Polyinosinic: Polycytidylic Acid and Murine Cytomegalovirus Modulate Expression of Murine IL-10 and IL-21 in White Adipose Tissue

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cells
2.3. Virus
2.4. In Vivo Poly(I:C) Injections and Organ Collection for Cytokine Evaluation
2.5. Adipose Tissue Explant Cultures and MCMV Infections
2.6. Cytokine Evaluation
2.7. Isolation of GWAT-Associated Stromal Vascular Fraction (SVF), Cell Staining, and Flow Cytometric Analysis
2.8. Normalisation of Cytokine and Breg Data
2.9. Statistics
3. Results
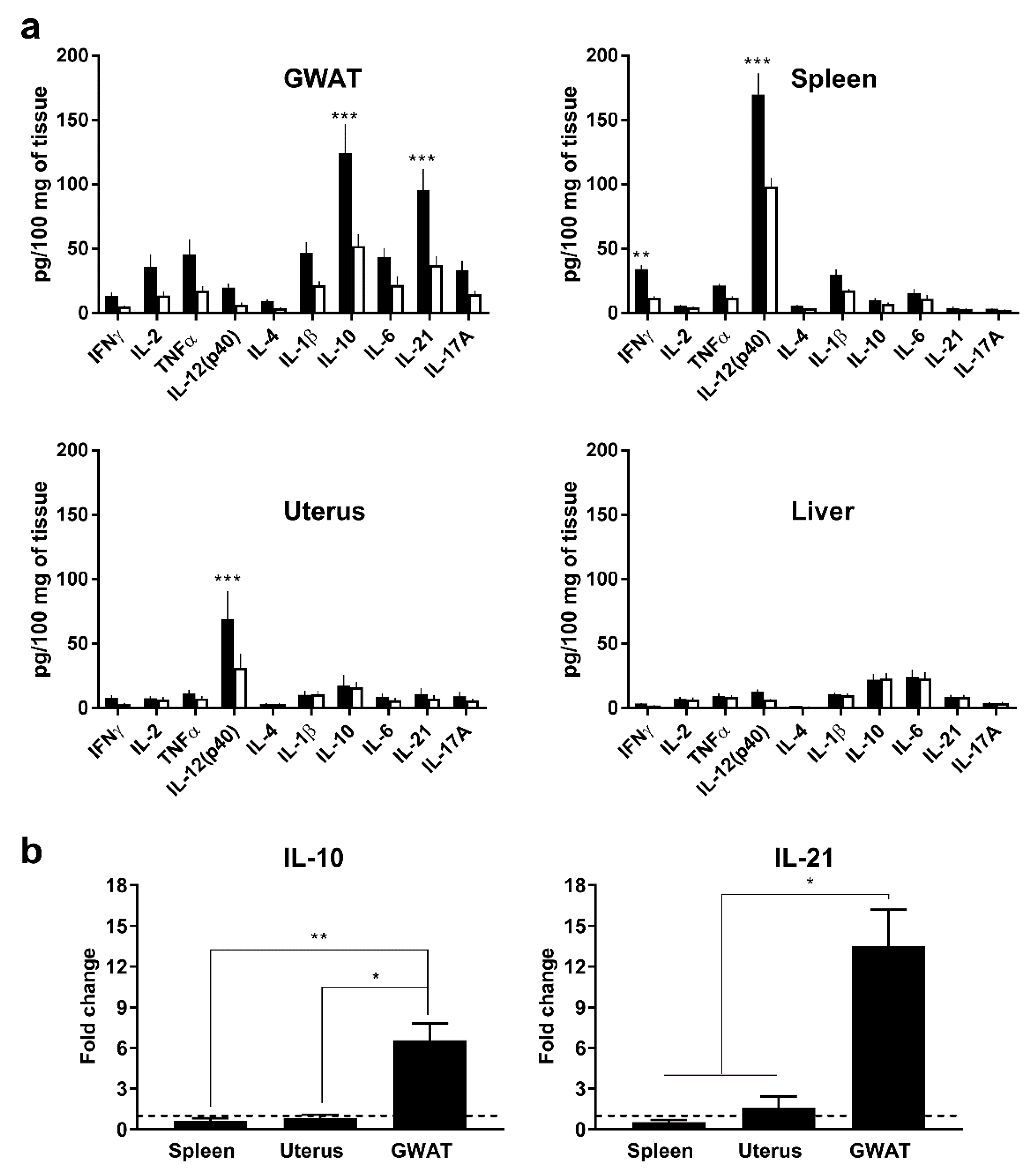
3.1. In Vivo, Poly(I:C) Induces High Expression of Murine IL-10 and IL-21 in GWAT but not in Other Organs
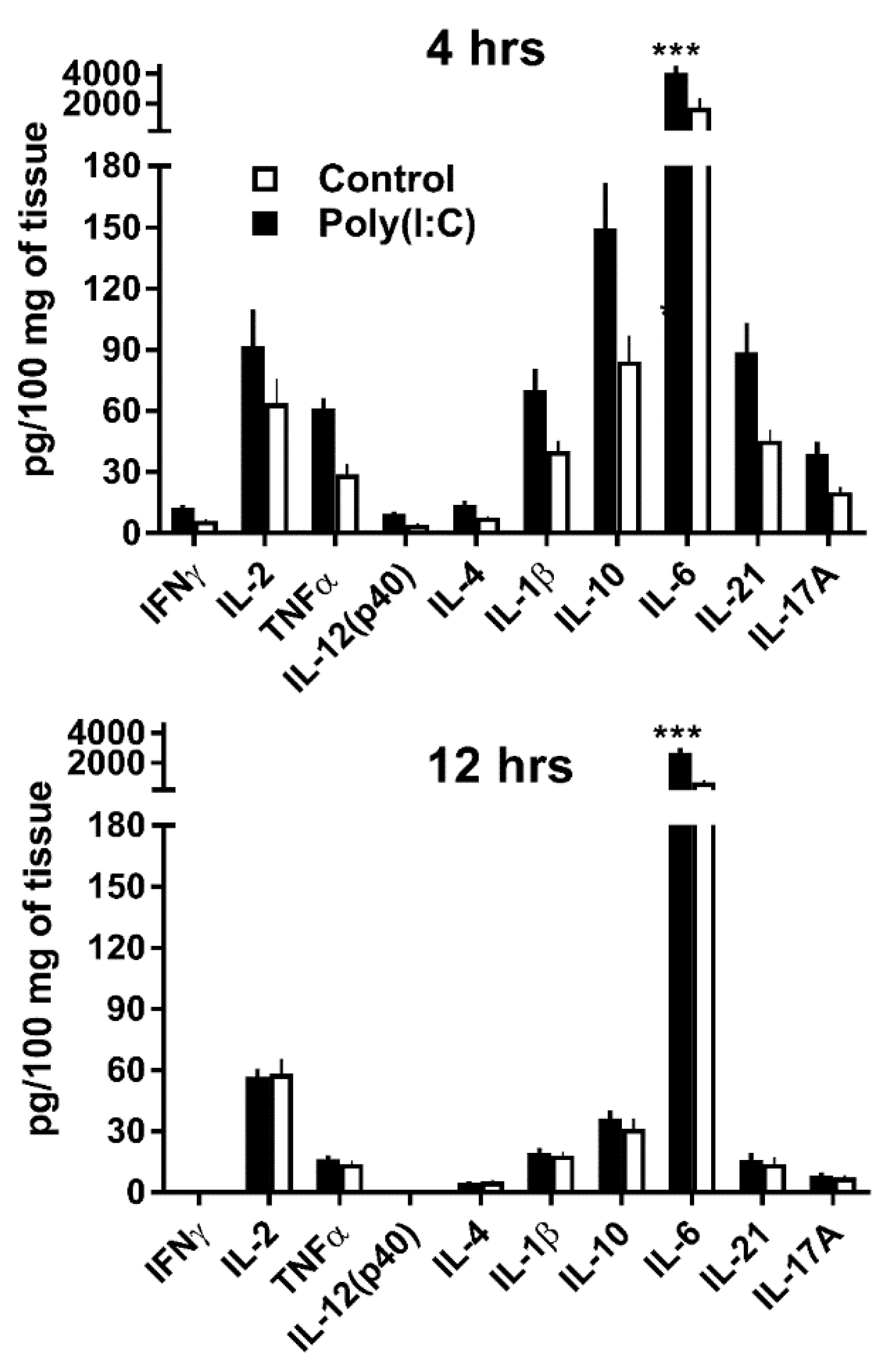
3.2. GWAT Explants Stimulated Ex Vivo with Poly(I:C) Reproduce Endogenous IL-10 and IL-21 Responses Observed In Vivo
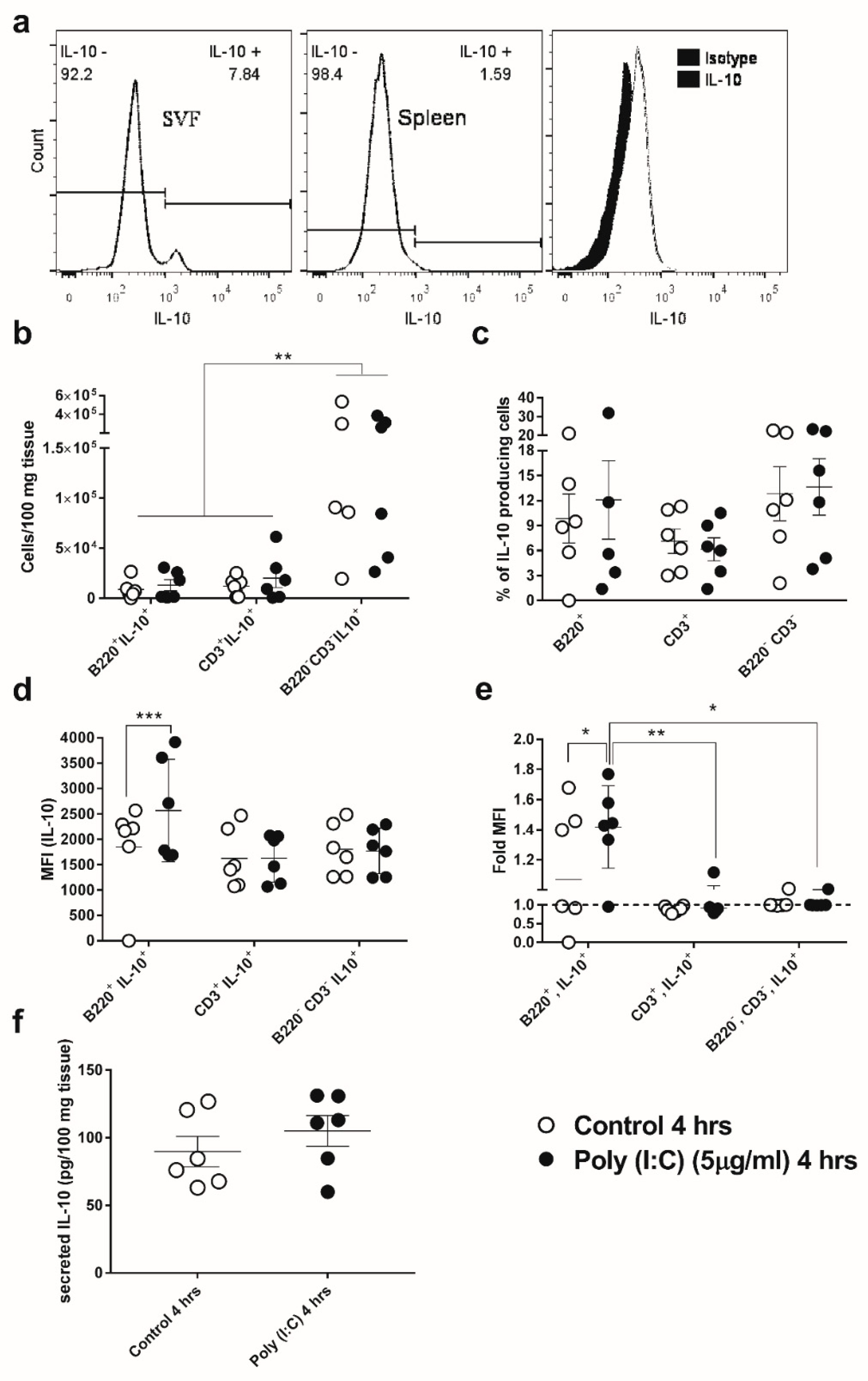
3.3. GWAT-Resident B-Cells Respond to Poly(I:C) by Increased Expression of IL-10
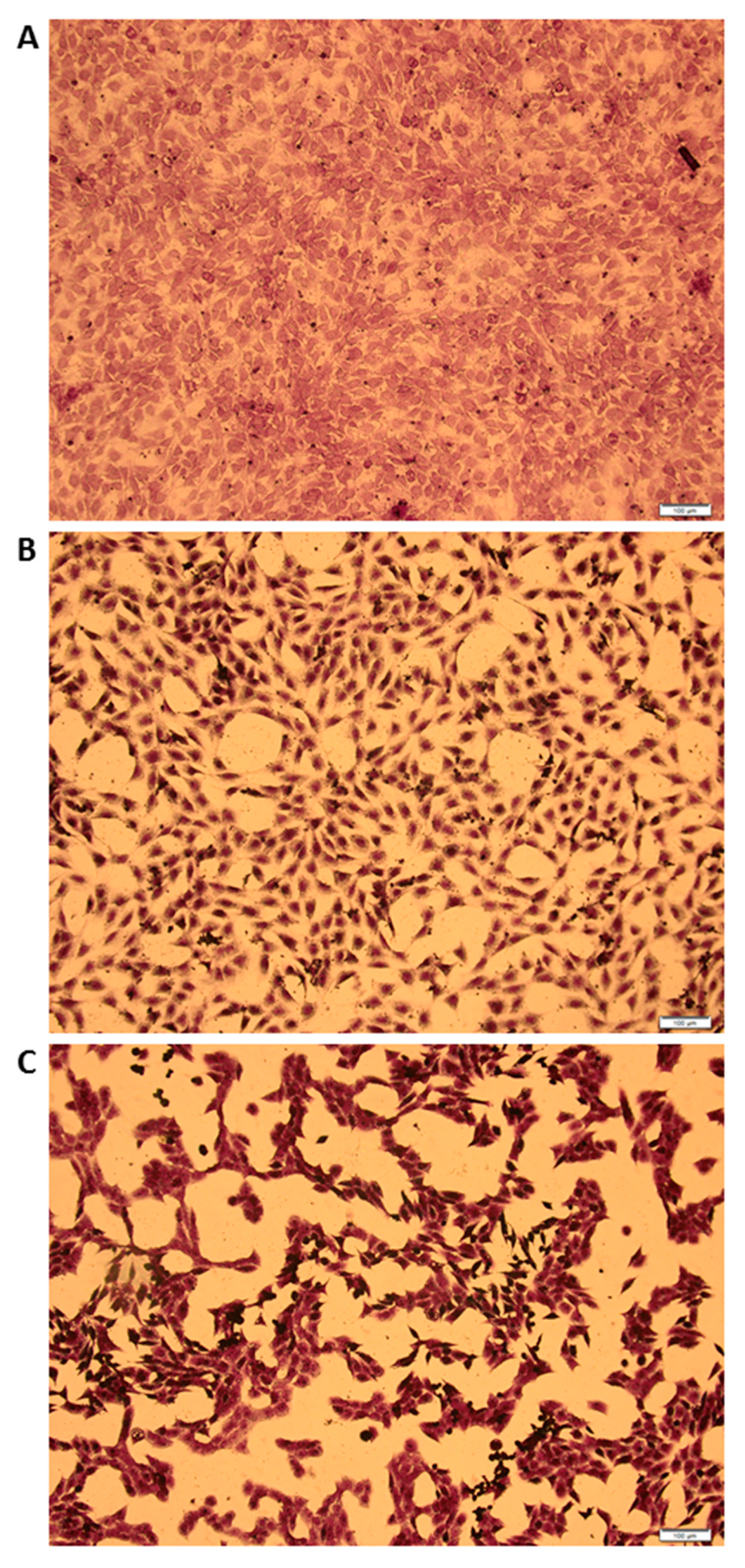
3.4. MCMV Replicates in Gonadal White Adipose Tissue (GWAT)
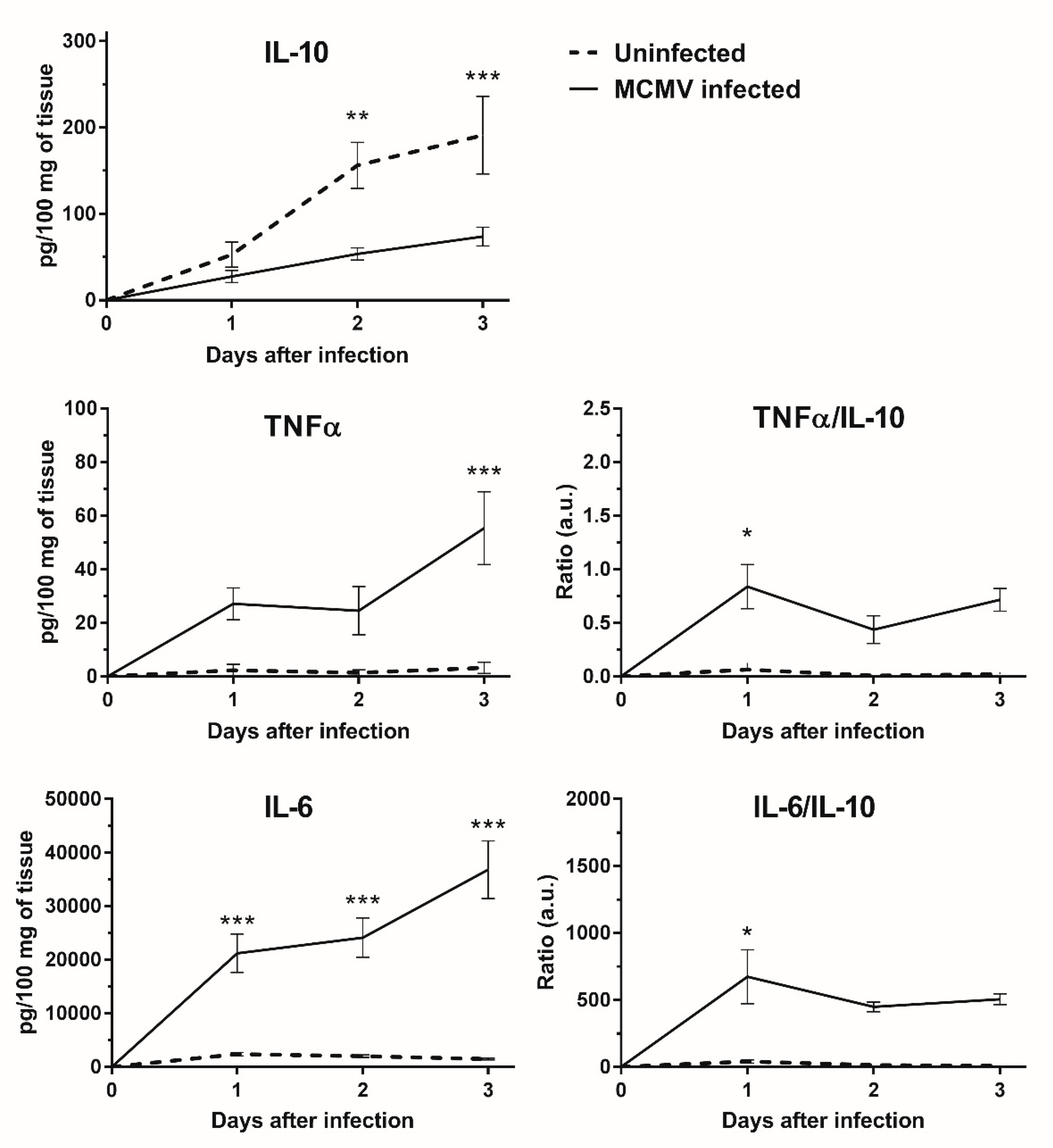
3.5. IL-10 Expression Is Down-regulated in MCMV Infected GWAT Explants
3.6. Both the Number and Relative Percentage of Bregs Are Decreased in GWAT after MCMV Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Grant, R.W.; Dixit, V.D. Adipose tissue as an immunological organ. Obesity 2015, 23, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Caspar-Bauguil, S.; Cousin, B.; Galinier, A.; Segafredo, C.; Nibbelink, M.; Andre, M.; Casteilla, L.; Penicaud, L. Adipose tissues as an ancestral immune organ: Site-specific change in obesity. FEBS Lett. 2005, 579, 3487–3492. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Obin, M.S. Obesity and the role of adipose tissue in inflammation and metabolism. Am. J. Clin. Nutr. 2006, 83, 461S–465S. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.H.; Scherer, P.E. Adipose tissue, inflammation, and cardiovascular disease. Circul. Res. 2005, 96, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, A.W., Jr. The immune cells in adipose tissue. Diabetes Obes. Metab. 2013, 15, 34–38. [Google Scholar] [CrossRef]
- St-Onge, M.P. Are normal-weight Americans over-fat? Obesity 2010, 18, 2067–2068. [Google Scholar] [CrossRef]
- Finlay, J.B.; Liu, X.; Ermel, R.W.; Adamson, T.W. Maternal Weight Gain as a Predictor of Litter Size in Swiss Webster, C57BL/6J, and BALB/cJ mice. J. Am. Assoc. Lab. Anim. 2015, 54, 694–699. [Google Scholar]
- Reed, D.R.; Bachmanov, A.A.; Tordoff, M.G. Forty mouse strain survey of body composition. Physiol. Behav. 2007, 91, 593–600. [Google Scholar] [CrossRef]
- Ballak, D.B.; van Asseldonk, E.J.P.; van Diepen, J.A.; Jansen, H.; Hijmans, A.; Joosten, L.A.B.; Tack, C.J.; Netea, M.G.; Stienstra, R. TLR-3 is Present in Human Adipocytes, but Its Signalling is Not Required for Obesity-Induced Inflammation in Adipose Tissue In Vivo. PLoS ONE 2015, 10, e0123152. [Google Scholar] [CrossRef]
- Damouche, A.; Lazure, T.; Avettand-Fenoel, V.; Huot, N.; Dejucq-Rainsford, N.; Satie, A.P.; Melard, A.; David, L.; Gommet, C.; Ghosn, J.; et al. Adipose Tissue Is a Neglected Viral Reservoir and an Inflammatory Site during Chronic HIV and SIV Infection. PLoS Pathog. 2015, 11, e1005153. [Google Scholar] [CrossRef]
- Hegde, V.; Dhurandhar, N.V. Microbes and obesity--interrelationship between infection, adipose tissue and the immune system. Clin. Microbiol. Infect. 2013, 19, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Juge-Aubry, C.E.; Somm, E.; Pernin, A.; Alizadeh, N.; Giusti, V.; Dayer, J.M.; Meier, C.A. Adipose tissue is a regulated source of interleukin-10. Cytokine 2005, 29, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Juge-Aubry, C.E.; Somm, E.; Giusti, V.; Pernin, A.; Chicheportiche, R.; Verdumo, C.; Rohner-Jeanrenaud, F.; Burger, D.; Dayer, J.-M.; Meier, C.A. Adipose Tissue Is a Major Source of Interleukin-1 Receptor Antagonist. Diabetes 2003, 52, 1104–1110. [Google Scholar] [CrossRef]
- Nishimura, S.; Manabe, I.; Takaki, S.; Nagasaki, M.; Otsu, M.; Yamashita, H.; Sugita, J.; Yoshimura, K.; Eto, K.; Komuro, I.; et al. Adipose Natural Regulatory B Cells Negatively Control Adipose Tissue Inflammation. Cell Metab. 2013, 18, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.; Abendroth, A.; Slobedman, B. A novel viral transcript with homology to human interleukin-10 is expressed during latent human cytomegalovirus infection. J. Virol. 2004, 78, 1440–1447. [Google Scholar] [CrossRef] [PubMed]
- Clement, M.; Marsden, M.; Stacey, M.A.; Abdul-Karim, J.; Gimeno Brias, S.; Costa Bento, D.; Scurr, M.J.; Ghazal, P.; Weaver, C.T.; Carlesso, G.; et al. Cytomegalovirus-Specific IL-10-Producing CD4+ T Cells Are Governed by Type-I IFN-Induced IL-27 and Promote Virus Persistence. PLoS Pathog. 2016, 12, e1006050. [Google Scholar] [CrossRef]
- Mandaric, S.; Walton, S.M.; Rulicke, T.; Richter, K.; Girard-Madoux, M.J.; Clausen, B.E.; Zurunic, A.; Kamanaka, M.; Flavell, R.A.; Jonjic, S.; et al. IL-10 suppression of NK/DC crosstalk leads to poor priming of MCMV-specific CD4 T cells and prolonged MCMV persistence. PLoS Pathog. 2012, 8, e1002846. [Google Scholar] [CrossRef]
- Poole, E.; Avdic, S.; Hodkinson, J.; Jackson, S.; Wills, M.; Slobedman, B.; Sinclair, J. Latency-associated viral interleukin-10 (IL-10) encoded by human cytomegalovirus modulates cellular IL-10 and CCL8 Secretion during latent infection through changes in the cellular microRNA hsa-miR-92a. J. Virol. 2014, 88, 13947–13955. [Google Scholar] [CrossRef]
- Rojas, J.M.; Avia, M.; Martin, V.; Sevilla, N. IL-10: A Multifunctional Cytokine in Viral Infections. J. Immunol. Res 2017, 2017, 6104054. [Google Scholar] [CrossRef]
- Goodrum, F. Human Cytomegalovirus Latency: Approaching the Gordian Knot. Annu. Rev. Virol. 2016, 3, 333–357. [Google Scholar] [CrossRef]
- Neighbour, P.A.; Fraser, L.R. Murine cytomegalovirus and fertility: Potential sexual transmission and the effect of this virus on fertilization in vitro. Fertil. Steril. 1978, 30, 216–222. [Google Scholar] [CrossRef]
- Murray, C.; Griffin, E.W.; O’Loughlin, E.; Lyons, A.; Sherwin, E.; Ahmed, S.; Stevenson, N.J.; Harkin, A.; Cunningham, C. Interdependent and independent roles of type I interferons and IL-6 in innate immune, neuroinflammatory and sickness behaviour responses to systemic poly I:C. Brain Behav. Immun. 2015, 48, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Kumagai, Y.; Kato, H.; Guo, Z.; Matsushita, K.; Satoh, T.; Kawagoe, T.; Kumar, H.; Jang, M.H.; Kawai, T.; et al. Poly I:C-induced activation of NK cells by CD8 alpha+ dendritic cells via the IPS-1 and TRIF-dependent pathways. J. Immunol. 2009, 183, 2522–2528. [Google Scholar] [CrossRef] [PubMed]
- Melkamu, T.; Kita, H.; O’Grady, S.M. TLR3 activation evokes IL-6 secretion, autocrine regulation of Stat3 signaling and TLR2 expression in human bronchial epithelial cells. J. Cell Commun. Signal. 2013, 7, 109–118. [Google Scholar] [CrossRef]
- Porowski, D.; Wirkowska, A.; Hryniewiecka, E.; Wyzgal, J.; Pacholczyk, M.; Paczek, L. Liver Failure Impairs the Intrahepatic Elimination of Interleukin-6, Tumor Necrosis Factor-Alpha, Hepatocyte Growth Factor, and Transforming Growth Factor-Beta. BioMed Res. Int. 2015, 2015, 934065. [Google Scholar] [CrossRef]
- Price, P.; Eddy, K.S.; Papadimitriou, J.M.; Robertson, T.A.; Shellam, G.R. Cytomegalovirus infection of adipose tissues induces steatitis in adult mice. Int. J. Exp. Pathol. 1990, 71, 557–571. [Google Scholar]
- Farrell, H.E.; Bruce, K.; Lawler, C.; Stevenson, P.G. Murine Cytomegalovirus Spread Depends on the Infected Myeloid Cell Type. J. Virol. 2019, 93, e00540-19. [Google Scholar] [CrossRef] [PubMed]
- Contreras, N.A.; Sitnik, K.M.; Jeftic, I.; Coplen, C.P.; Cicin-Sain, L.; Nikolich-Zugich, J. Life-long control of cytomegalovirus (CMV) by T resident memory cells in the adipose tissue results in inflammation and hyperglycemia. PLoS Pathog. 2019, 15, e1007890. [Google Scholar] [CrossRef]
- Yu, L.; Yan, K.; Liu, P.; Li, N.; Liu, Z.; Zhu, W.; Chen, Y.; Han, D. Pattern recognition receptor-initiated innate antiviral response in mouse adipose cells. Immunol. Cell Biol. 2014, 92, 105–115. [Google Scholar] [CrossRef]
- Fabrizi, M.; Marchetti, V.; Mavilio, M.; Marino, A.; Casagrande, V.; Cavalera, M.; Moreno-Navarrete, J.M.; Mezza, T.; Sorice, G.P.; Fiorentino, L.; et al. IL-21 is a major negative regulator of IRF4-dependent lipolysis affecting Tregs in adipose tissue and systemic insulin sensitivity. Diabetes 2014, 63, 2086–2096. [Google Scholar] [CrossRef]
- Somm, E.; Cettour-Rose, P.; Asensio, C.; Charollais, A.; Klein, M.; Theander-Carrillo, C.; Juge-Aubry, C.E.; Dayer, J.M.; Nicklin, M.J.; Meda, P.; et al. Interleukin-1 receptor antagonist is upregulated during diet-induced obesity and regulates insulin sensitivity in rodents. Diabetologia 2006, 49, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, A.; Ehrentraut, S.; Scharm, M.; Wang, H.; Hartig, R.; Morse, H.C., 3rd; Zenclussen, A.C. Plasma Cell Alloantigen 1 and IL-10 Secretion Define Two Distinct Peritoneal B1a B Cell Subsets With Opposite Functions, PC1(high) Cells Being Protective and PC1(low) Cells Harmful for the Growing Fetus. Front. Immunol. 2018, 9, 1045. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Genuino, R.M.; Eldi, P.; Garcia-Valtanen, P.; Hayball, J.D.; Diener, K.R. Uterine B Cells Exhibit Regulatory Properties During the Peri-Implantation Stage of Murine Pregnancy. Front. Immunol. 2019, 10, 2899. [Google Scholar] [CrossRef] [PubMed]
- Peckham, C.S. Cytomegalovirus infection: Congenital and neonatal disease. Scand. J. Infect. Dis. Suppl. 1991, 80, 82–87. [Google Scholar]
- Alford, C.A.; Stagno, S.; Pass, R.F.; Britt, W.J. Congenital and perinatal cytomegalovirus infections. Rev. Infect. Dis. 1990, 12, S745–S753. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Valtanen, P.; Guzman-Genuino, R.M.; Hayball, J.D.; Diener, K.R. Polyinosinic: Polycytidylic Acid and Murine Cytomegalovirus Modulate Expression of Murine IL-10 and IL-21 in White Adipose Tissue. Viruses 2020, 12, 569. https://doi.org/10.3390/v12050569
Garcia-Valtanen P, Guzman-Genuino RM, Hayball JD, Diener KR. Polyinosinic: Polycytidylic Acid and Murine Cytomegalovirus Modulate Expression of Murine IL-10 and IL-21 in White Adipose Tissue. Viruses. 2020; 12(5):569. https://doi.org/10.3390/v12050569
Chicago/Turabian StyleGarcia-Valtanen, Pablo, Ruth Marian Guzman-Genuino, John D. Hayball, and Kerrilyn R. Diener. 2020. "Polyinosinic: Polycytidylic Acid and Murine Cytomegalovirus Modulate Expression of Murine IL-10 and IL-21 in White Adipose Tissue" Viruses 12, no. 5: 569. https://doi.org/10.3390/v12050569
APA StyleGarcia-Valtanen, P., Guzman-Genuino, R. M., Hayball, J. D., & Diener, K. R. (2020). Polyinosinic: Polycytidylic Acid and Murine Cytomegalovirus Modulate Expression of Murine IL-10 and IL-21 in White Adipose Tissue. Viruses, 12(5), 569. https://doi.org/10.3390/v12050569