro and evidence for an adeno-associated virus replication function. J. Virol. 1984, 51, 611–619. [Google Scholar] [CrossRef]
- Chang, L.S.; Shi, Y.; Shenk, T. Adeno-associated virus P5 promoter contains an adenovirus E1A-inducible element and a binding site for the major late transcription factor. J. Virol. 1989, 63, 3479–3488. [Google Scholar] [CrossRef]
- Xiao, X.; Li, J.; Samulski, R.J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998, 72, 2224–2232. [Google Scholar] [CrossRef]
- Tratschin, J.D.; West, M.H.; Sandbank, T.; Carter, B.J. A human parvovirus, adeno-associated virus, as a eucaryotic vector: Transient expression and encapsidation of the procaryotic gene for chloramphenicol acetyltransferase. Mol. Cell Biol. 1984, 4, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Richardson, W.D.; Westphal, H. Requirement for either early region 1a or early region 1b adenovirus gene products in the helper effect for adeno-associated virus. J. Virol. 1984, 51, 404–410. [Google Scholar] [CrossRef]
- Shi, Y.; Seto, E.; Chang, L.S.; Shenk, T. Transcriptional repression by YY1, a human GLI-Kruppel-related protein, and relief of repression by adenovirus E1A protein. Cell 1991, 67, 377–388. [Google Scholar] [CrossRef]
- Samulski, R.J.; Shenk, T. Adenovirus E1B 55-Mr polypeptide facilitates timely cytoplasmic accumulation of adeno-associated virus mRNAs. J. Virol. 1988, 62, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Fisher, K.J.; Gao, G.P.; Weitzman, M.D.; DeMatteo, R.; Burda, J.F.; Wilson, J.M. Transduction with recombinant adeno-associated virus for gene therapy is limited by leading-strand synthesis. J. Virol. 1996, 70, 520–532. [Google Scholar] [CrossRef]
- Pilder, S.; Moore, M.; Logan, J.; Shenk, T. The adenovirus E1B-55K transforming polypeptide modulates transport or cytoplasmic stabilization of viral and host cell mRNAs. Mol. Cell Biol. 1986, 6, 470–476. [Google Scholar] [CrossRef]
- Ward, P.; Dean, F.B.; O’Donnell, M.E.; Berns, K.I. Role of the adenovirus DNA-binding protein in in vitro adeno-associated virus DNA replication. J. Virol. 1998, 72, 420–427. [Google Scholar] [CrossRef]
- Matsushita, T.; Elliger, S.; Elliger, C.; Podsakoff, G.; Villarreal, L.; Kurtzman, G.J.; Iwaki, Y.; Colosi, P. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene 1998, 5, 938–945. [Google Scholar] [CrossRef]
- Stracker, T.H.; Cassell, G.D.; Ward, P.; Loo, Y.-M.; van Breukelen, B.; Carrington-Lawrence, S.D.; Hamatake, R.K.; van der Vliet, P.C.; Weller, S.K.; Melendy, T.; et al. The Rep Protein of Adeno-Associated Virus Type 2 Interacts with Single-Stranded DNA-Binding Proteins That Enhance Viral Replication. J. Virol. 2004, 78, 441–453. [Google Scholar] [CrossRef]
- Carter, B.J.; Antoni, B.A.; Klessig, D.F. Adenovirus containing a deletion of the early region 2A gene allows growth of adeno-associated virus with decreased efficiency. Virology 1992, 191, 473–476. [Google Scholar] [CrossRef]
- Chang, L.S.; Shenk, T. The adenovirus DNA-binding protein stimulates the rate of transcription directed by adenovirus and adeno-associated virus promoters. J. Virol. 1990, 64, 2103–2109. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, F.K.; Samulski, T.; Shenk, T.; Samulski, R.J. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J. Virol. 1996, 70, 3227–3234. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.A.; Palacios, J.A.; Cassell, G.D.; Adam, S.; Giacca, M.; Weitzman, M.D. The Mre11/Rad50/Nbs1 complex limits adeno-associated virus transduction and replication. J. Virol. 2007, 81, 12936–12945. [Google Scholar] [CrossRef] [PubMed]
- Lentz, T.B.; Samulski, R.J. Insight into the mechanism of inhibition of adeno-associated virus by the Mre11/Rad50/Nbs1 complex. J. Virol. 2015, 89, 181–194. [Google Scholar] [CrossRef]
- Allen, J.M.; Halbert, C.L.; Miller, A.D. Improved adeno-associated virus vector production with transfection of a single helper adenovirus gene, E4orf6. Mol. Ther. 1 2000, 1, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Nayak, R.; Pintel, D.J. Adeno-associated viruses can induce phosphorylation of eIF2alpha via PKR activation, which can be overcome by helper adenovirus type 5 virus-associated RNA. J. Virol. 2007, 81, 11908–11916. [Google Scholar] [CrossRef]
- Janik, J.E.; Huston, M.M.; Cho, K.; Rose, J.A. Efficient synthesis of adeno-associated virus structural proteins requires both adenovirus DNA binding protein and VA I RNA. Virology 1989, 168, 320–329. [Google Scholar] [CrossRef]
- Matsushita, T.; Okada, T.; Inaba, T.; Mizukami, H.; Ozawa, K.; Colosi, P. The adenovirus E1A and E1B19K genes provide a helper function for transfection-based adeno-associated virus vector production. J. Gen. Virol. 2004, 85, 2209–2214. [Google Scholar] [CrossRef]
- Ostrove, J.M.; Berns, K.I. Adenovirus early region 1b gene function required for rescue of latent adeno-associated virus. Virology 1980, 104, 502–505. [Google Scholar] [CrossRef]
- Weitzman, M.D.; Fisher, K.J.; Wilson, J.M. Recruitment of wild-type and recombinant adeno-associated virus into adenovirus replication centers. J. Virol. 1996, 70, 1845–1854. [Google Scholar] [CrossRef]
- Thomson, B.J.; Efstathiou, S.; Honess, R.W. Acquisition of the human adeno-associated virus type-2 rep gene by human herpesvirus type-6. Nature 1991, 351, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Hermonat, P.L. Adeno-associated virus inhibits human papillomavirus type 16: A viral interaction implicated in cervical cancer. Cancer Res. 1994, 54, 2278–2281. [Google Scholar] [PubMed]
- Meyers, C.; Alam, S.; Mane, M.; Hermonat, P.L. Altered biology of adeno-associated virus type 2 and human papillomavirus during dual infection of natural host tissue. Virology 2001, 287, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Coker, A.L.; Russell, R.B.; Bond, S.M.; Pirisi, L.; Liu, Y.; Mane, M.; Kokorina, N.; Gerasimova, T.; Hermonat, P.L. Adeno-associated virus is associated with a lower risk of high-grade cervical neoplasia. Exp. Mol. Pathol. 2001, 70, 83–89. [Google Scholar] [CrossRef]
- Smith, J.; Herrero, R.; Erles, K.; Grimm, D.; Munoz, N.; Bosch, F.X.; Tafur, L.; Shah, K.V.; Schlehofer, J.R. Adeno-associated virus seropositivity and HPV-induced cervical cancer in Spain and Colombia. Int. J. Cancer 2001, 94, 520–526. [Google Scholar] [CrossRef]
- Walz, C.; Deprez, A.; Dupressoir, T.; Rabreau, M.; Schlehofer, J.R. Interaction of human papillomavirus type 16 and adeno-associated virus type 2 co-infecting human cervical epithelium. J. Gen. Virol. 1997, 78, 1441–1452. [Google Scholar] [CrossRef]
- Cao, M.; Zhu, H.; Bandyopadhyay, S.; You, H.; Hermonat, P.L. HPV-16 E1, E2 and E6 each complement the Ad5 helper gene set, increasing rAAV2 and wt AAV2 production. Gene 2012, 19, 418–424. [Google Scholar] [CrossRef]
- Cao, M.; Bandyopadhyay, S.; Zhu, H.; You, H.; Hermonat, P.L. The HPV16 E1 Carboxyl Domain Provides a Helper Function for Adeno-Associated Virus Replication. Intervirology 2018, 61, 185–192. [Google Scholar] [CrossRef]
- You, H.; Liu, Y.; Prasad, C.K.; Agrawal, N.; Zhang, D.; Bandyopadhyay, S.; Liu, H.; Kay, H.H.; Mehta, J.L.; Hermonat, P.L. Multiple human papillomavirus genes affect the adeno-associated virus life cycle. Virology 2006, 344, 532–540. [Google Scholar] [CrossRef]
- Guidry, J.T.; Scott, R.S. The interaction between human papillomavirus and other viruses. Virus Res. 2017, 231, 139–147. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Cao, M.; Liu, Y.; Hermonat, P.L. HPV E1 up-regulates replication-related biochemistries of AAV Rep78. Virology 2010, 402, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Raney, K.D.; Liu, Y.; Hermonat, P.L. AAV-2 Rep78 and HPV-16 E1 interact in vitro, modulating their ATPase activity. Biochemistry 2008, 47, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cheng, F.; Engelhardt, J.F.; Yan, Z.; Qiu, J. Development of a Novel Recombinant Adeno-Associated Virus Production System Using Human Bocavirus 1 Helper Genes. Mol. Methods Clin. Dev. 2018, 11, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Fakhiri, J.; Schneider, M.A.; Puschhof, J.; Stanifer, M.; Schildgen, V.; Holderbach, S.; Voss, Y.; El Andari, J.; Schildgen, O.; Boulant, S.; et al. Novel Chimeric Gene Therapy Vectors Based on Adeno-Associated Virus and Four Different Mammalian Bocaviruses. Mol. Methods Clin. Dev. 2019, 12, 202–222. [Google Scholar] [CrossRef]
- Bantel-Schaal, U.; Zur Hausen, H. Adeno-associated viruses inhibit SV40 DNA amplification and replication of herpes simplex virus in SV40-transformed hamster cells. Virology 1988, 164, 64–74. [Google Scholar] [CrossRef]
- Glauser, D.L.; Seyffert, M.; Strasser, R.; Franchini, M.; Laimbacher, A.S.; Dresch, C.; de Oliveira, A.P.; Vogel, R.; Buning, H.; Salvetti, A.; et al. Inhibition of Herpes Simplex Virus Type 1 Replication by Adeno-Associated Virus Rep Proteins Depends on Their Combined DNA-Binding and ATPase/Helicase Activities. J. Virol. 2010, 84, 3808–3824. [Google Scholar] [CrossRef]
- Heilbronn, R.; Bürkle, A.; Stephan, S.; zur Hausen, H. The adeno-associated virus rep gene suppresses herpes simplex virus-induced DNA amplification. J. Virol. 1990, 64, 3012–3018. [Google Scholar] [CrossRef]
- Seyffert, M.; Glauser, D.L.; Tobler, K.; Georgiev, O.; Vogel, R.; Vogt, B.; Agúndez, L.; Linden, M.; Büning, H.; Ackermann, M.; et al. Adeno-Associated Virus Type 2 Rep68 Can Bind to Consensus Rep-Binding Sites on the Herpes Simplex Virus 1 Genome. J. Virol. 2015, 89, 11150–11158. [Google Scholar] [CrossRef][Green Version]
- Franzoso, F.D.; Seyffert, M.; Vogel, R.; Yakimovich, A.; de Andrade Pereira, B.; Meier, A.F.; Sutter, S.O.; Tobler, K.; Vogt, B.; Greber, U.F.; et al. Cell Cycle-Dependent Expression of Adeno-Associated Virus 2 (AAV2) Rep in Coinfections with Herpes Simplex Virus 1 (HSV-1) Gives Rise to a Mosaic of Cells Replicating either AAV2 or HSV-1. J. Virol. 2017, 91, e00357-00317. [Google Scholar] [CrossRef]
- Cohen, G.H.; Vaughan, R.K.; Lawrence, W.C. Deoxyribonucleic Acid Synthesis in Synchronized Mammalian KB Cells Infected with Herpes Simplex Virus. J. Virol. 1971, 7, 783–791. [Google Scholar] [CrossRef]
- Hobbs, W.E.; DeLuca, N.A. Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. J. Virol. 1999, 73, 8245–8255. [Google Scholar] [CrossRef] [PubMed]
- Lomonte, P.; Everett, R.D. Herpes Simplex Virus Type 1 Immediate-Early Protein Vmw110 Inhibits Progression of Cells through Mitosis and from G1 into S Phase of the Cell Cycle. J. Virol. 1999, 73, 9456–9467. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Liu, J.J.; Yeh, K.-C.; Knipe, D.M. Herpes Simplex Virus Infection Blocks Events in the G1 Phase of the Cell Cycle. Virology 2000, 267, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Vogel, R.; Seyffert, M.; Strasser, R.; de Oliveira, A.P.; Dresch, C.; Glauser, D.L.; Jolinon, N.; Salvetti, A.; Weitzman, M.D.; Ackermann, M.; et al. Adeno-Associated Virus Type 2 Modulates the Host DNA Damage Response Induced by Herpes Simplex Virus 1 during Coinfection. J. Virol. 2012, 86, 143–155. [Google Scholar] [CrossRef]
- Casper, J.M.; Timpe, J.M.; Dignam, J.D.; Trempe, J.P. Identification of an adeno-associated virus Rep protein binding site in the adenovirus E2a promoter. J. Virol. 2005, 79, 28–38. [Google Scholar] [CrossRef]
- Needham, P.G.; Casper, J.M.; Kalman-Maltese, V.; Verrill, K.; Dignam, J.D.; Trempe, J.P. Adeno-associated virus rep protein-mediated inhibition of transcription of the adenovirus major late promoter in vitro. J. Virol. 2006, 80, 6207–6217. [Google Scholar] [CrossRef][Green Version]
- Timpe, J.M.; Verrill, K.C.; Trempe, J.P. Effects of adeno-associated virus on adenovirus replication and gene expression during coinfection. J. Virol. 2006, 80, 7807–7815. [Google Scholar] [CrossRef]
- Seyffert, M.; Glauser, D.L.; Schraner, E.M.; de Oliveira, A.-P.; Mansilla-Soto, J.; Vogt, B.; Büning, H.; Linden, R.M.; Ackermann, M.; Fraefel, C. Novel Mutant AAV2 Rep Proteins Support AAV2 Replication without Blocking HSV-1 Helpervirus Replication. PLoS ONE 2017, 12, e0170908. [Google Scholar] [CrossRef]
- Di Pasquale, G.; Chiorini, J.A. PKA/PrKX activity is a modulator of AAV/adenovirus interaction. Embo J. 2003, 22, 1716–1724. [Google Scholar] [CrossRef]
- Weger, S.; Hammer, E.; Gonsior, M.; Stutika, C.; Heilbronn, R. A Regulatory Element Near the 3’ End of the Adeno-Associated Virus rep Gene Inhibits Adenovirus Replication in cis by Means of p40 Promoter-Associated Short Transcripts. J. Virol. 2016, 90, 3981–3993. [Google Scholar] [CrossRef]
- Hermonat, P.L.; Plott, R.T.; Santin, A.D.; Parham, G.P.; Flick, J.T. Adeno-associated virus Rep78 inhibits oncogenic transformation of primary human keratinocytes by a human papillomavirus type 16-ras chimeric. Gynecol. Oncol. 1997, 66, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Zhan, D.; Santin, A.D.; Liu, Y.; Parham, G.P.; Li, C.; Meyers, C.; Hermonat, P.L. Binding of the human papillomavirus type 16 p97 promoter by the adeno-associated virus Rep78 major regulatory protein correlates with inhibition. J. Biol. Chem. 1999, 274, 31619–31624. [Google Scholar] [CrossRef] [PubMed]
- Walz, C.M.; Correa-Ochoa, M.M.; Muller, M.; Schlehofer, J.R. Adenoassociated virus type 2-induced inhibition of the human papillomavirus type 18 promoter in transgenic mice. Virology 2002, 293, 172–181. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zheng, Y.; Peng, Z.L.; Lou, J.Y.; Wang, H. Detection of physical status of human papillomavirus 16 in cervical cancer tissue and SiHa cell line by multiplex real-time polymerase chain reaction. Ai Zheng 2006, 25, 373–377. [Google Scholar] [PubMed]
- Antoni, B.A.; Rabson, A.B.; Miller, I.L.; Trempe, J.P.; Chejanovsky, N.; Carter, B.J. Adeno-associated virus Rep protein inhibits human immunodeficiency virus type 1 production in human cells. J. Virol. 1991, 65, 396–404. [Google Scholar] [CrossRef]
- Srivastava, A. In vivo tissue-tropism of adeno-associated viral vectors. Curr. Opin. Virol. 2016, 21, 75–80. [Google Scholar] [CrossRef]
- Kotin, R.M.; Siniscalco, M.; Samulski, R.J.; Zhu, X.D.; Hunter, L.; Laughlin, C.A.; McLaughlin, S.; Muzyczka, N.; Rocchi, M.; Berns, K.I. Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. USA 1990, 87, 2211–2215. [Google Scholar] [CrossRef]
- Surosky, R.T.; Urabe, M.; Godwin, S.G.; McQuiston, S.A.; Kurtzman, G.J.; Ozawa, K.; Natsoulis, G. Adeno-associated virus Rep proteins target DNA sequences to a unique locus in the human genome. J. Virol. 1997, 71, 7951–7959. [Google Scholar] [CrossRef]
- McCarty, D.M.; Monahan, P.E.; Samulski, R.J. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene 2001, 8, 1248–1254. [Google Scholar] [CrossRef]
- Grimm, D.; Kern, A.; Rittner, K.; Kleinschmidt, J.A. Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum. Gene 1998, 9, 2745–2760. [Google Scholar] [CrossRef]
- Collaco, R.F.; Cao, X.; Trempe, J.P. A helper virus-free packaging system for recombinant adeno-associated virus vectors. Gene 1999, 238, 397–405. [Google Scholar] [CrossRef]
- Grieger, J.C.; Soltys, S.M.; Samulski, R.J. Production of Recombinant Adeno-associated Virus Vectors Using Suspension HEK293 Cells and Continuous Harvest of Vector From the Culture Media for GMP FIX and FLT1 Clinical Vector. Mol. Ther. 2016, 24, 287–297. [Google Scholar] [CrossRef]
- Martin, J.; Frederick, A.; Luo, Y.; Jackson, R.; Joubert, M.; Sol, B.; Poulin, F.; Pastor, E.; Armentano, D.; Wadsworth, S.; et al. Generation and characterization of adeno-associated virus producer cell lines for research and preclinical vector production. Hum. Gene Methods 2013, 24, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.P.; Qu, G.; Faust, L.Z.; Engdahl, R.K.; Xiao, W.; Hughes, J.V.; Zoltick, P.W.; Wilson, J.M. High-titer adeno-associated viral vectors from a Rep/Cap cell line and hybrid shuttle virus. Hum. Gene 1998, 9, 2353–2362. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.E.; Rhys, C.M.; Zolotukhin, I.; Zolotukhin, S.; Muzyczka, N.; Hayward, G.S.; Byrne, B.J. High-titer recombinant adeno-associated virus production utilizing a recombinant herpes simplex virus type I vector expressing AAV-2 Rep and Cap. Gene 1999, 6, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Clement, N.; Knop, D.R.; Byrne, B.J. Large-scale adeno-associated viral vector production using a herpesvirus-based system enables manufacturing for clinical studies. Hum. Gene 2009, 20, 796–806. [Google Scholar] [CrossRef]
- Thomas, D.L.; Wang, L.; Niamke, J.; Liu, J.; Kang, W.; Scotti, M.M.; Ye, G.J.; Veres, G.; Knop, D.R. Scalable recombinant adeno-associated virus production using recombinant herpes simplex virus type 1 coinfection of suspension-adapted mammalian cells. Hum. Gene 2009, 20, 861–870. [Google Scholar] [CrossRef]
- Flotte, T.R.; Trapnell, B.C.; Humphries, M.; Carey, B.; Calcedo, R.; Rouhani, F.; Campbell-Thompson, M.; Yachnis, A.T.; Sandhaus, R.A.; McElvaney, N.G.; et al. Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing alpha1-antitrypsin: Interim results. Hum. Gene 2011, 22, 1239–1247. [Google Scholar] [CrossRef]
- Virag, T.; Cecchini, S.; Kotin, R.M. Producing recombinant adeno-associated virus in foster cells: Overcoming production limitations using a baculovirus-insect cell expression strategy. Hum. Gene 2009, 20, 807–817. [Google Scholar] [CrossRef]
- Smith, R.H.; Levy, J.R.; Kotin, R.M. A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Mol. Ther. 2009, 17, 1888–1896. [Google Scholar] [CrossRef]
- Kohlbrenner, E.; Aslanidi, G.; Nash, K.; Shklyaev, S.; Campbell-Thompson, M.; Byrne, B.J.; Snyder, R.O.; Muzyczka, N.; Warrington, K.H., Jr.; Zolotukhin, S. Successful production of pseudotyped rAAV vectors using a modified baculovirus expression system. Mol. Ther. 2005, 12, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, M.A.; Holkers, M.; van Nierop, G.P.; Wieringa, R.; Pau, M.G.; de Vries, A.A. Targeted chromosomal insertion of large DNA into the human genome by a fiber-modified high-capacity adenovirus-based vector system. PLoS ONE 2008, 3, e3084. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, M.A.; van Nierop, G.P.; Tijssen, M.R.; Lefesvre, P.; Knaan-Shanzer, S.; van der Velde, I.; van Bekkum, D.W.; Valerio, D.; de Vries, A.A. Transfer of the full-length dystrophin-coding sequence into muscle cells by a dual high-capacity hybrid viral vector with site-specific integration ability. J. Virol. 2005, 79, 3146–3162. [Google Scholar] [CrossRef] [PubMed]
- Heister, T.; Heid, I.; Ackermann, M.; Fraefel, C. Herpes simplex virus type 1/adeno-associated virus hybrid vectors mediate site-specific integration at the adeno-associated virus preintegration site, AAVS1, on human chromosome 19. J. Virol. 2002, 76, 7163–7173. [Google Scholar] [CrossRef] [PubMed]
- Recchia, A.; Perani, L.; Sartori, D.; Olgiati, C.; Mavilio, F. Site-specific integration of functional transgenes into the human genome by adeno/AAV hybrid vectors. Mol. Ther. 2004, 10, 660–670. [Google Scholar] [CrossRef]
- Oehmig, A.; Cortes, M.L.; Perry, K.F.; Sena-Esteves, M.; Fraefel, C.; Breakefield, X.O. Integration of active human beta-galactosidase gene (100 kb) into genome using HSV/AAV amplicon vector. Gene 2007, 14, 1078–1091. [Google Scholar] [CrossRef]
- Fraefel, C.; Bittermann, A.G.; Bueler, H.; Heid, I.; Bachi, T.; Ackermann, M. Spatial and Temporal Organization of Adeno-Associated Virus DNA Replication in Live Cells. J. Virol. 2004, 78, 389–398. [Google Scholar] [CrossRef][Green Version]
- Burnett, J.R.; Hooper, A.J. Alipogene tiparvovec, an adeno-associated virus encoding the Ser(447)X variant of the human lipoprotein lipase gene for the treatment of patients with lipoprotein lipase deficiency. Curr. Opin. Mol. 2009, 11, 681–691. [Google Scholar]
- Deverman, B.E.; Ravina, B.M.; Bankiewicz, K.S.; Paul, S.M.; Sah, D.W.Y. Gene therapy for neurological disorders: Progress and prospects. Nat. Rev. Drug Discov. 2018, 17, 767. [Google Scholar] [CrossRef]
- Bass-Stringer, S.; Bernardo, B.C.; May, C.N.; Thomas, C.J.; Weeks, K.L.; McMullen, J.R. Adeno-Associated Virus Gene Therapy: Translational Progress and Future Prospects in the Treatment of Heart Failure. Heart Lung Circ. 2018, 27, 1285–1300. [Google Scholar] [CrossRef]
- Grimm, D.; Büning, H. Small But Increasingly Mighty: Latest Advances in AAV Vector Research, Design, and Evolution. Hum. Gene Ther. 2017, 28, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K.A. Immune responses to AAV in clinical trials. Curr. Gene 2011, 11, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, V.; Petry, H.; Salmon, F. Immune Responses to AAV-Vectors, the Glybera Example from Bench to Bedside. Front. Immunol. 2014, 5, 82. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.E.; Liu, J.M.; Xiao, X.; Young, N.S.; Nienhuis, A.W.; Samulski, R.J. Regulated high level expression of a human gamma-globin gene introduced into erythroid cells by an adeno-associated virus vector. Proc. Natl. Acad. Sci. USA 1992, 89, 7257–7261. [Google Scholar] [CrossRef] [PubMed]
- Balague, C.; Kalla, M.; Zhang, W.W. Adeno-associated virus Rep78 protein and terminal repeats enhance integration of DNA sequences into the cellular genome. J. Virol. 1997, 71, 3299–3306. [Google Scholar] [CrossRef] [PubMed]
- Urcelay, E.; Ward, P.; Wiener, S.M.; Safer, B.; Kotin, R.M. Asymmetric replication in vitro from a human sequence element is dependent on adeno-associated virus Rep protein. J. Virol. 1995, 69, 2038–2046. [Google Scholar] [CrossRef]
- Weitzman, M.D.; Kyostio, S.R.; Kotin, R.M.; Owens, R.A. Adeno-associated virus (AAV) Rep proteins mediate complex formation between AAV DNA and its integration site in human DNA. Proc. Natl. Acad. Sci. USA 1994, 91, 5808–5812. [Google Scholar] [CrossRef]
- Zeltner, N.; Kohlbrenner, E.; Clement, N.; Weber, T.; Linden, R.M. Near-perfect infectivity of wild-type AAV as benchmark for infectivity of recombinant AAV vectors. Gene 2010, 17, 872–879. [Google Scholar] [CrossRef]


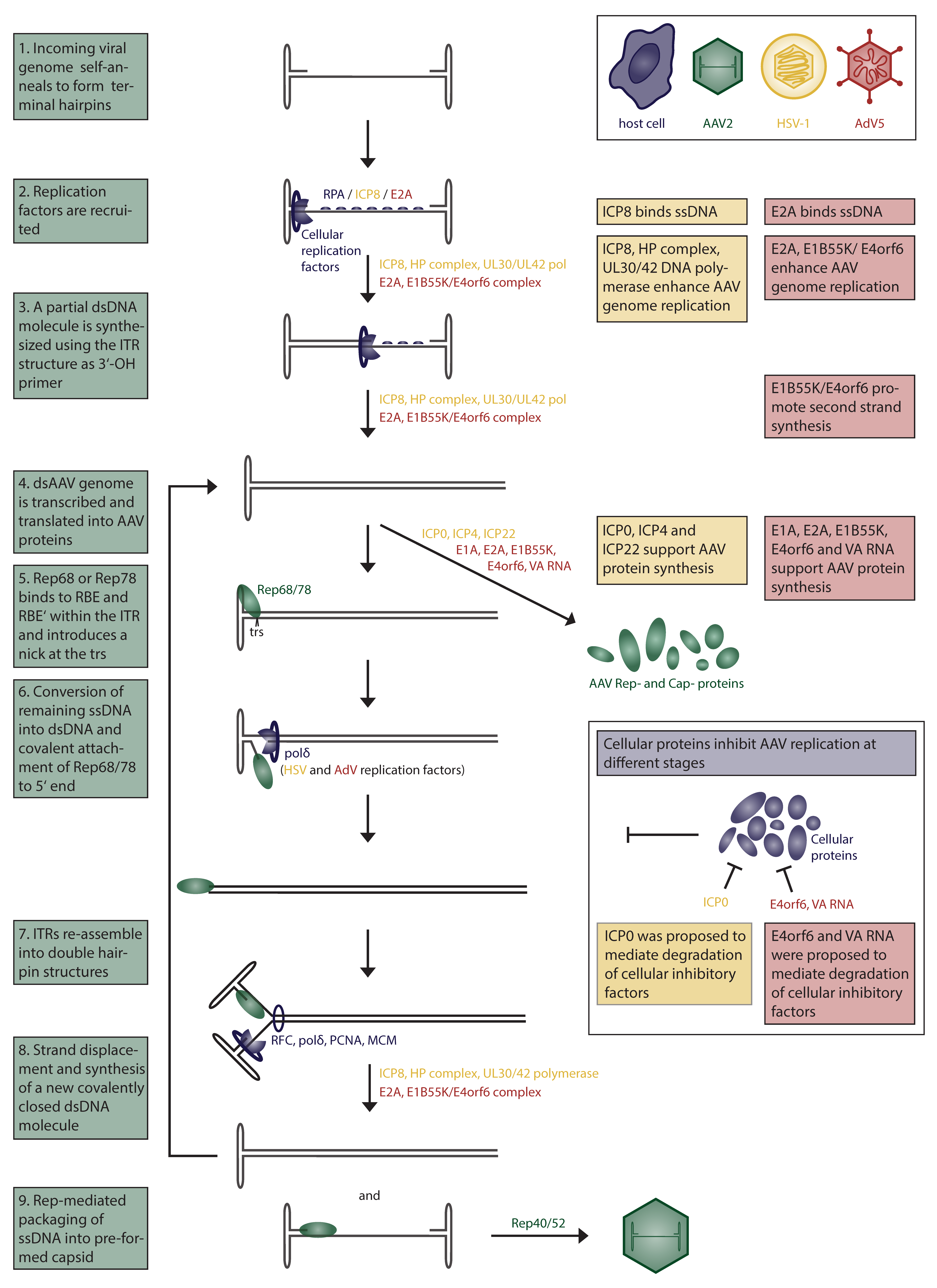{kind=link}
| Helper Virus | Gene Product | Native Function | Helper Function | Essential For AAV | References |
|---|---|---|---|---|---|
| HSV-1 | UL5 | HP complex helicase subunit, unwinds DNA at replication fork | Promotes AAV genome replication, requires UL5 helicase activity | Yes | [132,135] |
| UL8 | HP complex subunit, stimulates enzymatic activity of UL5 and UL52 | Promotes AAV genome replication | Yes | [132,135] | |
| UL52 | HP complex primase subunit, primase activity during DNA replication | Promotes AAV genome replication | Yes | [132,135] | |
| ICP8 | ssDNA binding protein, required for HSV-1 genome replication | Promotes AAV genome replication, binds Rep78 and AAV ssDNA | Yes | [132,133] | |
| ICP0 | E3 ubiquitin ligase, trans-activator of HSV-1 gene expression | Supports AAV Rep-expression | No | [139] | |
| ICP4 | Major viral transcription factor | Supports AAV Rep-expression | No | [139] | |
| ICP22 | Transcriptional regulator | Supports AAV Rep-expression | No | [139] | |
| UL30 | HSV-1 polymerase catalytic subunit | Enhances AAV genome replication | No | [139,148] | |
| UL42 | HSV-1 polymerase subunit | Enhances AAV genome replication | No | [139,148] | |
| AdV5 | E1A | General transcription factor, AdV early promoter activation, oncogene | AAV promoter activation, drives cells to S-phase | Yes | [190,192,194,195,211] |
| E1B19K | Inhibits proapoptotic Bcl-2 homologs (Bax and Bak), induces autophagy, oncogene | Enhances AAV vector titers | No | [211,212] | |
| E1B55K | In complex with E4orf6 it prevents E1A mediated p53 stabilization, oncogene | Involved in AAV mRNA export, promotes AAV second-strand synthesis | Yes | [153,175,197,199,211,212] | |
| protein IX | Minor component of the AdV capsid, capsid stability | Molecular function unclear, enhances AAV vector titers | No | [211] | |
| E2A | ssDNA binding protein, viral DNA replication & mRNA processing | AAV promoter regulation, AAV genome replication, Rep splicing, capsid protein production | Yes | [193,200,201,202,203,204,211] | |
| E4orf6 | In complex with E1B55K it prevents E1A mediated p53 stabilization, supports viral DNA replication and RNA processing | Promotes AAV second-strand synthesis, inhibits the MRN complex | Yes | [29,153,193,198,201,205,208] | |
| VA RNA | inhibits the eIF-2 protein kinase, promotes viral protein translation | Prevents E4orf6/E1B mediated degradation of AAV capsids & Rep52 | Yes | [29,189,193,201,210] | |
| HPV-16 | E1 | HPV-16 DNA replication, binds origin of DNA replication on the HPV-16 genome | Binds AAV Rep-ITR, nicking activity, complements AdV5 helper factors, increases rAAV titers, augments rep and cap expression | No | [220,221,222,224,225] |
| E2 | Increases p53 levels, activates transcription, inhibits E6 | Enhances AAV titers | No | [10,222] | |
| E6 | Oncogene | Complements AdV5 helper factors, increases rAAV titers, augments rep and cap expression | No | [219,220,222] | |
| HBoV1 | NS2 | Trans-activation of viral promoters | Promotes AAV second-strand synthesis, not essential when AAV genome is a duplex | (Yes) | [12,226] |
| NS4 | Trans-activation of viral promoters | Activation of AAV promoter | (Yes/No) 1 | [12,226] | |
| NP1 | Processing of viral pre-mRNA, trans-activation of viral promoters | Activation of AAV promoter | Yes | [12,226] | |
| BocaSR | Regulates NS1, NS2, NS3, NP1 expression and viral DNA replication | Not known | Yes | [12,226] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meier, A.F.; Fraefel, C.; Seyffert, M. The Interplay between Adeno-Associated Virus and Its Helper Viruses. Viruses 2020, 12, 662. https://doi.org/10.3390/v12060662
Meier AF, Fraefel C, Seyffert M. The Interplay between Adeno-Associated Virus and Its Helper Viruses. Viruses. 2020; 12(6):662. https://doi.org/10.3390/v12060662
Chicago/Turabian StyleMeier, Anita F., Cornel Fraefel, and Michael Seyffert. 2020. "The Interplay between Adeno-Associated Virus and Its Helper Viruses" Viruses 12, no. 6: 662. https://doi.org/10.3390/v12060662
APA StyleMeier, A. F., Fraefel, C., & Seyffert, M. (2020). The Interplay between Adeno-Associated Virus and Its Helper Viruses. Viruses, 12(6), 662. https://doi.org/10.3390/v12060662