Genomic Sequencing and Analysis of Eight Camel-Derived Middle East Respiratory Syndrome Coronavirus (MERS-CoV) Isolates in Saudi Arabia

 ,
,  , , ,
, , ,
Abstract
1. Introduction
2. Results
2.1. Genome Organization
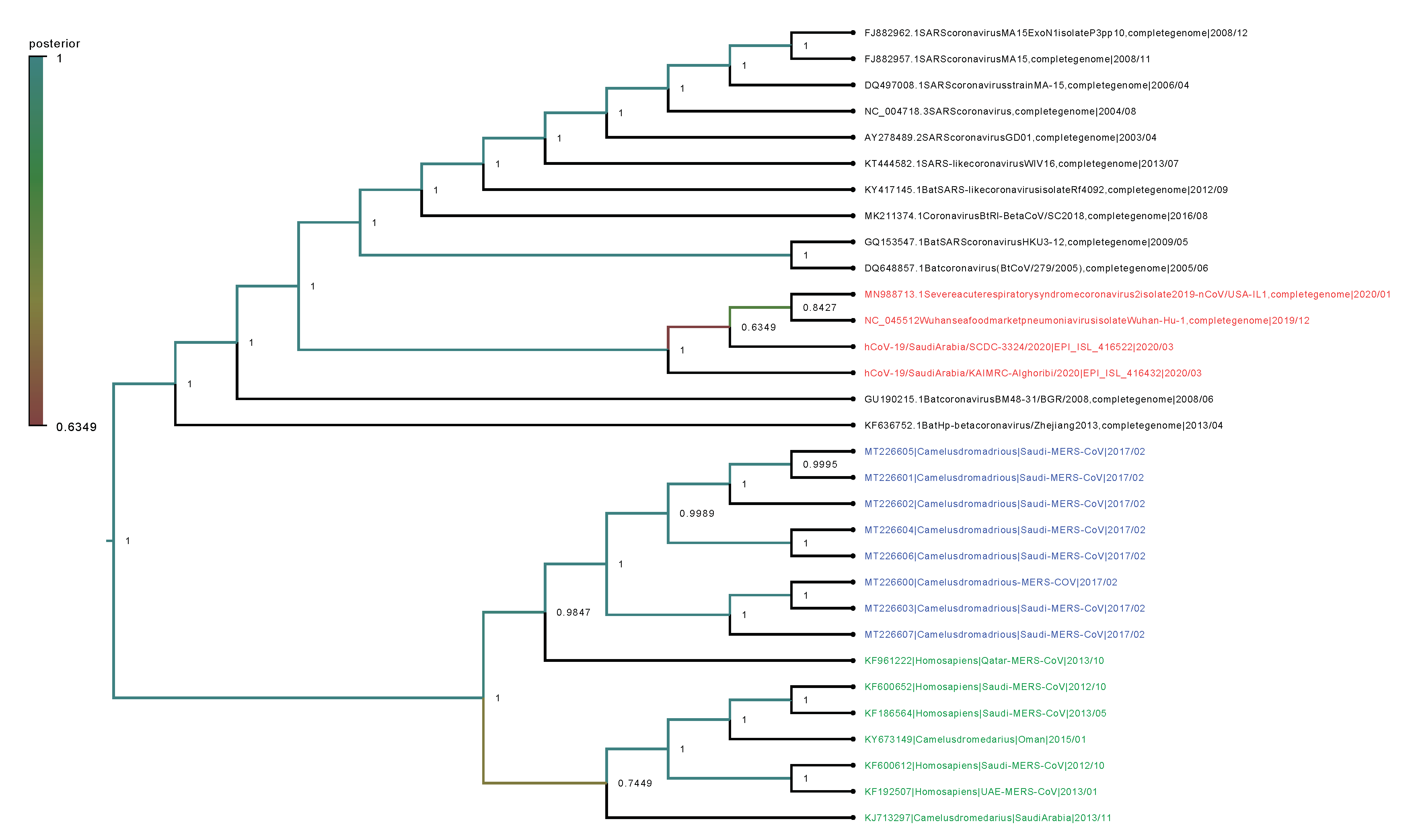
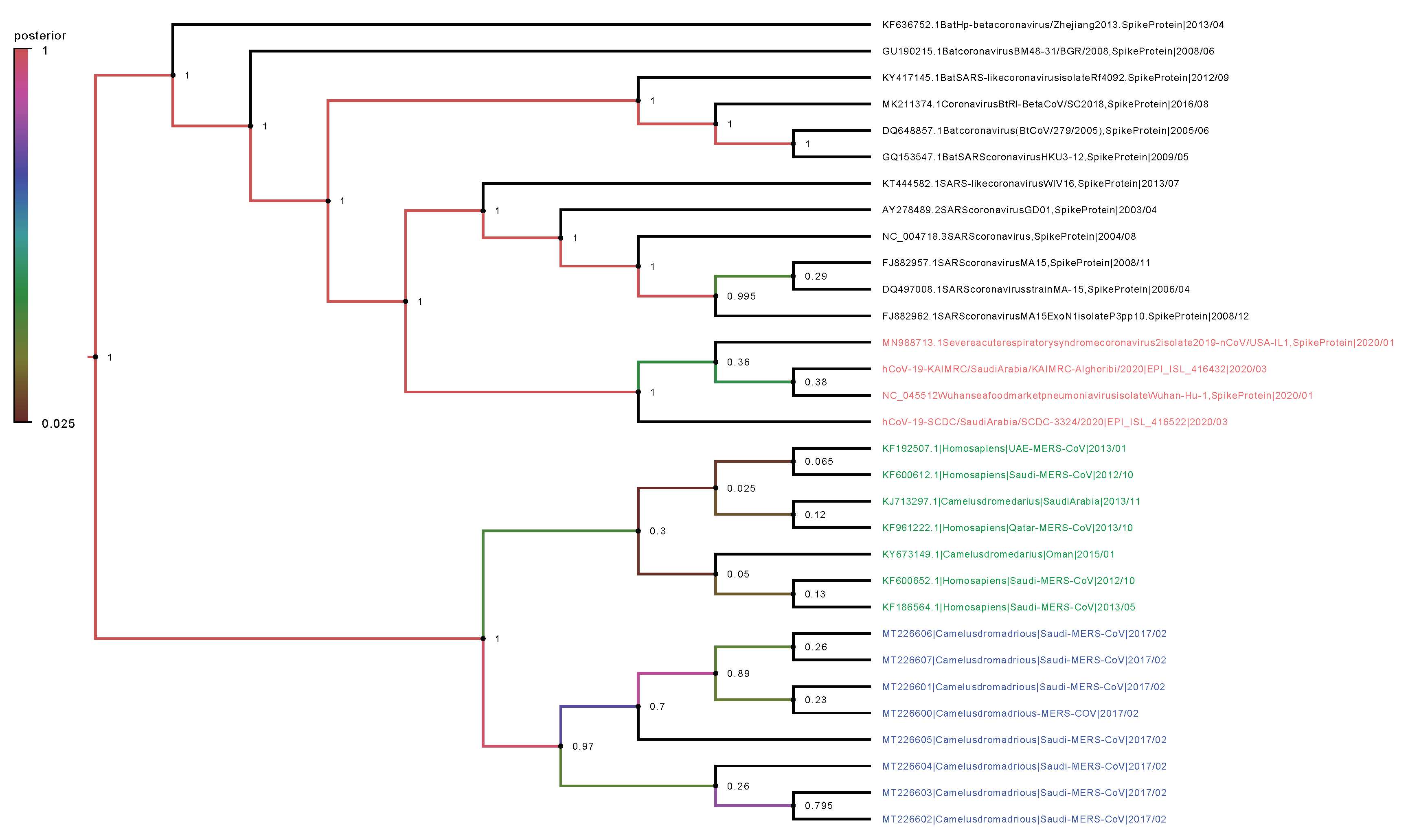
2.2. Phylogenetic Analysis
2.3. MERS-CoV B Cell Epitope Analysis
3. Discussion
4. Materials and Methods
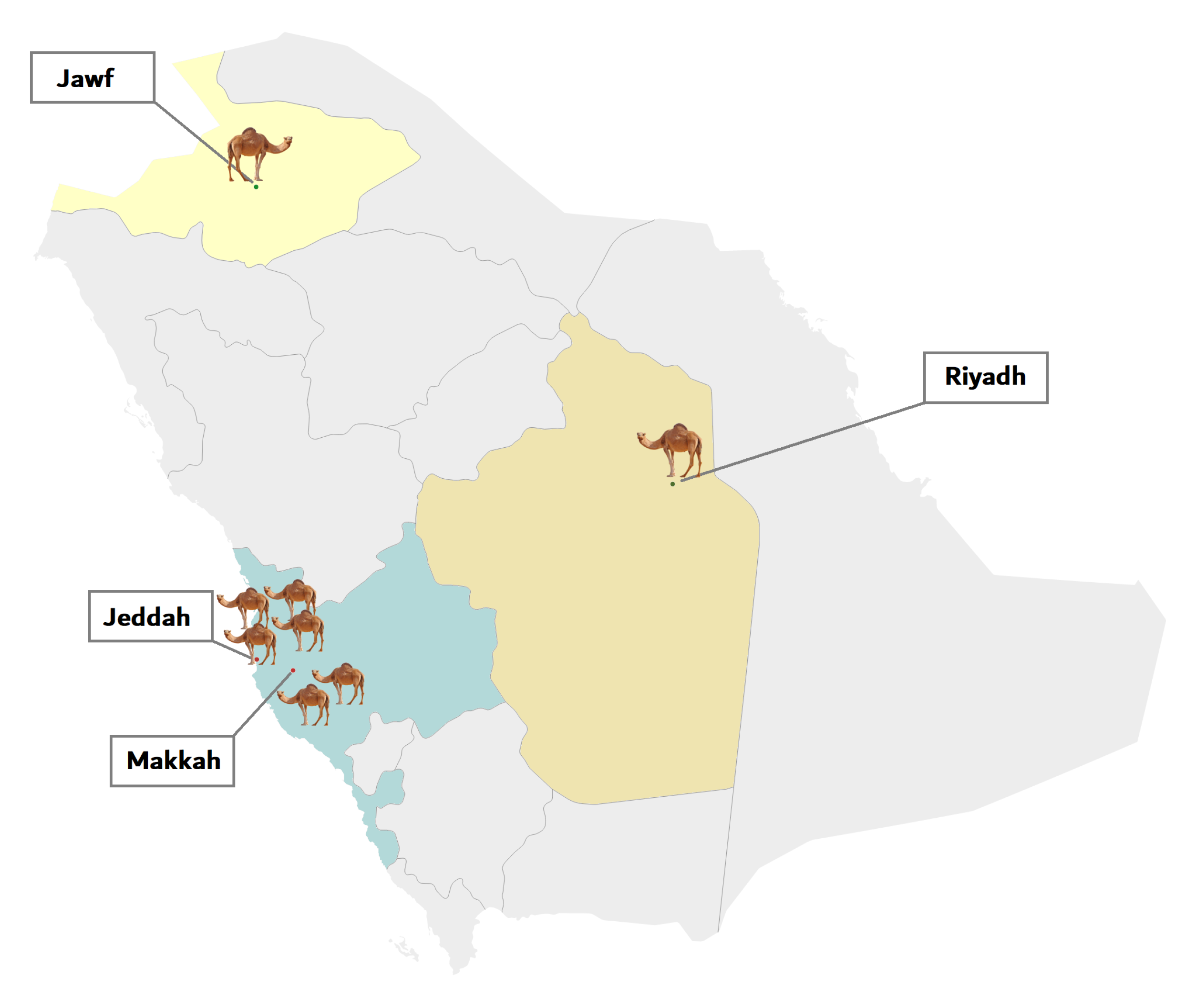
4.1. Sample Collection
4.2. RNA Extraction
4.3. Sequence Preparation
4.4. RNA Quantification and cDNA Conversion
4.5. Library Preparation and Sequencing
4.6. Gap Filling and Sanger Sequencing
4.7. Phylogenetic Trees and Evolutionary Dynamics
4.8. Prediction of MERS-CoV B Cell Epitopes
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Miłek, J.; Blicharz-Domańska, K. Coronaviruses in avian species–review with focus on epidemiology and diagnosis in wild birds. J. Vet. Res. 2018, 62, 249–255. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, X.; Irwin, D.M.; Shen, Y. Emergence of SARS-like coronavirus poses new challenge in China. J. Infect. 2020, 80, 350–371. [Google Scholar] [CrossRef]
- Li, F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed]
- Millet, J.K.; Séron, K.; Labitt, R.N.; Danneels, A.; Palmer, K.E.; Whittaker, G.R.; Dubuisson, J.; Belouzard, S. Middle East respiratory syndrome coronavirus infection is inhibited by griffithsin. Antivir. Res. 2016, 133, 1–8. [Google Scholar] [CrossRef]
- Zaki, A.M.; Van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- World Health Organization. Middle East Respiratory Syndrome Coronavirus (MERS-CoV). 2018. Available online: http://www.who.int/news-room/fact-sheets/detail/middle-east-respiratory-syndrome-coronavirus-(mers-cov) (accessed on 28 August 2018).
- Hemida, M.G.; Al-Naeem, A.; Perera, R.A.; Chin, A.W.; Poon, L.L.; Peiris, M. Lack of Middle East respiratory syndrome coronavirus transmission from infected camels. Emerg. Infect. Dis. 2015, 21, 699. [Google Scholar] [CrossRef] [PubMed]
- Hemida, M.G.; Chu, D.K.; Poon, L.L.; Perera, R.A.; Alhammadi, M.A.; Ng, H.Y.; Siu, L.Y.; Guan, Y.; Alnaeem, A.; Peiris, M. MERS coronavirus in dromedary camel herd, Saudi Arabia. Emerg. Infect. Dis. 2014, 20, 1231. [Google Scholar] [CrossRef]
- Perera, R.; Wang, P.; Gomaa, M.; El-Shesheny, R.; Kandeil, A.; Bagato, O.; Siu, L.; Shehata, M.; Kayed, A.; Moatasim, Y.; et al. Seroepidemiology for MERS coronavirus using microneutralisation and pseudoparticle virus neutralisation assays reveal a high prevalence of antibody in dromedary camels in Egypt, June 2013. Eurosurveillance 2013, 18, 20574. [Google Scholar] [CrossRef]
- Müller, M.A.; Corman, V.M.; Jores, J.; Meyer, B.; Younan, M.; Liljander, A.; Bosch, B.J.; Lattwein, E.; Hilali, M.; Musa, B.E.; et al. MERS coronavirus neutralizing antibodies in camels, Eastern Africa, 1983–1997. Emerg. Infect. Dis. 2014, 20, 2093. [Google Scholar] [CrossRef]
- Zumla, A.; Hui, D.S.; Perlman, S. Middle East respiratory syndrome. Lancet 2015, 386, 995–1007. [Google Scholar] [CrossRef]
- Van Doremalen, N.; Munster, V.J. Animal models of Middle East respiratory syndrome coronavirus infection. Antivir. Res. 2015, 122, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Memish, Z.A.; Cotten, M.; Meyer, B.; Watson, S.J.; Alsahafi, A.J.; Al Rabeeah, A.A.; Corman, V.M.; Sieberg, A.; Makhdoom, H.Q.; Assiri, A.; et al. Human infection with MERS coronavirus after exposure to infected camels, Saudi Arabia, 2013. Emerg. Infect. Dis. 2014, 20, 1012. [Google Scholar] [CrossRef] [PubMed]
- Alagaili, A.N.; Briese, T.; Mishra, N.; Kapoor, V.; Sameroff, S.C.; de Wit, E.; Munster, V.J.; Hensley, L.E.; Zalmout, I.S.; Kapoor, A.; et al. Middle East respiratory syndrome coronavirus infection in dromedary camels in Saudi Arabia. MBio 2014, 5, e00884-14. [Google Scholar] [CrossRef] [PubMed]
- Azhar, E.I.; El-Kafrawy, S.A.; Farraj, S.A.; Hassan, A.M.; Al-Saeed, M.S.; Hashem, A.M.; Madani, T.A. Evidence for camel-to-human transmission of MERS coronavirus. N. Engl. J. Med. 2014, 370, 2499–2505. [Google Scholar] [CrossRef]
- Wu, H.; Guang, X.; Al-Fageeh, M.B.; Cao, J.; Pan, S.; Zhou, H.; Zhang, L.; Abutarboush, M.H.; Xing, Y.; Xie, Z.; et al. Camelid genomes reveal evolution and adaptation to desert environments. Nat. Commun. 2014, 5, 5188. [Google Scholar] [CrossRef]
- Fowler, M. Medicine and Surgery of Camelids; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Conzade, R.; Grant, R.; Malik, M.; Elkholy, A.; Elhakim, M.; Samhouri, D.; Ben Embarek, P.; Van Kerkhove, M. Reported direct and indirect contact with dromedary camels among laboratory-confirmed MERS-CoV cases. Viruses 2018, 10, 425. [Google Scholar] [CrossRef]
- Cauchemez, S.; Nouvellet, P.; Cori, A.; Jombart, T.; Garske, T.; Clapham, H.; Moore, S.; Mills, H.L.; Salje, H.; Collins, C.; et al. Unraveling the drivers of MERS-CoV transmission. Proc. Natl. Acad. Sci. USA 2016, 113, 9081–9086. [Google Scholar] [CrossRef]
- Al-Tawfiq, J.A.; Memish, Z.A. Middle East respiratory syndrome coronavirus: Transmission and phylogenetic evolution. Trends Microbiol. 2014, 22, 573–579. [Google Scholar] [CrossRef]
- Kim, J.I.; Kim, Y.J.; Lemey, P.; Lee, I.; Park, S.; Bae, J.Y.; Kim, D.; Kim, H.; Jang, S.I.; Yang, J.S.; et al. The recent ancestry of Middle East respiratory syndrome coronavirus in Korea has been shaped by recombination. Sci. Rep. 2016, 6, 18825. [Google Scholar] [CrossRef]
- Du, L.; Yang, Y.; Zhou, Y.; Lu, L.; Li, F.; Jiang, S. MERS-CoV spike protein: A key target for antivirals. Expert Opin. Ther. Targets 2017, 21, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Ul Qamar, M.T.; Saleem, S.; Ashfaq, U.A.; Bari, A.; Anwar, F.; Alqahtani, S. Epitope-based peptide vaccine design and target site depiction against Middle East Respiratory Syndrome Coronavirus: An immune-informatics study. J. Transl. Med. 2019, 17, 362. [Google Scholar] [CrossRef] [PubMed]
- Goo, J.; Jeong, Y.; Park, Y.S.; Yang, E.; Jung, D.I.; Rho, S.; Park, U.; Sung, H.; Park, P.G.; Choi, J.A.; et al. Characterization of novel monoclonal antibodies against MERS-coronavirus spike protein. Virus Res. 2020, 197863. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Lee, S.I.; Bae, J.Y.; Park, M.S.; Lee, Y.; Kwon, H.J. Production of a Monoclonal Antibody targeting the M protein of MERS-CoV for detection of MERS-CoV using a synthetic peptide epitope formulated with a CpG–DNA–liposome complex. Int. J. Pept. Res. Ther. 2019, 25, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Paden, C.; Yusof, M.; Al Hammadi, Z.; Queen, K.; Tao, Y.; Eltahir, Y.; Elsayed, E.; Marzoug, B.; Bensalah, O.; Khalafalla, A.; et al. Zoonotic origin and transmission of Middle East respiratory syndrome coronavirus in the UAE. Zoonoses Public Health 2018, 65, 322–333. [Google Scholar] [CrossRef]
- Wernery, U.; Lau, S.K.; Woo, P.C. Middle East respiratory syndrome (MERS) coronavirus and dromedaries. Vet. J. 2017, 220, 75–79. [Google Scholar] [CrossRef]
- Killerby, M.E.; Biggs, H.M.; Midgley, C.M.; Gerber, S.I.; Watson, J.T. Middle East respiratory syndrome coronavirus transmission. Emerg. Infect. Dis. 2020, 26, 191. [Google Scholar] [CrossRef]
- Nowotny, N.; Kolodziejek, J. Middle East respiratory syndrome coronavirus (MERS-CoV) in dromedary camels, Oman, 2013. Eurosurveillance 2014, 19, 20781. [Google Scholar] [CrossRef]
- Yusof, M.F.; Eltahir, Y.M.; Serhan, W.S.; Hashem, F.M.; Elsayed, E.A.; Marzoug, B.A.; Abdelazim, A.S.; Bensalah, O.K.A.; Al Muhairi, S.S. Prevalence of Middle East respiratory syndrome coronavirus (MERS-CoV) in dromedary camels in Abu Dhabi emirate, United Arab Emirates. Virus Genes 2015, 50, 509–513. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Hemida, M.G.; Abduallah, M.M.B. The SARS-CoV-2 outbreak from a one health perspective. One Health 2020, 100127. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Suchard, M.; Xie, D.; Drummond, A. Tracer v1. 6. Available online: http://tree.bio.ed.ac.uk/software/tracer (accessed on 26 May 2020).
- Peters, B.; Sidney, J.; Bourne, P.; Bui, H.H.; Buus, S.; Doh, G.; Fleri, W.; Kronenberg, M.; Kubo, R.; Lund, O.; et al. The immune epitope database and analysis resource: From vision to blueprint. PLoS Biol. 2005, 3, e91. [Google Scholar] [CrossRef] [PubMed]
- EL-Manzalawy, Y.; Dobbs, D.; Honavar, V. Predicting linear B-cell epitopes using string kernels. J. Mol. Recognit. Interdiscip. J. 2008, 21, 243–255. [Google Scholar] [CrossRef]
- Yao, B.; Zheng, D.; Liang, S.; Zhang, C. Conformational B-cell epitope prediction on antigen protein structures: A review of current algorithms and comparison with common binding site prediction methods. PLoS ONE 2013, 8, e62249. [Google Scholar] [CrossRef]
- Haste Andersen, P.; Nielsen, M.; Lund, O. Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci. 2006, 15, 2558–2567. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism Name | Accession Number | S Protein Identity (%) | S Gene Identity (%) |
|---|---|---|---|
| MERS-COV | MT226600 * | 99.85 | 82.06 |
| MERS-COV | MT226601 * | 99.85 | 81.97 |
| MERS-COV | MT226602 * | 99.85 | 82.9 |
| MERS-COV | MT226603 * | 99.70 | 82.31 |
| MERS-COV | MT226604 * | 100.00 | 100.00 |
| MERS-COV | MT226605 * | 99.93 | 82.53 |
| MERS-COV | MT226606 * | 99.85 | 83.37 |
| MERS-COV | MT226607 * | 99.85 | 82.51 |
| MERS-COV | KJ713297 | 99.78 | 82.46 |
| MERS-COV | KY673149 | 99.63 | 82.36 |
| MERS-COV | KF600612 | 99.78 | 82.16 |
| MERS-COV | KF186564 | 99.78 | 83.00 |
| MERS-COV | KF600652 | 99.78 | 82.31 |
| MERS-COV | KF961222 | 99.78 | 82.71 |
| MERS-COV | KF192507 | 99.70 | 83.74 |
| 2019-nCOV | EPI_ISL_416432 | 27.44 | 39.16 |
| 2019-nCOV | EPI_ISL_416522 | 27.44 | 40.43 |
| 2019-nCOV | NC_045512 | 27.44 | 40.46 |
| 2019-nCOV | MN988713.1 | 27.44 | 39.98 |
| Batcoronavirus | KF636752.1 | 27.88 | 39.27 |
| Batcoronavirus | GU190215.1 | 26.90 | 39.95 |
| Batcoronavirus | DQ648857.1 | 26.99 | 39.42 |
| Batcoronavirus | MK211374.1 | 26.94 | 39.27 |
| Bat SARS coronavirus HKU3-12 | GQ153547.1 | 66.32 | 65.78 |
| Bat SARS-like coronavirus | KY417145.1 | 27.32 | 39.51 |
| SARS-like coronavirus | KT444582.1 | 27.01 | 39.98 |
| SARS-like coronavirus | AY278489.2 | 26.87 | 39.83 |
| SARS coronavirus | NC_004718.3 | 26.87 | 39.22 |
| SARS coronavirus | DQ497008.1 | 26.94 | 39.88 |
| SARS coronavirus | FJ882957.1 | 26.94 | 39.44 |
| SARS coronavirus | FJ882962.1 | 26.94 | 39.88 |
| Strain | Start | End | Peptide | Length | Score | 3D Structure |
|---|---|---|---|---|---|---|
| MERS-CoV | 523 | 566 | YSPCVSIVPSTVWEDGDYYRKQLSPLEGGGWLVASGSTVAMTEQ | 44 | 0.898 | A |
| 702 | 743 | STYGPLQTPVGCVLGLVNSSLFVEDCKLPLGQSLCALPDTPS | 42 | 0.88 | B | |
| 485 | 518 | PHNLTTITKPLKYSYINKCSRLLSDDRTEVPQLV | 34 | 0.848 | C | |
| 90 | 99 | GHATGTTPQK | 10 | 0.839 | D | |
| 618 | 628 | QNCTAVGVRQQ | 11 | 0.827 | E | |
| 18 | 32 | YVDVGPDSVKSACIE | 15 | 0.801 | F | |
| 2019-nCov | 1071 | 1147 | QEKNFTTAPAICHDGKAHFPREGVFVSNGTHWFVTQRNFYEPQIITTDNTFVSGNCDVVIGIVNNTVYDPLQPELDS | 77 | 0.88 | - |
| 92 | 192 | FASTEKSNIIRGWIFGTTLDSKTQSLLIVNNATNVVIKVCEFQFCNDPFLGVNCTFEYVSFKNLREF | 67 | 0.788 | - | |
| 328 | 364 | RFPNITNLCPFGEVFNATRFASVYAWNRKRISNCVAD | 37 | 0.767 | - | |
| 553 | 564 | TESNKKFLPFQQ | 12 | 0.721 | - | |
| SARS-CoV | 684 | 702 | DSSIAYSNNTIAIPTNFSI | 19 | 0.712 | - |
| 315 | 355 | RFPNITNLCPFGEVFNATKFPSVYAWERKKISNCVADYSVL | 41 | 0.692 | - | |
| 765 | 797 | AQVKQMYKTPTLKYFGGFNFSQILPDPLKPTKR | 33 | 0.638 | - |
| Organism Name | Host | Country | Accession Number |
|---|---|---|---|
| MERS-COV | Camelus dromedarius | Saudi Arabia | MT226600 * |
| MERS-COV | Camelus dromedarius | Saudi Arabia | MT226601 * |
| MERS-COV | Camelus dromedarius | Saudi Arabia | MT226602 * |
| MERS-COV | Camelus dromedarius | Saudi Arabia | MT226603 * |
| MERS-COV | Camelus dromedarius | Saudi Arabia | MT226604 * |
| MERS-COV | Camelus dromedarius | Saudi Arabia | MT226605 * |
| MERS-COV | Camelus dromedarius | Saudi Arabia | MT226606 * |
| MERS-COV | Camelus dromedarius | Saudi Arabia | MT226607 * |
| MERS-COV | Camelus dromedarius | Saudi Arabia | KJ713297 |
| MERS-COV | Camelus dromedarius | Oman | KY673149 |
| MERS-COV | Homo sapiens | Saudi Arabia | KF600612 |
| MERS-COV | Homo sapiens | Saudi Arabia | KF186564 |
| MERS-COV | Homo sapiens | Saudi Arabia | KF600652 |
| MERS-COV | Homo sapiens | Qatar | KF961222 |
| MERS-COV | Homo sapiens | United Arab Emirates | KF192507 |
| 2019-nCOV | Homo sapiens | Saudi Arabia | EPI_ISL_416432 |
| 2019-nCOV | Homo sapiens | Saudi Arabia | EPI_ISL_416522 |
| 2019-nCOV | Homo sapiens | China | NC_045512 |
| 2019-nCOV | Homo sapiens | USA | MN988713.1 |
| Batcoronavirus | Hipposideros pratti | China | KF636752.1 |
| Batcoronavirus | Rhinolophus blasii | China | GU190215.1 |
| Batcoronavirus | Rhinolophus macrotis | China | DQ648857.1 |
| Batcoronavirus | Rhinolophus sp. | China | MK211374.1 |
| Bat SARS coronavirus HKU3-12 | Homo sapiens | China | GQ153547.1 |
| Bat SARS-like coronavirus | Rhinilophus ferrumequinum | China | KY417145.1 |
| SARS-like coronavirus | Rhinolophus sinicus | China | KT444582.1 |
| SARS-like coronavirus | Homo sapiens | China | AY278489.2 |
| SARS coronavirus | Homo sapiens | Canada | NC_004718.3 |
| SARS coronavirus | Mus musculus | USA | DQ497008.1 |
| SARS coronavirus | Mus musculus | USA | FJ882957.1 |
| SARS coronavirus | Mus musculus | USA | FJ882962.1 |
| Primer Name | Sequence |
|---|---|
| Gap1_F1 | CCTCGTTCTCTTGCAGAACTT |
| Gap1_R1 | CCGGACGAAACCGTGTATT |
| Gap1_F2 | CACTTCCCCTCGTTCTCTTG |
| Gap1_R2 | ACCGTGTATTGTGACCGAGA |
| Gap2_F1 | ACCAATTGGCTTATAGCTCTAGT |
| Cap2_R1 | CCTTTAGGATCCGCCTCAAA |
| Gap3_F1 | GGGATTACCCTAAGTGTGATAGAG |
| Gap3_R1 | GTGCACTGACATTAGCAGTTG |
| Gap4_F1 | CTGGGTTGTACCCAACCATT |
| Gap4_R1 | CGTGCTGTAGGGTAGTAAATCG |
| Gap5_F1 | TTTGCCAGGTTGTGATGG |
| Gap5_R1 | ATACTCTCTGTACTCTGTAGCAT |
| Gap6_F1 | TTCTTTACTTACCTGTGTAACCTCA |
| Gap6_R1 | TCATAGGAGTGGAATTTCTCCAAA |
| Gap7_F1 | CAGCATCAGCTCGTGATCTT |
| Gap7_R1 | CTCCCAGAGCCTGATTAAACTTAT |
| Gap8_F1 | CTCCTTTGGCCGTAGATGTT |
| Gap8_R1 | CCGCTAGCAGGAATGTATGT |
| Gap8_F2 | CCTTTGGCCGTAGATGTTGT |
| Gap8_R2 | GCCGCTAGCAGGAATGTATG |
| Gap9_F1 | AGATCGCGGCAATCGTT |
| Gap9_R1 | GGCACTGTTCACTTGCAATC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Shomrani, B.M.; Manee, M.M.; Alharbi, S.N.; Altammami, M.A.; Alshehri, M.A.; Nassar, M.S.; Bakhrebah, M.A.; Al-Fageeh, M.B. Genomic Sequencing and Analysis of Eight Camel-Derived Middle East Respiratory Syndrome Coronavirus (MERS-CoV) Isolates in Saudi Arabia. Viruses 2020, 12, 611. https://doi.org/10.3390/v12060611
Al-Shomrani BM, Manee MM, Alharbi SN, Altammami MA, Alshehri MA, Nassar MS, Bakhrebah MA, Al-Fageeh MB. Genomic Sequencing and Analysis of Eight Camel-Derived Middle East Respiratory Syndrome Coronavirus (MERS-CoV) Isolates in Saudi Arabia. Viruses. 2020; 12(6):611. https://doi.org/10.3390/v12060611
Chicago/Turabian StyleAl-Shomrani, Badr M., Manee M. Manee, Sultan N. Alharbi, Mussad A. Altammami, Manal A. Alshehri, Majed S. Nassar, Muhammed A. Bakhrebah, and Mohamed B. Al-Fageeh. 2020. "Genomic Sequencing and Analysis of Eight Camel-Derived Middle East Respiratory Syndrome Coronavirus (MERS-CoV) Isolates in Saudi Arabia" Viruses 12, no. 6: 611. https://doi.org/10.3390/v12060611
APA StyleAl-Shomrani, B. M., Manee, M. M., Alharbi, S. N., Altammami, M. A., Alshehri, M. A., Nassar, M. S., Bakhrebah, M. A., & Al-Fageeh, M. B. (2020). Genomic Sequencing and Analysis of Eight Camel-Derived Middle East Respiratory Syndrome Coronavirus (MERS-CoV) Isolates in Saudi Arabia. Viruses, 12(6), 611. https://doi.org/10.3390/v12060611