Rous Sarcoma Virus Genomic RNA Dimerization Capability In Vitro Is Not a Prerequisite for Viral Infectivity

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of Proteins and RNAs
2.2. XL-SHAPE, Primer Extension and Capillary Electrophoresis
2.3. XL-SHAPE Data Analysis and RNA Structure Modeling
2.4. RNA Dimerization Assays
2.5. Fluorescence Anisotropy (FA)-Based Salt Titration Binding Assays
2.6. Virus Infectivity Assays
3. Results
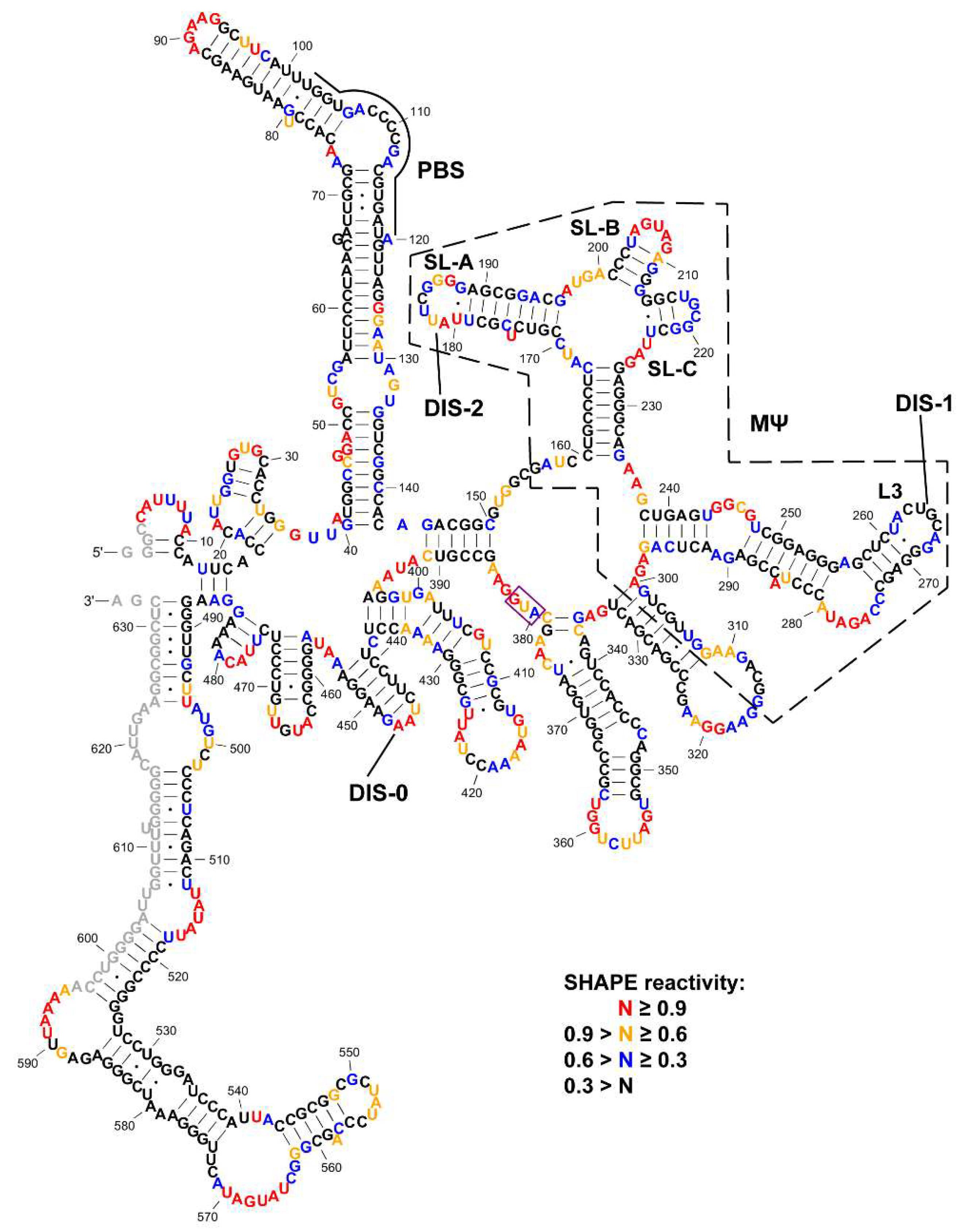
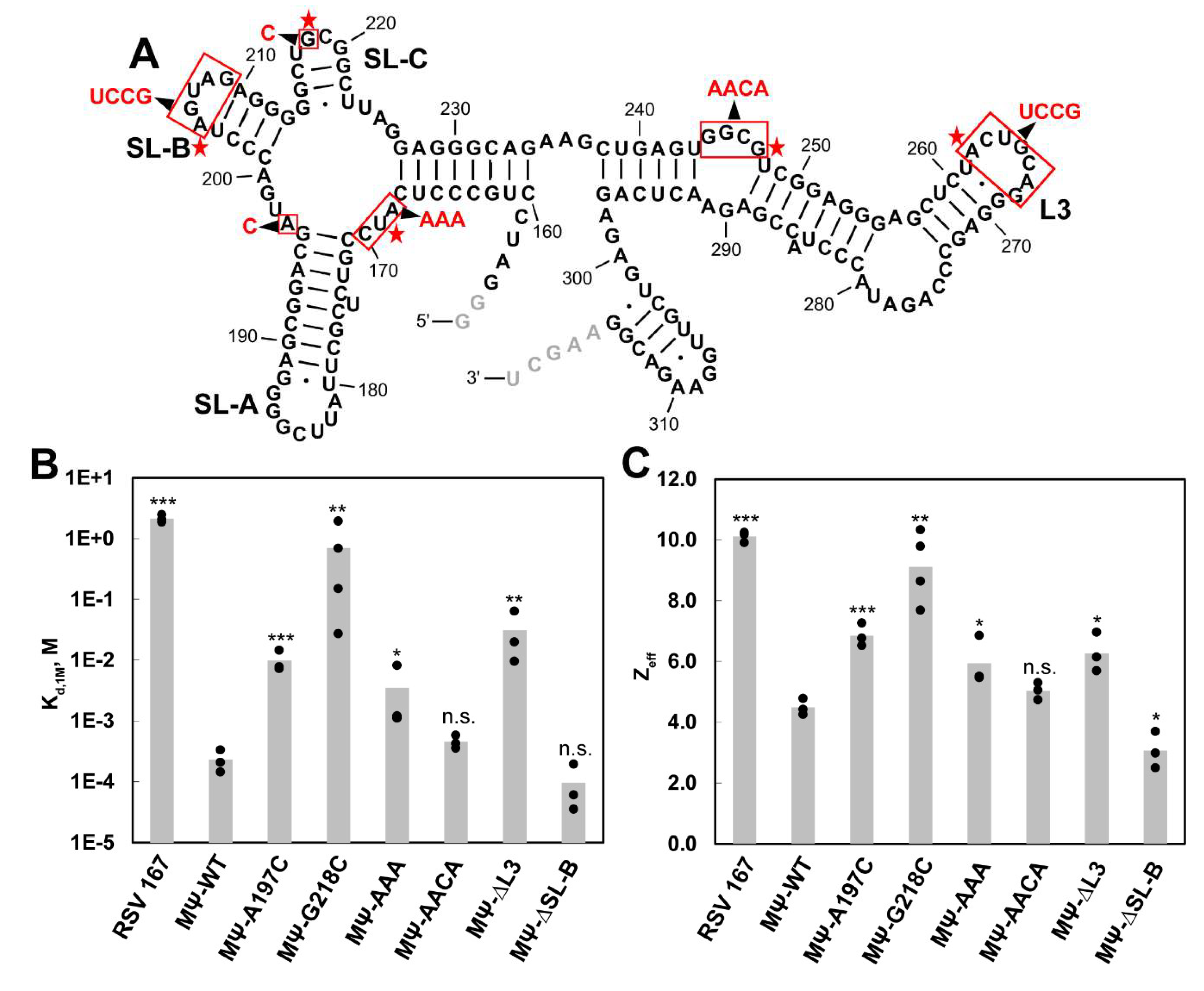
3.1. SHAPE Probing and Secondary Structure Analysis of RSV 5′-Leader
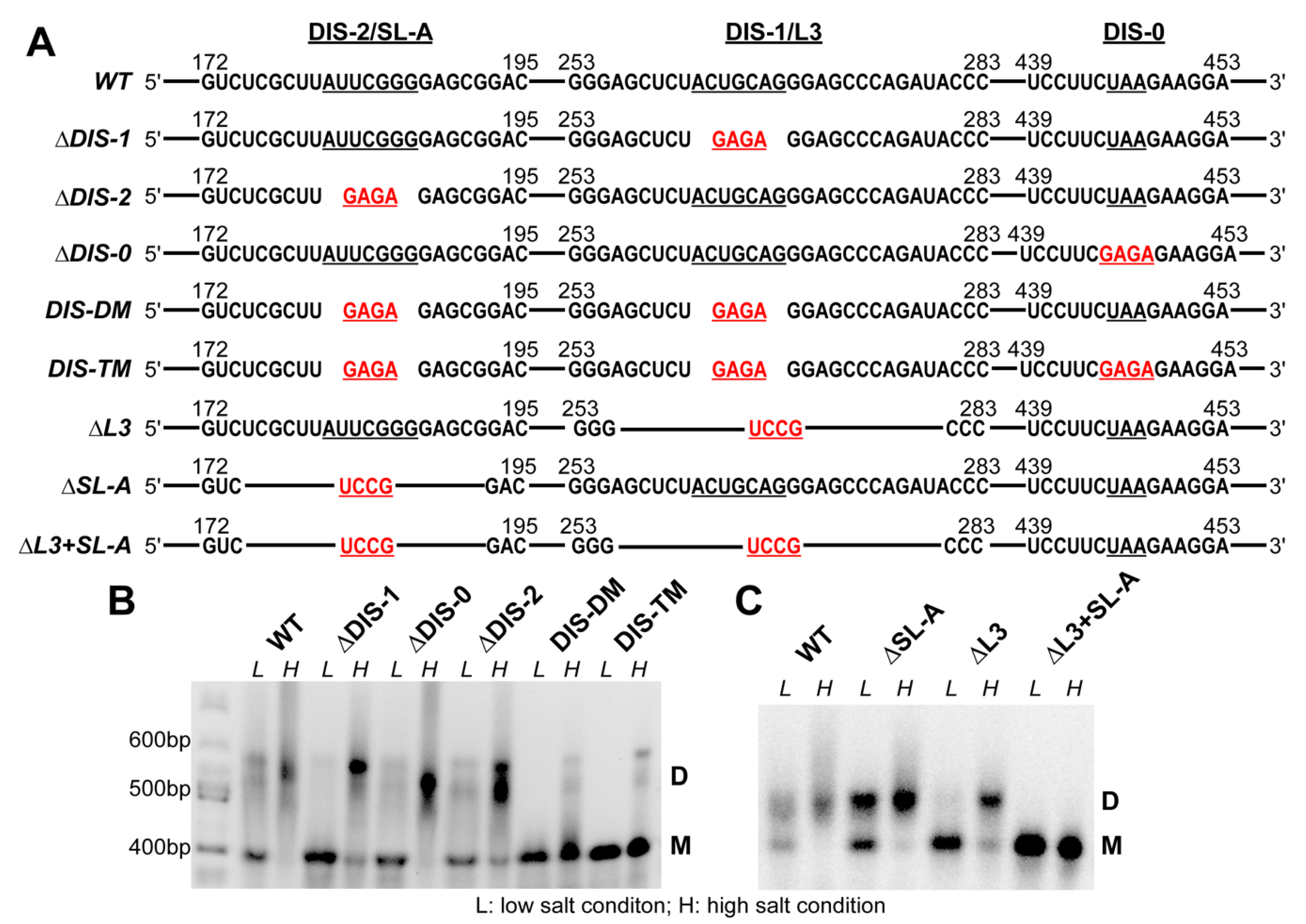
3.2. The Dimerization Site of RSV 5′-Leader RNA is Bipartite
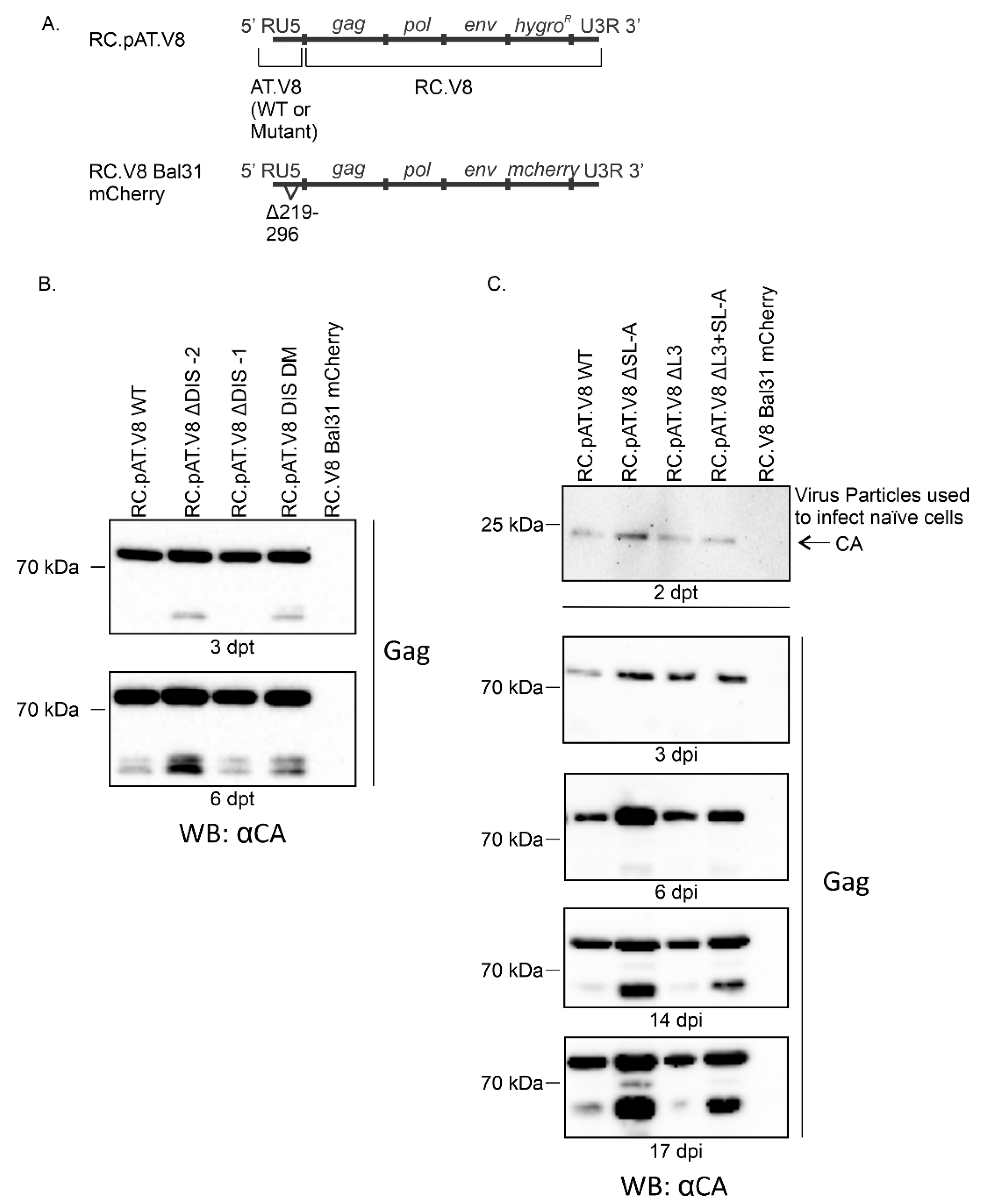
3.3. DIS Mutations Do not Have a Negative Effect on Virus Infectivity in Culture
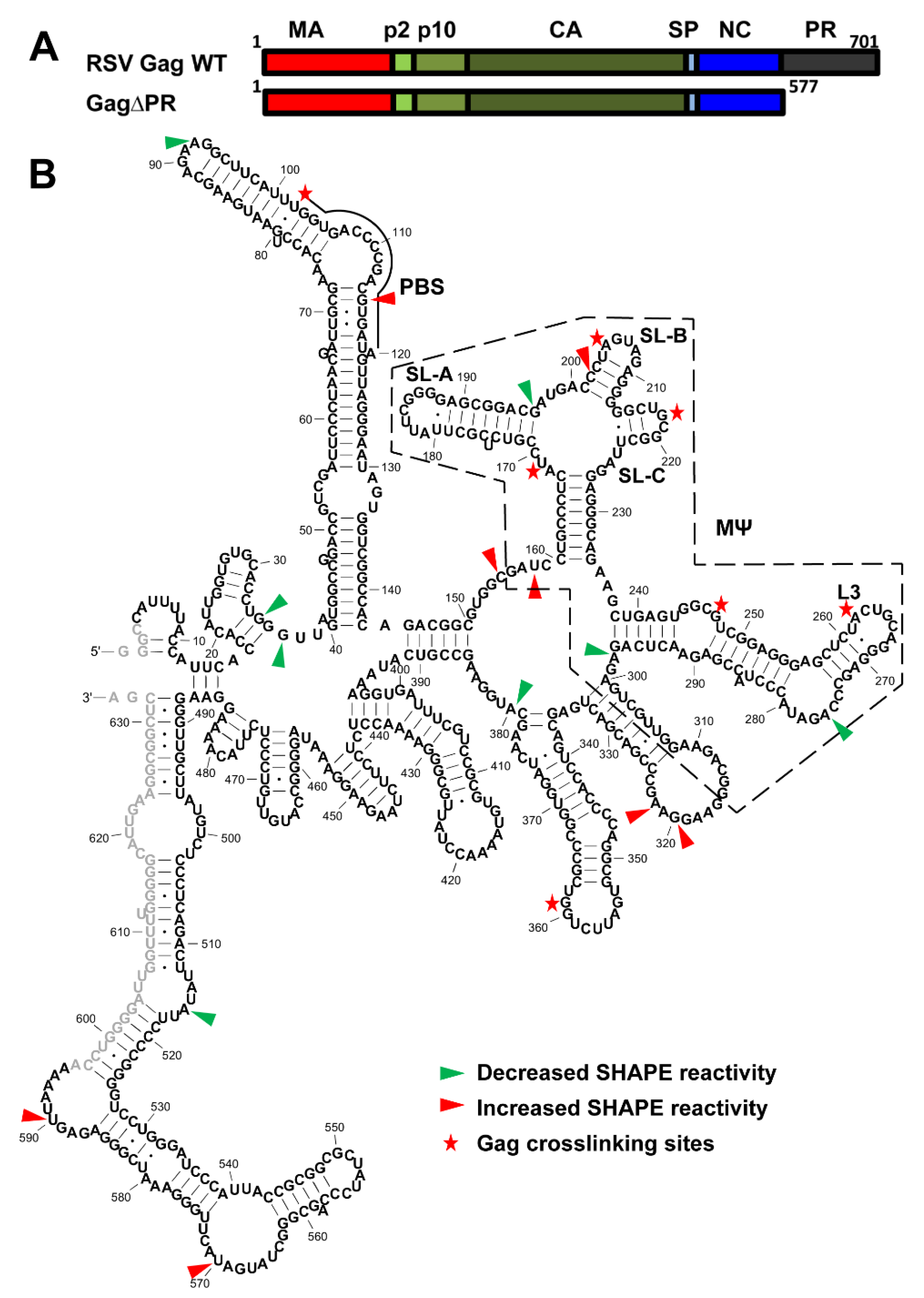
3.4. Identification of RSV Gag∆PR-Induced 5′-Leader RNA Conformational Changes and RSV Gag∆PR-Binding Sites
3.5. Mutations of Identified Crosslinked Residues in RSV MΨ Showed Distinct Effects on Gag∆PR Binding Specificity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brickell, P.M. The p60c-src family of protein-tyrosine kinases: Structure, regulation, and function. Crit. Rev. Oncog. 1992, 3, 401–446. [Google Scholar] [PubMed]
- Kaddis Maldonado, R.J.; Parent, L.J. Orchestrating the selection and packaging of genomic RNA by retroviruses: An ensemble of viral and host factors. Viruses 2016, 8, 257. [Google Scholar] [CrossRef] [PubMed]
- Meric, C.; Spahr, P.F. Rous sarcoma virus nucleic acid-binding protein p12 is necessary for viral 70S RNA dimer formation and packaging. J. Virol. 1986, 60, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Katz, R.A.; Terry, R.W.; Skalka, A.M. A conserved cis-acting sequence in the 5′ leader of avian sarcoma virus RNA is required for packaging. J. Virol. 1986, 59, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Heng, X.; Summers, M.F. Structural determinants and mechanism of HIV-1 genome packaging. J. Mol. Biol. 2011, 410, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Banks, J.D.; Yeo, A.; Green, K.; Cepeda, F.; Linial, M.L. A minimal avian retroviral packaging sequence has a complex structure. J. Virol. 1998, 72, 6190–6194. [Google Scholar] [CrossRef]
- Banks, J.D.; Kealoha, B.O.; Linial, M.L. An Mpsi-containing heterologous RNA, but not env mRNA, is efficiently packaged into avian retroviral particles. J. Virol. 1999, 73, 8926–8933. [Google Scholar] [CrossRef]
- Banks, J.D.; Linial, M.L. Secondary structure analysis of a minimal avian leukosis-sarcoma virus packaging signal. J. Virol. 2000, 74, 456–464. [Google Scholar] [CrossRef]
- Lee, E.G.; Linial, M.L. Yeast three-hybrid screening of rous sarcoma virus mutants with randomly mutagenized minimal packaging signals reveals regions important for gag interactions. J. Virol. 2000, 74, 9167–9174. [Google Scholar] [CrossRef]
- Heng, X.; Kharytonchyk, S.; Garcia, E.L.; Lu, K.; Divakaruni, S.S.; LaCotti, C.; Edme, K.; Telesnitsky, A.; Summers, M.F. Identification of a minimal region of the HIV-1 5′-leader required for RNA dimerization, NC binding, and packaging. J. Mol. Biol. 2012, 417, 224–239. [Google Scholar] [CrossRef]
- Keane, S.C.; Heng, X.; Lu, K.; Kharytonchyk, S.; Ramakrishnan, V.; Carter, G.; Barton, S.; Hosic, A.; Florwick, A.; Santos, J.; et al. RNA structure. Structure of the HIV-1 RNA packaging signal. Science 2015, 348, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Nikolaitchik, O.A.; Rahman, S.A.; Chen, J.; Pathak, V.K.; Hu, W.S. HIV-1 sequence necessary and sufficient to package non-viral RNAs into HIV-1 Particles. J. Mol. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, R.D.; Hammarskjold, M.L.; Helga-Maria, C.; Rekosh, D.; Goff, S.P. 5′ regions of HIV-1 RNAs are not sufficient for encapsidation: Implications for the HIV-1 packaging signal. Virology 1995, 212, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Dick, R.A.; Vogt, V.M. Membrane interaction of retroviral gag proteins. Front. Microbiol. 2014, 5, 187. [Google Scholar] [CrossRef]
- Dick, R.A.; Datta, S.A.; Nanda, H.; Fang, X.; Wen, Y.; Barros, M.; Wang, Y.X.; Rein, A.; Vogt, V.M. Hydrodynamic and membrane binding properties of purified rous sarcoma virus gag protein. J. Virol. 2015, 89, 10371–10382. [Google Scholar] [CrossRef]
- Rye-McCurdy, T.D.; Nadaraia-Hoke, S.; Gudleski-O’Regan, N.; Flanagan, J.M.; Parent, L.J.; Musier-Forsyth, K. Mechanistic differences between nucleic acid chaperone activities of the Gag proteins of Rous sarcoma virus and human immunodeficiency virus type 1 are attributed to the MA domain. J. Virol. 2014, 88, 7852–7861. [Google Scholar] [CrossRef]
- Nandhagopal, N.; Simpson, A.A.; Johnson, M.C.; Francisco, A.B.; Schatz, G.W.; Rossmann, M.G.; Vogt, V.M. Dimeric rous sarcoma virus capsid protein structure relevant to immature gag assembly. J. Mol. Biol. 2004, 335, 275–282. [Google Scholar] [CrossRef]
- Bush, D.L.; Vogt, V.M. In Vitro Assembly of Retroviruses. Annu. Rev. Virol. 2014, 1, 561–580. [Google Scholar] [CrossRef]
- Craven, R.C.; Parent, L.J. Dynamic interactions of the gag polyprotein. Curr. Top. Microbiol. Immunol. 1996, 214, 65–94. [Google Scholar] [CrossRef]
- Prats, A.C.; Sarih, L.; Gabus, C.; Litvak, S.; Keith, G.; Darlix, J.L. Small finger protein of avian and murine retroviruses has nucleic acid annealing activity and positions the replication primer tRNA onto genomic RNA. EMBO J. 1988, 7, 1777–1783. [Google Scholar] [CrossRef]
- Stewart, L.; Schatz, G.; Vogt, V.M. Properties of avian retrovirus particles defective in viral protease. J. Virol. 1990, 64, 5076–5092. [Google Scholar] [CrossRef] [PubMed]
- Wills, J.W.; Cameron, C.E.; Wilson, C.B.; Xiang, Y.; Bennett, R.P.; Leis, J. An assembly domain of the Rous sarcoma virus gag protein required late in budding. J. Virol. 1994, 68, 6605–6618. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Cameron, C.E.; Wills, J.W.; Leis, J. Fine mapping and characterization of the Rous sarcoma virus Pr76gag late assembly domain. J. Virol. 1996, 70, 5695–5700. [Google Scholar] [CrossRef] [PubMed]
- Scheifele, L.Z.; Kenney, S.P.; Cairns, T.M.; Craven, R.C.; Parent, L.J. Overlapping roles of the Rous sarcoma virus Gag p10 domain in nuclear export and virion core morphology. J. Virol. 2007, 81, 10718–10728. [Google Scholar] [CrossRef] [PubMed]
- Bush, D.L.; Monroe, E.B.; Bedwell, G.J.; Prevelige, P.E., Jr.; Phillips, J.M.; Vogt, V.M. Higher-order structure of the Rous sarcoma virus SP assembly domain. J. Virol. 2014, 88, 5617–5629. [Google Scholar] [CrossRef][Green Version]
- Craven, R.C.; Leure-duPree, A.E.; Erdie, C.R.; Wilson, C.B.; Wills, J.W. Necessity of the spacer peptide between CA and NC in the Rous sarcoma virus gag protein. J. Virol. 1993, 67, 6246–6252. [Google Scholar] [CrossRef]
- Meric, C.; Gouilloud, E.; Spahr, P.F. Mutations in Rous sarcoma virus nucleocapsid protein p12 (NC): Deletions of Cys-His boxes. J. Virol. 1988, 62, 3328–3333. [Google Scholar] [CrossRef]
- Lee, E.; Yeo, A.; Kraemer, B.; Wickens, M.; Linial, M.L. The gag domains required for avian retroviral RNA encapsidation determined by using two independent assays. J. Virol. 1999, 73, 6282–6292. [Google Scholar] [CrossRef]
- Dupraz, P.; Oertle, S.; Meric, C.; Damay, P.; Spahr, P.F. Point mutations in the proximal Cys-His box of Rous sarcoma virus nucleocapsid protein. J. Virol. 1990, 64, 4978–4987. [Google Scholar] [CrossRef]
- Lee, E.G.; Linial, M.L. Basic residues of the retroviral nucleocapsid play different roles in gag-gag and Gag-Psi RNA interactions. J. Virol. 2004, 78, 8486–8495. [Google Scholar] [CrossRef]
- Lee, E.G.; Alidina, A.; May, C.; Linial, M.L. Importance of basic residues in binding of rous sarcoma virus nucleocapsid to the RNA packaging signal. J. Virol. 2003, 77, 2010–2020. [Google Scholar] [CrossRef]
- Zhou, J.; McAllen, J.K.; Tailor, Y.; Summers, M.F. High affinity nucleocapsid protein binding to the muPsi RNA packaging signal of Rous sarcoma virus. J. Mol. Biol. 2005, 349, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Bean, R.L.; Vogt, V.M.; Summers, M. Solution structure of the Rous sarcoma virus nucleocapsid protein: MuPsi RNA packaging signal complex. J. Mol. Biol. 2007, 365, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Doria-Rose, N.A.; Vogt, V.M. In vivo selection of Rous sarcoma virus mutants with randomized sequences in the packaging signal. J. Virol. 1998, 72, 8073–8082. [Google Scholar] [CrossRef]
- Knight, J.B.; Si, Z.H.; Stoltzfus, C.M. A base-paired structure in the avian sarcoma virus 5′ leader is required for efficient encapsidation of RNA. J. Virol. 1994, 68, 4493–4502. [Google Scholar] [CrossRef] [PubMed]
- Dubois, N.; Marquet, R.; Paillart, J.C.; Bernacchi, S. Retroviral RNA Dimerization: From Structure to Functions. Front. Microbiol. 2018, 9, 527. [Google Scholar] [CrossRef] [PubMed]
- Murti, K.G.; Bondurant, M.; Tereba, A. Secondary structural features in the 70S RNAs of Moloney murine leukemia and Rous sarcoma viruses as observed by electron microscopy. J. Virol. 1981, 37, 411–419. [Google Scholar] [CrossRef]
- Bieth, E.; Gabus, C.; Darlix, J.L. A study of the dimer formation of Rous sarcoma virus RNA and of its effect on viral protein synthesis in vitro. Nucleic Acids Res. 1990, 18, 119–127. [Google Scholar] [CrossRef]
- Lear, A.L.; Haddrick, M.; Heaphy, S. A study of the dimerization of Rous sarcoma virus RNA in vitro and in vivo. Virology 1995, 212, 47–57. [Google Scholar] [CrossRef]
- Polge, E.; Darlix, J.L.; Paoletti, J.; Fosse, P. Characterization of loose and tight dimer forms of avian leukosis virus RNA. J. Mol. Biol. 2000, 300, 41–56. [Google Scholar] [CrossRef]
- Fosse, P.; Motte, N.; Roumier, A.; Gabus, C.; Muriaux, D.; Darlix, J.L.; Paoletti, J. A short autocomplementary sequence plays an essential role in avian sarcoma-leukosis virus RNA dimerization. Biochemistry 1996, 35, 16601–16609. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, J.C.; Prestwood, L.J.; Lever, A.M. A novel combined RNA-protein interaction analysis distinguishes HIV-1 Gag protein binding sites from structural change in the viral RNA leader. Sci. Rep. 2015, 5, 14369. [Google Scholar] [CrossRef] [PubMed]
- Katz, R.A.; Omer, C.A.; Weis, J.H.; Mitsialis, S.A.; Faras, A.J.; Guntaka, R.V. Restriction endonuclease and nucleotide sequence analyses of molecularly cloned unintegrated avian tumor virus DNA: Structure of large terminal repeats in circle junctions. J. Virol. 1982, 42, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.E.; Tizard, R.; Gilbert, W. Nucleotide sequence of Rous sarcoma virus. Cell 1983, 32, 853–869. [Google Scholar] [CrossRef]
- Rye-McCurdy, T.; Olson, E.D.; Liu, S.; Binkley, C.; Reyes, J.P.; Thompson, B.R.; Flanagan, J.M.; Parent, L.J.; Musier-Forsyth, K. Functional Equivalence of Retroviral MA Domains in Facilitating Psi RNA Binding Specificity by Gag. Viruses 2016, 8, 256. [Google Scholar] [CrossRef]
- Milligan, J.F.; Groebe, D.R.; Witherell, G.W.; Uhlenbeck, O.C. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987, 15, 8783–8798. [Google Scholar] [CrossRef]
- Chiu, J.; March, P.E.; Lee, R.; Tillett, D. Site-directed, Ligase-Independent Mutagenesis (SLIM): A single-tube methodology approaching 100% efficiency in 4 h. Nucleic Acids Res. 2004, 32, e174. [Google Scholar] [CrossRef]
- Rye-McCurdy, T.; Rouzina, I.; Musier-Forsyth, K. Fluorescence anisotropy-based salt-titration approach to characterize protein-nucleic acid interactions. Methods Mol. Biol. 2015, 1259, 385–402. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A., 3rd; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Garbitt, R.A.; Albert, J.A.; Kessler, M.D.; Parent, L.J. trans-acting inhibition of genomic RNA dimerization by Rous sarcoma virus matrix mutants. J. Virol. 2001, 75, 260–268. [Google Scholar] [CrossRef]
- Wu, W.; Hatterschide, J.; Syu, Y.C.; Cantara, W.A.; Blower, R.J.; Hanson, H.M.; Mansky, L.M.; Musier-Forsyth, K. Human T-cell leukemia virus type 1 Gag domains have distinct RNA-binding specificities with implications for RNA packaging and dimerization. J. Biol. Chem. 2018, 293, 16261–16276. [Google Scholar] [CrossRef] [PubMed]
- Cantara, W.A.; Hatterschide, J.; Wu, W.; Musier-Forsyth, K. RiboCAT: A new capillary electrophoresis data analysis tool for nucleic acid probing. RNA 2016. [Google Scholar] [CrossRef] [PubMed]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef] [PubMed]
- Steen, K.A.; Rice, G.M.; Weeks, K.M. Fingerprinting noncanonical and tertiary RNA structures by differential SHAPE reactivity. J. Am. Chem. Soc. 2012, 134, 13160–13163. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.; Leis, J. Changes in Rous sarcoma virus RNA secondary structure near the primer binding site upon tRNATrp primer annealing. J. Virol. 1999, 73, 6307–6318. [Google Scholar] [CrossRef]
- Wilkinson, K.A.; Merino, E.J.; Weeks, K.M. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): Quantitative RNA structure analysis at single nucleotide resolution. Nat. Protoc. 2006, 1, 1610–1616. [Google Scholar] [CrossRef]
- Merino, E.J.; Wilkinson, K.A.; Coughlan, J.L.; Weeks, K.M. RNA structure analysis at single nucleotide resolution by selective 2′-hydroxyl acylation and primer extension (SHAPE). J. Am. Chem. Soc. 2005, 127, 4223–4231. [Google Scholar] [CrossRef]
- Abbink, T.E.; Berkhout, B. A novel long distance base-pairing interaction in human immunodeficiency virus type 1 RNA occludes the Gag start codon. J. Biol. Chem. 2003, 278, 11601–11611. [Google Scholar] [CrossRef]
- Lu, K.; Heng, X.; Garyu, L.; Monti, S.; Garcia, E.L.; Kharytonchyk, S.; Dorjsuren, B.; Kulandaivel, G.; Jones, S.; Hiremath, A.; et al. NMR detection of structures in the HIV-1 5′-leader RNA that regulate genome packaging. Science 2011, 334, 242–245. [Google Scholar] [CrossRef]
- Kalloush, R.M.; Vivet-Boudou, V.; Ali, L.M.; Mustafa, F.; Marquet, R.; Rizvi, T.A. Packaging of Mason-Pfizer monkey virus (MPMV) genomic RNA depends upon conserved long-range interactions (LRIs) between U5 and gag sequences. RNA 2016, 22, 905–919. [Google Scholar] [CrossRef]
- Aktar, S.J.; Vivet-Boudou, V.; Ali, L.M.; Jabeen, A.; Kalloush, R.M.; Richer, D.; Mustafa, F.; Marquet, R.; Rizvi, T.A. Structural basis of genomic RNA (gRNA) dimerization and packaging determinants of mouse mammary tumor virus (MMTV). Retrovirology 2014, 11, 96. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.A.; Jones, C.P.; Parent, L.J.; Rouzina, I.; Musier-Forsyth, K. Distinct binding interactions of HIV-1 Gag to Psi and non-Psi RNAs: Implications for viral genomic RNA packaging. RNA 2013, 19, 1078–1088. [Google Scholar] [CrossRef] [PubMed]
- Hackett, P.B.; Dalton, M.W.; Johnson, D.P.; Petersen, R.B. Phylogenetic and physical analysis of the 5′ leader RNA sequences of avian retroviruses. Nucleic Acids Res. 1991, 19, 6929–6934. [Google Scholar] [CrossRef] [PubMed]
- Ly, H.; Parslow, T.G. Bipartite signal for genomic RNA dimerization in Moloney murine leukemia virus. J. Virol. 2002, 76, 3135–3144. [Google Scholar] [CrossRef][Green Version]
- Moore, M.D.; Hu, W.S. HIV-1 RNA dimerization: It takes two to tango. AIDS Rev. 2009, 11, 91–102. [Google Scholar]
- Hill, M.K.; Shehu-Xhilaga, M.; Campbell, S.M.; Poumbourios, P.; Crowe, S.M.; Mak, J. The dimer initiation sequence stem-loop of human immunodeficiency virus type 1 is dispensable for viral replication in peripheral blood mononuclear cells. J. Virol. 2003, 77, 8329–8335. [Google Scholar] [CrossRef]
- Kenyon, J.C.; Ghazawi, A.; Cheung, W.K.; Phillip, P.S.; Rizvi, T.A.; Lever, A.M. The secondary structure of the 5′ end of the FIV genome reveals a long-range interaction between R/U5 and gag sequences, and a large, stable stem-loop. RNA 2008, 14, 2597–2608. [Google Scholar] [CrossRef]
- Kutluay, S.B.; Bieniasz, P.D. Analysis of the initiating events in HIV-1 particle assembly and genome packaging. PLoS Pathog. 2010, 6, e1001200. [Google Scholar] [CrossRef]
- Jouvenet, N.; Simon, S.M.; Bieniasz, P.D. Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. USA 2009, 106, 19114–19119. [Google Scholar] [CrossRef]
- Garbitt-Hirst, R.; Kenney, S.P.; Parent, L.J. Genetic evidence for a connection between Rous sarcoma virus gag nuclear trafficking and genomic RNA packaging. J. Virol. 2009, 83, 6790–6797. [Google Scholar] [CrossRef]
- Gudleski, N.; Flanagan, J.M.; Ryan, E.P.; Bewley, M.C.; Parent, L.J. Directionality of nucleocytoplasmic transport of the retroviral gag protein depends on sequential binding of karyopherins and viral RNA. Proc. Natl. Acad. Sci. USA 2010, 107, 9358–9363. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, R.J.K.; Rice, B.; Chen, E.C.; Tuffy, K.M.; Chiari, E.F.; Fahrbach, K.M.; Hope, T.J.; Parent, L.J. Visualizing association of the retroviral gag protein with unspliced viral RNA in the nucleus. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Meisenheimer, K.M.; Koch, T.H. Photocross-linking of nucleic acids to associated proteins. Crit. Rev. Biochem. Mol. Biol. 1997, 32, 101–140. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.P.; Cantara, W.A.; Olson, E.D.; Musier-Forsyth, K. Small-angle X-ray scattering-derived structure of the HIV-1 5′ UTR reveals 3D tRNA mimicry. Proc. Natl. Acad. Sci. USA 2014, 111, 3395–3400. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNA | Kd,1M 1 | Zeff 2 | Relative Specificity 3 |
|---|---|---|---|
| RSV 167 | (2.1 ± 0.3) × 100 | 10.1 ± 0.2 | 0.00011 |
| MΨ-WT | (2.3 ± 1.0) × 10−4 | 4.5 ± 0.3 | 1.0 |
| MΨ-A197C | (9.8 ± 4.0) × 10−3 | 6.8 ± 0.4 | 0.023 |
| MΨ-G218C | (7.0 ± 8.8) × 10−1 | 9.1 ± 1.2 | 0.00033 |
| MΨ-AAA | (3.5 ± 4.1) × 10−3 | 5.9 ± 0.8 | 0.066 |
| MΨ-AACA | (4.5 ± 1.1) × 10−4 | 5.0 ± 0.3 | 0.51 |
| MΨ-∆L3 | (3.1 ± 2.9) × 10−2 | 6.3 ± 0.6 | 0.0074 |
| MΨ-∆SL-B | (9.6 ± 8.5) × 10−5 | 3.1 ± 0.6 | 2.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Kaddis Maldonado, R.; Rye-McCurdy, T.; Binkley, C.; Bah, A.; Chen, E.C.; Rice, B.L.; Parent, L.J.; Musier-Forsyth, K. Rous Sarcoma Virus Genomic RNA Dimerization Capability In Vitro Is Not a Prerequisite for Viral Infectivity. Viruses 2020, 12, 568. https://doi.org/10.3390/v12050568
Liu S, Kaddis Maldonado R, Rye-McCurdy T, Binkley C, Bah A, Chen EC, Rice BL, Parent LJ, Musier-Forsyth K. Rous Sarcoma Virus Genomic RNA Dimerization Capability In Vitro Is Not a Prerequisite for Viral Infectivity. Viruses. 2020; 12(5):568. https://doi.org/10.3390/v12050568
Chicago/Turabian StyleLiu, Shuohui, Rebecca Kaddis Maldonado, Tiffiny Rye-McCurdy, Christiana Binkley, Aissatou Bah, Eunice C. Chen, Breanna L. Rice, Leslie J. Parent, and Karin Musier-Forsyth. 2020. "Rous Sarcoma Virus Genomic RNA Dimerization Capability In Vitro Is Not a Prerequisite for Viral Infectivity" Viruses 12, no. 5: 568. https://doi.org/10.3390/v12050568
APA StyleLiu, S., Kaddis Maldonado, R., Rye-McCurdy, T., Binkley, C., Bah, A., Chen, E. C., Rice, B. L., Parent, L. J., & Musier-Forsyth, K. (2020). Rous Sarcoma Virus Genomic RNA Dimerization Capability In Vitro Is Not a Prerequisite for Viral Infectivity. Viruses, 12(5), 568. https://doi.org/10.3390/v12050568