Quality Control of Next-Generation Sequencing-Based HIV-1 Drug Resistance Data in Clinical Laboratory Information Systems Framework

 ,
,
Abstract
1. Introduction
2. Quality Control Management with Laboratory Information Systems
2.1. Pre-Analytical
2.2. Analytical
2.2.1. Reagent Tracking and Inventory
2.2.2. Instrument Integration and Automation
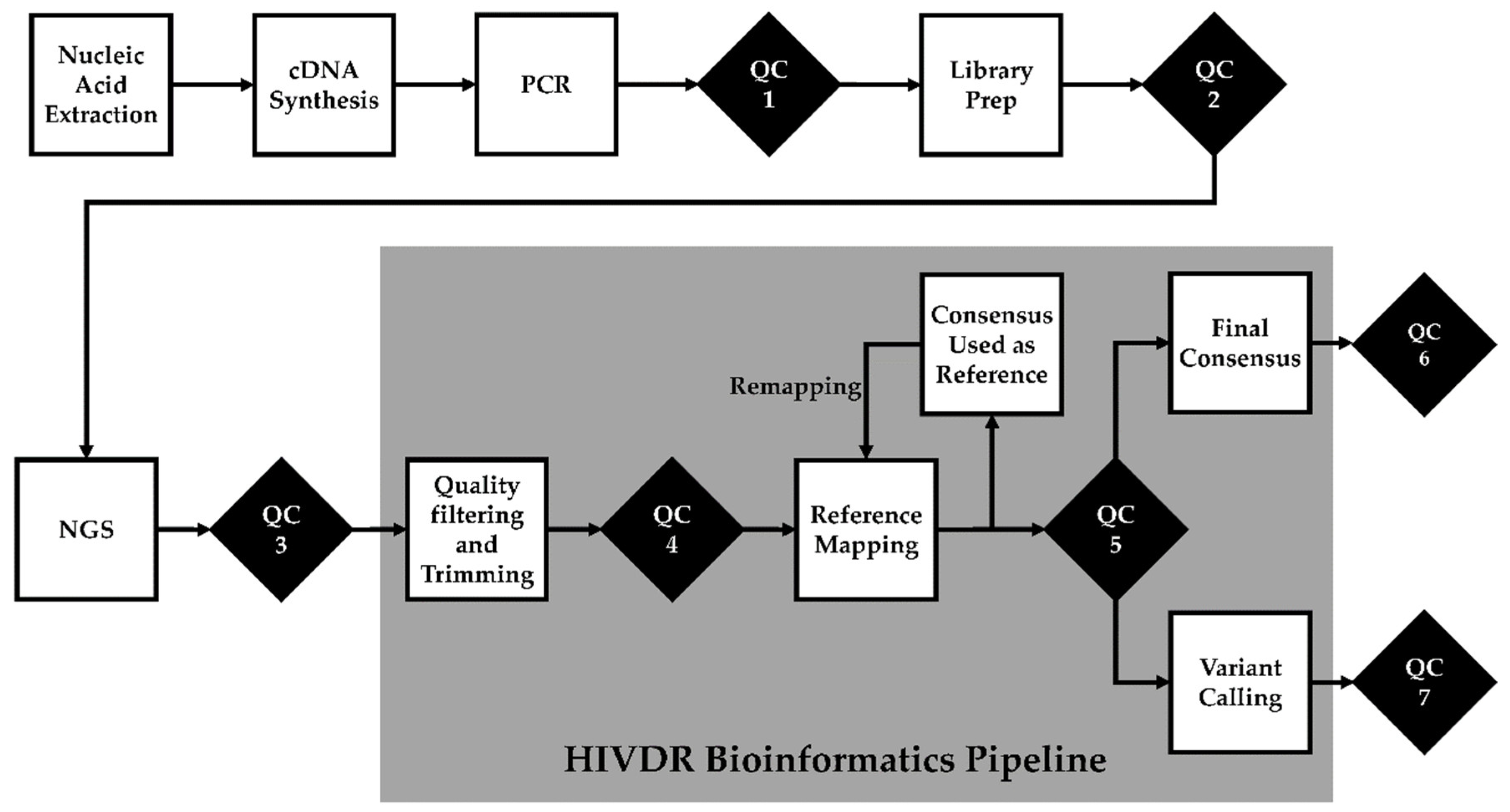
2.2.3. Quality Control Checks and Tractability
QC Checkpoint 1: Post-PCR Amplification
QC Checkpoint 2: Library Preparation
QC Checkpoint 3: Post-Sequencing Run
QC Checkpoint 4: Pre-Processing
QC Checkpoint 5: Post-Reference Mapping
QC Checkpoint 6: Sample Mislabeling and Contamination
QC Checkpoint 7: “Bad” Mutations
Clonal and Repeated Sample Check
Turn-Around Time Check
2.2.4. Data Review, Results Authorization, and Release
2.3. Post-Analytical
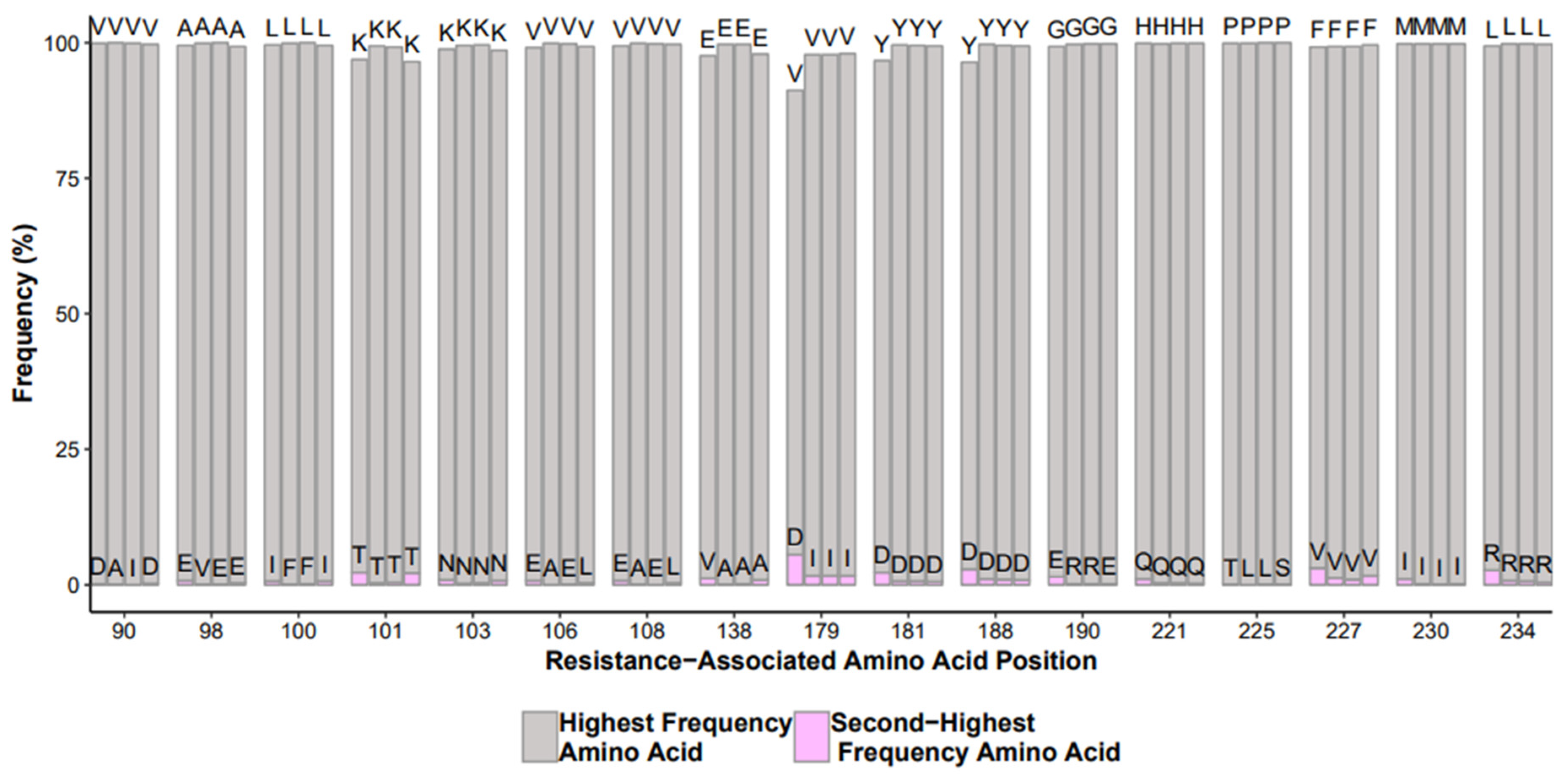
- Performance comparison of different reagent lot numbers, equipment, operators, and test controls run in different batches. A useful control to monitor is a mixture of clonal samples with known nucleotide mixtures and the comparison of the frequency of those mixtures. A display of histograms of mutations in test controls or repeated samples with definable flags of signification deviation from historic mutation frequencies;
- Automated scheduling of equipment maintenance and alerts staff of appropriate QC tasks;
- Automated tracking and stock management of reagent and consumables;
- Automated notification to lab manager of specimens with increased turnaround time;
- Automated notification of low specimen volumes and identified bottle necks;
- The system should allow monitoring of equipment performance, such temperature logs, frequency of failed runs, environmental conditions, and any documentations required by accrediting bodies;
- The system should provide summaries of QC reports to supervisors for review, corrective and preventive actions;
- Trend interesting results that are of interest to public health, such as the identification of genetic or transmission clusters, or changes in the prevalence of certain drug-resistant mutations [54].
- Frequently update or investigate new bioinformatics software which cannot be locked down to traditional Information Technology (IT) change control processes often associated with universal software applications used in office settings;
- Have IT security experts imbedded within scientific computers to ensure the hardware and software are secure, protected and monitored against threats that could compromise the security of the data they hold;
- Facilitate evolution of laboratory test for HIV drug resistance. Changes in the status quo often require a business analyst, programmer and infrastructure personnel to analyze the requirement, develop/modify the application and maintain the infrastructure without impacting business continuity;
- Reduce licensing costs by eliminating redundant LIS in an organization.
3. Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV infection. Nat. Rev. Dis. Prim. 2015, 1, 15060. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Enns, E.; Brumme, C.J.; Parkin, N.; Howison, M.; Lee, E.R.; Capina, R.; Marinier, E.; Avila-Rios, S.; Sandstrom, P.; et al. Bioinformatic data processing pipelines in support of next-generation sequencing-based HIV drug resistance testing: The Winnipeg Consensus. J. Int. AIDS Soc. 2018, 21, e25193. [Google Scholar] [CrossRef]
- World Health Organization. Global Action Plan. on HIV Drug Resistance 2017–2021; World Health Organization: Geneva, Switzerland, 2017; ISBN 9789241512848. [Google Scholar]
- Lee, E.R.; Parkin, N.; Jennings, C.; Brumme, C.J.; Enns, E.; Casadellà, M.; Howison, M.; Coetzer, M.; Avila-Rios, S.; Capina, R.; et al. Performance comparison of next generation sequencing analysis pipelines for HIV-1 drug resistance testing. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vrancken, B.; Trovão, N.; Baele, G.; van Wijngaerden, E.; Vandamme, A.-M.; van Laethem, K.; Lemey, P. Quantifying Next Generation Sequencing Sample Pre-Processing Bias in HIV-1 Complete Genome Sequencing. Viruses 2016, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Gargis, A.S.; Kalman, L.; Lubin, I.M. Assuring the quality of next-generation sequencing in clinical microbiology and public health laboratories. J. Clin. Microbiol. 2016, 54, 2857–2865. [Google Scholar] [CrossRef]
- Gargis, A.S.; Kalman, L.; Berry, M.W.; Bick, D.P.; Dimmock, D.P.; Hambuch, T.; Lu, F.; Lyon, E.; Voelkerding, K.V.; Zehnbauer, B.A.; et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat. Biotechnol. 2012, 30, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, R.J.; Phan, K.L.; Saboor, A.; Miller, J.D.; Muehlenbachs, A. Practical guidance to implementing quality management systems in public health laboratories performing next-generation sequencing: Personnel, equipment, and process management (Phase 1). J. Clin. Microbiol. 2019, 57, e00261. [Google Scholar] [CrossRef]
- MM09A2: Nucleic Acid Sequencing Methods in Lab Medicine. Available online: https://clsi.org/standards/products/molecular-diagnostics/documents/mm09/ (accessed on 25 March 2020).
- Vani, K.; Sompuram, S.R.; Naber, S.P.; Goldsmith, J.D.; Fulton, R.; Bogen, S.A.; Bogen, S. Levey-Jennings Analysis Uncovers Unsuspected Causes Of Immunohistochemistry Stain Variability HHS Public Access. Appl. Immunohistochem. Mol. Morphol. 2016, 24, 688–694. [Google Scholar] [CrossRef]
- Nelson, L.S. Shewhart Control Chart—Tests for Special Causes. J. Qual. Technol. 1984, 16, 237–239. [Google Scholar] [CrossRef]
- Infectious Disease Next Generation Sequencing Based Diagnostic Devices: Microbial Identification and Detection of Antimicrobial Resistance and Virulence Markers|FDA. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/infectious-disease-next-generation-sequencing-based-diagnostic-devices-microbial-identification-and (accessed on 25 March 2020).
- FDA Authorizes Marketing of First Next-Generation Sequencing Test for Detecting HIV-1 Drug Resistance Mutations|FDA. Available online: https://www.fda.gov/news-events/press-announcements/fda-authorizes-marketing-first-next-generation-sequencing-test-detecting-hiv-1-drug-resistance (accessed on 27 February 2020).
- Noguera-Julian, M.; Edgil, D.; Harrigan, P.R.; Sandstrom, P.; Godfrey, C.; Paredes, R. Immunodeficiency Virus Sequencing for Patient Management and Drug Resistance Surveillance. J. Infect. Dis. 2017, 216, S829–S833. [Google Scholar] [CrossRef]
- Introduction to Lab Information Management Systems. Available online: https://www.illumina.com/informatics/sample-experiment-management/lims.html (accessed on 25 March 2020).
- Laboratory Information Systems Project Management: A Guidebook for International Implementations. Available online: https://www.aphl.org/aboutAPHL/publications/Documents/GH-2019May-LIS-Guidebook-web.pdf (accessed on 25 March 2020).
- Harsono, D.; Galletly, C.L.; O’Keefe, E.; Lazzarini, Z. Criminalization of HIV Exposure: A Review of Empirical Studies in the United States. AIDS Behav. 2017, 21, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, J.L.; Young, D.S. The ideal laboratory information system. Arch. Pathol. Lab. Med. 2013, 137, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Q.; Vickers, K.; Zhao, S.; Wang, J.; Samuels, D.C.; Koues, O.; Shyr, Y.; Guo, Y. Multi-perspective quality control of Illumina RNA sequencing data analysis. Brief. Funct. Genom. 2017, 16, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Troubleshooting Demultiplexing Issues Using MiSeq Reporter. Available online: https://support.illumina.com/bulletins/2016/08/troubleshooting-demultiplexing-issues-using-miseq-reporter.html (accessed on 25 March 2020).
- Howison, M.; Coetzer, M.; Kantor, R. Measurement error and variant-calling in deep Illumina sequencing of HIV. Bioinformatics 2019, 35, 2029–2035. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.; Metzner, K.J.; Geissberger, F.D.; Shah, C.; Leemann, C.; Klimkait, T.; Böni, J.; Trkola, A.; Zagordi, O. MinVar: A rapid and versatile tool for HIV-1 drug resistance genotyping by deep sequencing. J. Virol. Methods 2017, 240, 7–13. [Google Scholar] [CrossRef]
- Doring, M.; Buch, J.; Friedrich, G.; Pironti, A.; Kalaghatgi, P.; Knops, E.; Heger, E.; Obermeier, M.; Däumer, M.; Thielen, A.; et al. geno2pheno[ngs-freq]: A genotypic interpretation system for identifying viral drug resistance using next-generation sequencing data. Nucleic Acids Res. 2018, 46, W271–W277. [Google Scholar] [CrossRef]
- HyDRA Web. Available online: https://hydra.canada.ca/ (accessed on 25 March 2020).
- GitHub-Cfe-Lab/MiCall: Pipeline for Processing FASTQ Data from an Illumina MiSeq to Genotype Human RNA Viruses Like HIV and Hepatitis C. Available online: https://github.com/cfe-lab/MiCall (accessed on 26 March 2020).
- PASEQ. Available online: https://paseq.org/ (accessed on 25 March 2020).
- Exatype. Available online: https://exatype.com/ (accessed on 26 March 2020).
- Garcia-Diaz, A.; McCormick, A.; Booth, C.; Gonzalez, D.; Sayada, C.; Haque, T.; Johnson, M.; Webster, D. Analysis of transmitted HIV-1 drug resistance using 454 ultra-deep-sequencing and the DeepChek®-HIV system. J. Int. AIDS Soc. 2014, 17, 19752. [Google Scholar] [CrossRef]
- Babraham Bioinformatics—FastQC A Quality Control tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 26 March 2020).
- WHO/Hivresnet Hiv Drug Resistance Laboratory Operational Framework. Available online: https://apps.who.int/iris/bitstream/handle/10665/259731/9789241512879-eng.pdf;jsessionid=622FC41FF2556EF9C7C50298B84EC30D?sequence=1 (accessed on 26 March 2020).
- Taylor, T.; Lee, E.R.; Nykoluk, M.; Enns, E.; Liang, B.; Capina, R.; Gauthier, M.K.; Van Domselaar, G.; Sandstrom, P.; Brooks, J.; et al. A MiSeq-HyDRA platform for enhanced HIV drug resistance genotyping and surveillance. Sci. Rep. 2019, 9, 8970. [Google Scholar] [CrossRef]
- Borst, A.; Box, A.T.A.; Fluit, A.C. False-positive results and contamination in nucleic acid amplification assays: Suggestions for a prevent and destroy strategy. Eur. J. Clin. Microbiol. Infect. Dis. 2004, 23, 289–299. [Google Scholar] [CrossRef]
- Woods, C.K.; Brumme, C.J.; Liu, T.F.; Chui, C.K.S.; Chu, A.L.; Wynhoven, B.; Hall, T.A.; Trevino, C.; Shafer, R.W.; Harrigan, P.R. Automating HIV drug resistance genotyping with RECall, a freely accessible sequence analysis tool. J. Clin. Microbiol. 2012, 50, 1936–1942. [Google Scholar] [CrossRef]
- WHO Resistance Quality Control Tool. Available online: https://pssm.cfenet.ubc.ca/who_qc (accessed on 2 March 2020).
- Poon, A.F.Y.; Joy, J.B.; Woods, C.K.; Shurgold, S.; Colley, G.; Brumme, C.J.; Hogg, R.S.; Montaner, J.S.G.; Harrigan, P.R. The impact of clinical, demographic and risk factors on rates of HIV transmission: A population-based phylogenetic analysis in British Columbia, Canada. J. Infect. Dis. 2015, 211, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Hightower, G.K.; May, S.J.; Pérez-Santiago, J.; Pacold, M.E.; Wagner, G.A.; Little, S.J.; Richman, D.D.; Mehta, S.R.; Smith, D.M.; Pond, S.L.K. HIV-1 Clade B pol Evolution following Primary Infection. PLoS ONE 2013, 8, e68188. [Google Scholar] [CrossRef] [PubMed]
- Brumme, C.J.; Poon, A.F.Y. Promises and pitfalls of Illumina sequencing for HIV resistance genotyping. Virus Res. 2017, 239, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Olivo, A.; Laeyendecker, O.; Forberg, K.; Ndembi, N.; Mbanya, D.; Kaptue, L.; Quinn, T.C.; Cloherty, G.A.; Rodgers, M.A.; et al. Universal Target Capture of HIV Sequences From NGS Libraries. Front. Microbiol. 2018, 9, 2150. [Google Scholar] [CrossRef]
- Zanini, F.; Brodin, J.; Thebo, L.; Lanz, C.; Bratt, G.; Albert, J.; Neher, R.A. Population genomics of intrapatient HIV-1 evolution. Elife 2015, 4, 13239. [Google Scholar] [CrossRef]
- GitHub—MicrobialGenomics/ViCroSeq: A Tool for the Removal of Viral Cross-Contamination in Sequencing—ViCroSeq. Available online: https://github.com/MicrobialGenomics/ViCroSeq (accessed on 25 March 2020).
- Tzou, P.L.; Kosakovsky Pond, S.L.; Avila-Rios, S.; Holmes, S.P.; Kantor, R.; Shafer, R.W. Analysis of unusual and signature APOBEC-mutations in HIV-1 pol next-generation sequences. PLoS ONE 2020, 15, e0225352. [Google Scholar] [CrossRef]
- HIV Sequence Database. Available online: https://www.hiv.lanl.gov/content/sequence/NEWALIGN/align.html (accessed on 2 March 2020).
- Noguera-Julian, M.; Cozzi-Lepri, A.; Di Giallonardo, F.; Schuurman, R.; Däumer, M.; Aitken, S.; Ceccherini-Silberstein, F.; Monforte, A.; Geretti, A.M.; Booth, C.L.; et al. Contribution of APOBEC3G/F activity to the development of low-abundance drug-resistant human immunodeficiency virus type 1 variants. Clin. Microbiol. Infect. 2016, 22, 191–200. [Google Scholar] [CrossRef]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef]
- Clutter, D.S.; Zhou, S.; Varghese, V.; Rhee, S.-Y.; Pinsky, B.A.; Fessel, W.J.; Klein, D.B.; Spielvogel, E.; Holmes, S.P.; Hurley, L.B.; et al. Prevalence of Drug-Resistant Minority Variants in Untreated HIV-1-Infected Individuals With and Those Without Transmitted Drug Resistance Detected by Sanger Sequencing. J. Infect. Dis. Br. Rep. 2017, 2017, 387–391. [Google Scholar] [CrossRef]
- Dauwe, K.; Staelens, D.; Vancoillie, L.; Mortier, V.; Verhofstede, C. Deep sequencing of HIV-1 RNA and DNA in newly diagnosed patients with baseline drug resistance showed no indications for hidden resistance and is biased by strong interference of hypermutation. J. Clin. Microbiol. 2016, 54, 1605–1615. [Google Scholar] [CrossRef]
- Delviks-Frankenberry, K.A.; Nikolaitchik, O.A.; Burdick, R.C.; Gorelick, R.J.; Keele, B.F.; Hu, W.S.; Pathak, V.K. Minimal Contribution of APOBEC3-Induced G-to-A Hypermutation to HIV-1 Recombination and Genetic Variation. PLoS Pathog. 2016, 12, e1005646. [Google Scholar] [CrossRef] [PubMed]
- Wensing, A.M.; Calvez, V.; Ceccherini-Silberstein, F.; Charpentier, C.; Günthard, H.F.; Paredes, R.; Shafer, R.W.; Richman, D.D. 2019 update of the drug resistance mutations in HIV-1. Top. Antivir. Med 2019, 27, 111–121. [Google Scholar] [PubMed]
- Wymant, C.; Blanquart, F.; Golubchik, T.; Gall, A.; Bakker, M.; Bezemer, D.; Croucher, N.J.; Hall, M.; Hillebregt, M.; Ong, S.H.; et al. Easy and accurate reconstruction of whole HIV genomes from short-read sequence data with shiver. Virus Evol. 2018, 4. [Google Scholar] [CrossRef]
- V-pipe|Virus NGS Pipeline—Bioinformatics Pipeline for the Analysis of Next-Generation Sequencing Data Derived from Intra-Host Viral Populations. Available online: https://cbg-ethz.github.io/V-pipe/ (accessed on 26 March 2020).
- Oosterhuis, W.P.; Ulenkate, H.J.L.M.; Goldschmidt, H.M.J. Evaluation of LabRespond, a new automated validation system for clinical laboratory test results. Clin. Chem. 2000, 46, 1811–1817. [Google Scholar] [CrossRef] [PubMed]
- Goldschmidt, H.M.J. A review of autovalidation software in laboratory medicine. Accredit. Qual. Assur. 2002, 7, 431–440. [Google Scholar] [CrossRef]
- Westgard, J.O.; Barry, P.L.; Burnett, R.W.; Nipper, H.; Hunt, M.R.; Groth, T. A Multi-Rule Shewhart Chart for Quality Control in Clinical Chemistry. Clin. Chem. 1981, 27, 493–501. [Google Scholar] [CrossRef]
- Poon, A.F.Y.; Gustafson, R.; Daly, P.; Zerr, L.; Demlow, S.E.; Wong, J.; Woods, C.K.; Hogg, R.S.; Krajden, M.; Moore, D.; et al. Near real-time monitoring of HIV transmission hotspots from routine HIV genotyping: An implementation case study. Lancet HIV 2016, 3, e231–e238. [Google Scholar] [CrossRef]
- Clinical Laboratory Improvement Amendments (CLIA)|CDC. Available online: https://www.cdc.gov/clia/ (accessed on 3 March 2020).
- Luh, F.; Yen, Y. FDA guidance for next generation sequencing-based testing: Balancing regulation and innovation in precision medicine. Npj Genom. Med. 2018, 3, 28. [Google Scholar] [CrossRef]
- Inzaule, S.C.; Hamers, R.L.; Noguera-Julian, M.; Casadellà, M.; Parera, M.; Kityo, C.; Steegen, K.; Naniche, D.; Clotet, B.; Rinke de Wit, T.F.; et al. Clinically relevant thresholds for ultrasensitive HIV drug resistance testing: A multi-country nested case-control study. Lancet HIV 2018, 5, e638–e646. [Google Scholar] [CrossRef]
- Grimes, S.M.; Ji, H.P. MendeLIMS: A web-based laboratory information management system for clinical genome sequencing. BMC Bioinform. 2014, 15, 290. [Google Scholar] [CrossRef]
- Goldberg, B.; Sichtig, H.; Geyer, C.; Ledeboer, N.; Weinstock, G.M. Making the leap from research laboratory to clinic: Challenges and opportunities for next-generation sequencing in infectious disease diagnostics. MBio 2015, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Matthijs, G.; Souche, E.; Alders, M.; Corveleyn, A.; Eck, S.; Feenstra, I.; Race, V.; Sistermans, E.; Sturm, M.; Weiss, M.; et al. Guidelines for diagnostic next-generation sequencing. Eur. J. Hum. Genet. 2016, 24, 1515. [Google Scholar] [CrossRef] [PubMed]
- Yohe, S.; Thyagarajan, B. Review Articles Review of Clinical Next-Generation Sequencing. Arch. Pathol Lab. Med. 2017, 141, 1544–1557. [Google Scholar] [CrossRef]
- Endrullat, C.; Glökler, J.; Franke, P.; Frohme, M. Standardization and quality management in next-generation sequencing. Appl. Transl. Genom. 2016, 10, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Wymant, C.; Hall, M.; Ratmann, O.; Bonsall, D.; Golubchik, T.; De Cesare, M.; Gall, A.; Cornelissen, M.; Fraser, C. PHYLOSCANNER: Inferring transmission from within- and between-host pathogen genetic diversity. Mol. Biol. Evol. 2018, 35, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Courtney, C.R.; Mayr, L.; Nanfack, A.J.; Banin, A.N.; Tuen, M.; Pan, R.; Jiang, X.; Kong, X.P.; Kirkpatrick, A.R.; Bruno, D.; et al. Contrasting antibody responses to intrasubtype superinfection with CRF02-AG. PLoS ONE 2017, 12, e0173705. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Metric/Threshold | Sample Expected Value | Sample QC Tool |
|---|---|---|
| QC1: Post-PCR | ||
| Amplicon | Negative control: no bandPositive control: band at correct size | Gel/Capillary electrophoresis |
| QC2: Library Preparation | ||
| Library size | Normal distribution around 300–500 bp | Bioanalyzer/Tapestation 1 |
| Library concentration | 0.2 ng/μL | Bioanalyzer/Tapestation |
| QC3: Post-Sequencing Run | See Hutchins et al. [8] | SAV 2 |
| QC4: Pre-processing | See Hutchins et al. [8] | FastQC 3 |
| QC5: Post-Reference Mapping (performed after final remapping) | ||
| Sequence Coverage | PR: codon 10–93 RT: codon 41–238 IN: codon 51–263 | HIVDR Pipeline, Tablet 4, UGENE 5 |
| Mean read depth | ≥1000 | HIVDR Pipeline, Tablet, UGENE |
| QC6: Mislabeling/Contamination (Check for genetic relatedness) | ||
| Nucleotide mixture | <3.5% nucleotide positions | MEGA 6 |
| Sequences from same patient | <2.5% genetic dissimilarity | WHO BCCFE HIVDR QC 7 |
| Intra-batch sample vs other sample | ≥0.5% genetic dissimilarity | WHO BCCFE HIVDR QC |
| Sample vs control strain | ≥0.5% genetic dissimilarity | WHO BCCFE HIVDR QC |
| Across-batch sample vs other sample | ≥0.5% genetic dissimilarity | WHO BCCFE HIVDR QC |
| QC7: “Bad” Mutations/Variant Calls | ||
| “Unusual” mutations | <1.0% | HIVdb-NGS 8 |
| Signature APOBEC hypermutations | <3 | HIVdb-NGS |
| APOBEC-context DRMs | <2 | HIVdb-NGS |
| Stop codons | 0 | HIVdb-NGS |
| Codon insertion/deletion | 0 | HIVdb-NGS |
| Frameshift insertion/deletion | 0 | HIVdb-NGS |
| Variant Calling | ||
| Position depth | ≥100 reads | HIVDR Pipeline |
| Q score | Q≥30 | HIVDR Pipeline |
| Variant count | ≥5 reads | HIVDR Pipeline |
| Turnaround Time | 5–6 Days | N/A |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capina, R.; Li, K.; Kearney, L.; Vandamme, A.-M.; Harrigan, P.R.; Van Laethem, K. Quality Control of Next-Generation Sequencing-Based HIV-1 Drug Resistance Data in Clinical Laboratory Information Systems Framework. Viruses 2020, 12, 645. https://doi.org/10.3390/v12060645
Capina R, Li K, Kearney L, Vandamme A-M, Harrigan PR, Van Laethem K. Quality Control of Next-Generation Sequencing-Based HIV-1 Drug Resistance Data in Clinical Laboratory Information Systems Framework. Viruses. 2020; 12(6):645. https://doi.org/10.3390/v12060645
Chicago/Turabian StyleCapina, Rupert, Katherine Li, Levon Kearney, Anne-Mieke Vandamme, P. Richard Harrigan, and Kristel Van Laethem. 2020. "Quality Control of Next-Generation Sequencing-Based HIV-1 Drug Resistance Data in Clinical Laboratory Information Systems Framework" Viruses 12, no. 6: 645. https://doi.org/10.3390/v12060645
APA StyleCapina, R., Li, K., Kearney, L., Vandamme, A.-M., Harrigan, P. R., & Van Laethem, K. (2020). Quality Control of Next-Generation Sequencing-Based HIV-1 Drug Resistance Data in Clinical Laboratory Information Systems Framework. Viruses, 12(6), 645. https://doi.org/10.3390/v12060645