An Elvitegravir Nanoformulation Crosses the Blood–Brain Barrier and Suppresses HIV-1 Replication in Microglia


 , ,
, , 

 and
and 
Abstract
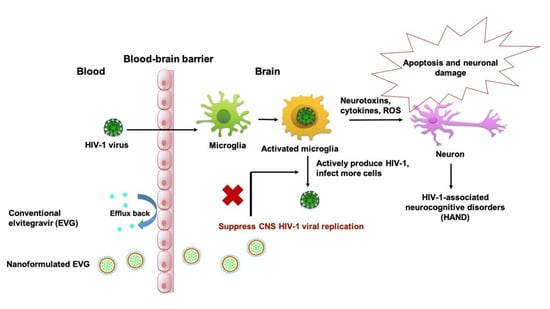
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
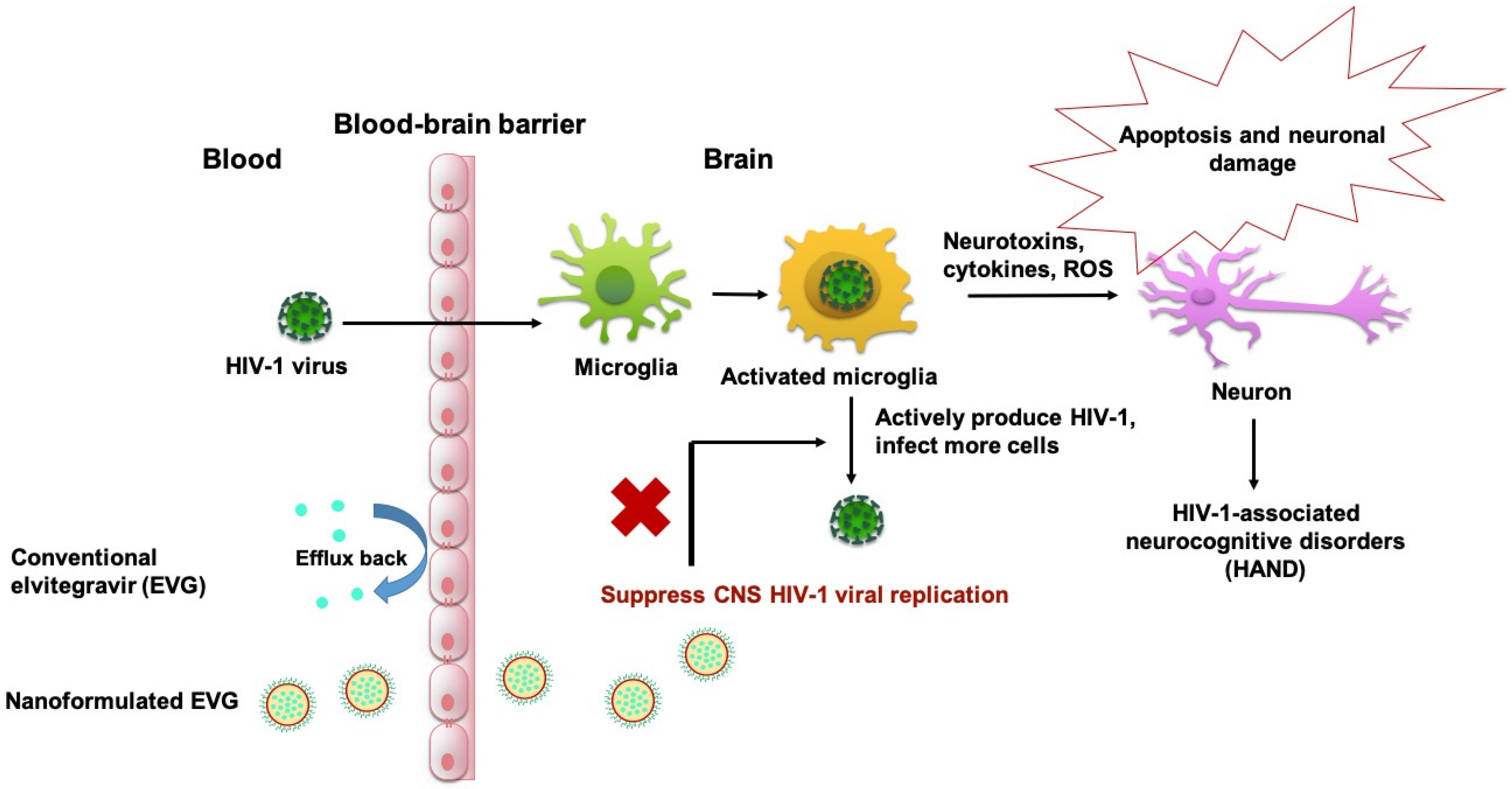
1. Introduction
2. Material and Methods
2.1. Materials
2.2. Preparation of PLGA-EVG NPs
2.3. Generation of HIV-1-Infected Monocytes-Derived Microglia-Like Cells (MMG)
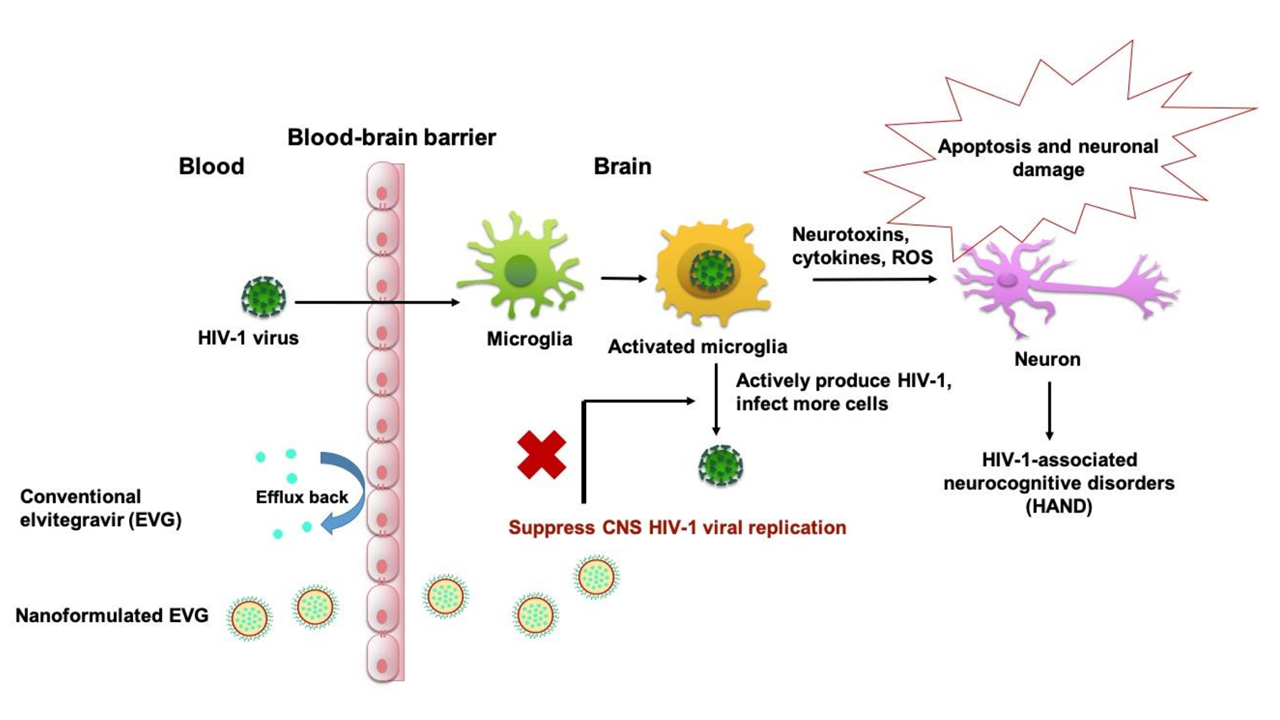
2.4. Biocompatibility Assay with MMG
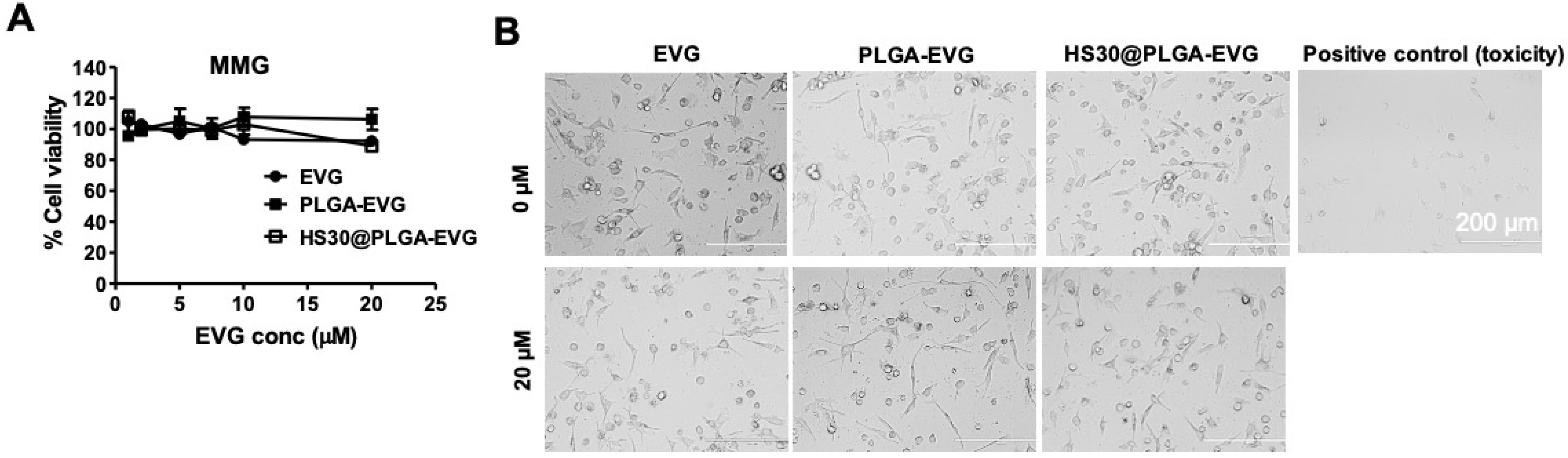
2.5. Cellular Uptake, Subcellular Localization, and Internalization Mechanism of PLGA NPs
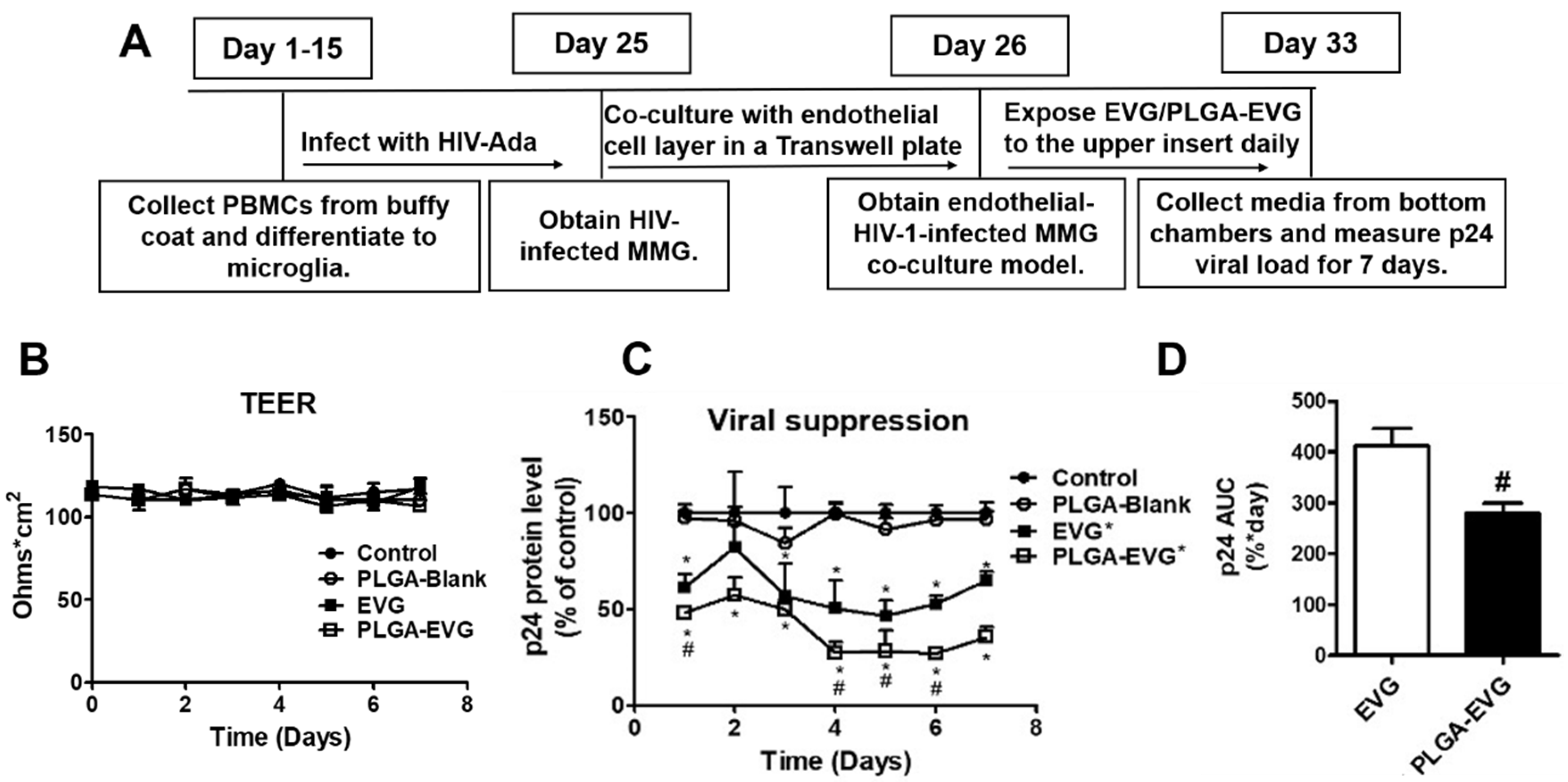
2.6. Viral Suppression of PLGA-EVG NPs in MMG after Crossing an In Vitro BBB Model
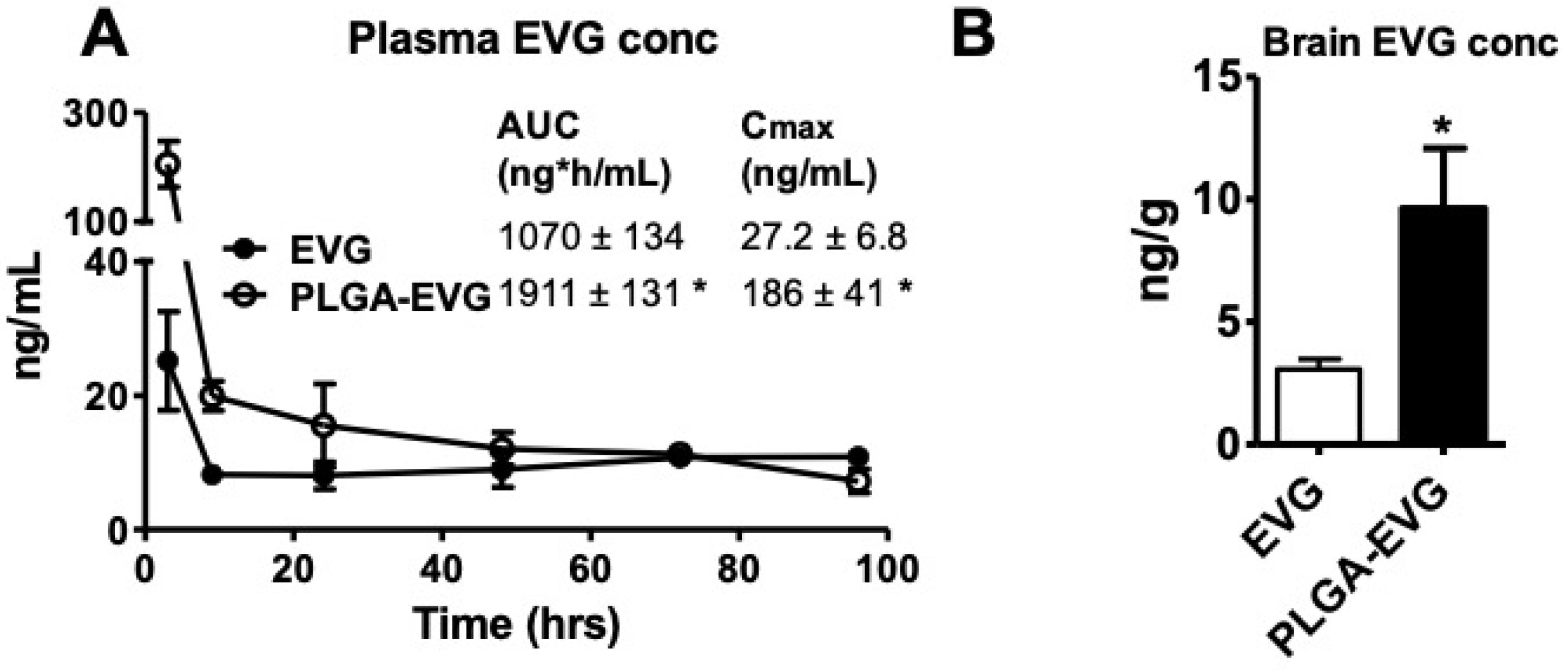
2.7. EVG Level in the In Vivo Mouse Model
2.8. Quantification of EVG, Using LC–MS/MS
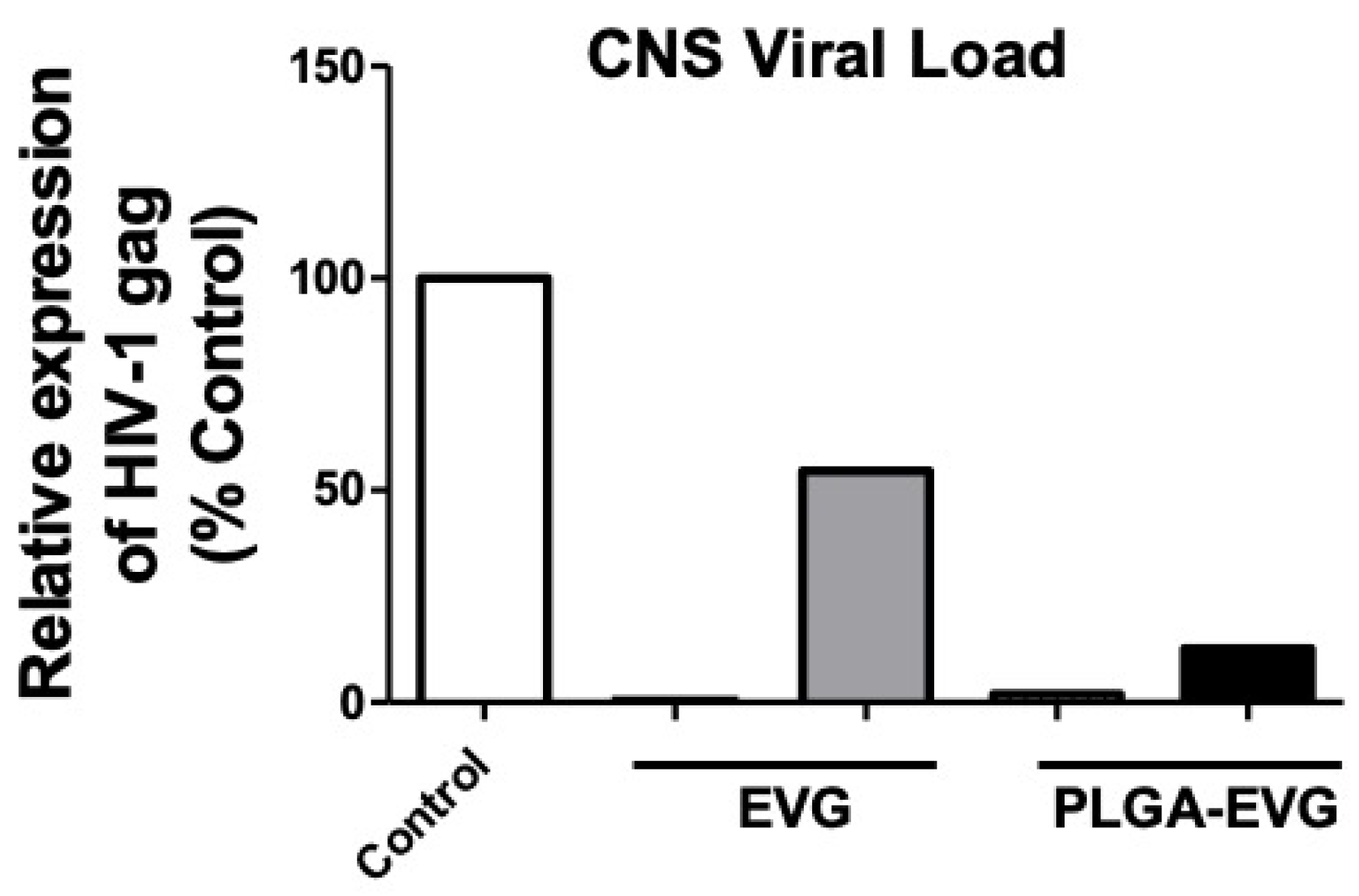
2.9. Viral Suppression of PLGA-EVG NPs in HIV-1 Encephalitic (HIVE) Mice
2.10. Statistical Analysis
3. Results
3.1. Biocompatibility of PLGA-EVG NPs in MMG
3.2. Internalization Mechanism of the PLGA NPs in MMG
3.3. Improved Viral Suppression in HIV-1-Infected MMG after Crossing the In Vitro BBB
3.4. EVG Levels in Mice
3.5. Improved Viral Suppression in an HIV-1 Encephalitis (HIVE) Mouse Model
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cohen, M.S.; Chen, Y.Q.; McCauley, M.; Gamble, T.; Hosseinipour, M.C.; Kumarasamy, N.; Hakim, J.G.; Kumwenda, J.; Grinsztejn, B.; Pilotto, J.H.; et al. Prevention of HIV-1 infection with early antiretroviral therapy. N. Engl. J. Med. 2011, 365, 493–505. [Google Scholar] [CrossRef]
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy. CHARTER Study 2010, 75, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef]
- Verma, A.S.; Singh, U.P.; Dwivedi, P.D.; Singh, A. Contribution of CNS cells in NeuroAIDS. J. Pharm. Bioallied Sci. 2010, 2, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E. Novel mechanisms of central nervous system damage in HIV infection. HIV/AIDS Res. Palliat. Care 2010, 2, 39. [Google Scholar] [CrossRef]
- Kedzierska, K.; Crowe, S.M. The role of monocytes and macrophages in the pathogenesis of HIV-1 infection. Curr. Med. Chem. 2002, 9, 1893–1903. [Google Scholar] [CrossRef]
- Burdo, T.H.; Lackner, A.; Williams, K.C. Monocyte/macrophages and their role in HIV neuropathogenesis. Immunol. Rev. 2013, 254, 102–113. [Google Scholar] [CrossRef]
- Koenig, S.; Gendelman, H.; Orenstein, J.; Canto, M.D.; Pezeshkpour, G.; Yungbluth, M.; Janotta, F.; Aksamit, A.; Martin, M.; Fauci, A. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science 1986, 233, 1089–1093. [Google Scholar] [CrossRef]
- Wiley, C.A.; Schrier, R.D.; Nelson, J.A.; Lampert, P.W.; Oldstone, M.B. Cellular localization of human immunodeficiency virus infection within the brains of acquired immune deficiency syndrome patients. Proc. Natl. Acad. Sci. USA 1986, 83, 7089–7093. [Google Scholar] [CrossRef]
- Mocchetti, I.; Bachis, A.; Avdoshina, V. Neurotoxicity of human immunodeficiency virus-1: Viral proteins and axonal transport. Neurotox. Res. 2011, 21, 79–89. [Google Scholar] [CrossRef]
- Kovalevich, J.; Langford, D. Neuronal toxicity in HIV CNS disease. Futur. Virol. 2012, 7, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Clifford, D.B.; Ances, B.M. HIV-associated neurocognitive disorder. Lancet Infect. Dis. 2013, 13, 976–986. [Google Scholar] [CrossRef]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder—Pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, A.; Price, R.W.; Gisslén, M. Antiretroviral drug treatment of CNS HIV-1 infection. J. Antimicrob. Chemother. 2011, 67, 299–311. [Google Scholar] [CrossRef]
- Haas, D.W.; Johnson, B.; Nicotera, J.; Bailey, V.L.; Harris, V.L.; Bowles, F.B.; Raffanti, S.; Schranz, J.; Finn, T.S.; Saah, A.J.; et al. Effects of Ritonavir on Indinavir Pharmacokinetics in Cerebrospinal Fluid and Plasma. Antimicrob. Agents Chemother. 2003, 47, 2131–2137. [Google Scholar] [CrossRef][Green Version]
- Decloedt, E.H.; Rosenkranz, B.; Maartens, G.; Joska, J. Central Nervous System Penetration of Antiretroviral Drugs: Pharmacokinetic, Pharmacodynamic and Pharmacogenomic Considerations. Clin. Pharmacokinet. 2015, 54, 581–598. [Google Scholar] [CrossRef]
- Morris, A.; Marsden, M.; Halcrow, K.; Hughes, E.S.; Brettle, R.P.; Bell, J.E.; Simmonds, P. Mosaic Structure of the Human Immunodeficiency Virus Type 1 Genome Infecting Lymphoid Cells and the Brain: Evidence for Frequent In Vivo Recombination Events in the Evolution of Regional Populations. J. Virol. 1999, 73, 8720–8731. [Google Scholar] [CrossRef]
- Albright, A.V.; Shieh, J.T.C.; Itoh, T.; Lee, B.; Pleasure, D.; O’Connor, M.J.; Doms, R.W.; González-Scarano, F. Microglia Express CCR5, CXCR4, and CCR3, but of These, CCR5 Is the Principal Coreceptor for Human Immunodeficiency Virus Type 1 Dementia Isolates. J. Virol. 1999, 73, 205–213. [Google Scholar] [CrossRef]
- Glass, J.D.; Wesselingh, S.L. Microglia in HIV-associated neurological diseases. Microsc. Res. Tech. 2001, 54, 95–105. [Google Scholar] [CrossRef]
- Shieh, J.T.C.; Albright, A.V.; Sharron, M.; Gartner, S.; Strizki, J.; Doms, R.W.; González-Scarano, F. Chemokine Receptor Utilization by Human Immunodeficiency Virus Type 1 Isolates That Replicate in Microglia. J. Virol. 1998, 72, 4243–4249. [Google Scholar] [CrossRef]
- Garden, G. Microglia in human immunodeficiency virus-associated neurodegeneration. Glia 2002, 40, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Fiala, M.; Looney, D.J.; Stins, M.; Way, D.D.; Zhang, L.; Gan, X.; Chiappelli, F.; Schweitzer, E.S.; Shapshak, P.; Weinand, M.; et al. TNF-α Opens a Paracellular Route for HIV-1 Invasion across the Blood-Brain Barrier. Mol. Med. 1997, 3, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Collman, R.G. CNS inflammation and macrophage/microglial biology associated with HIV-1 infection. J. Neuroimmune Pharmacol. 2009, 4, 430–447. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.; Schlichter, L.; Bendayan, R. Multidrug Resistance Protein (MRP) 4- and MRP 5-Mediated Efflux of 9-(2-Phosphonylmethoxyethyl)adenine by Microglia. J. Pharmacol. Exp. Ther. 2004, 309, 1221–1229. [Google Scholar] [CrossRef]
- Prasad, B.; Unadkat, J. The concept of fraction of drug transported (ft) with special emphasis on BBB efflux of CNS and antiretroviral drugs. Clin. Pharmacol. Ther. 2015, 97, 320–323. [Google Scholar] [CrossRef]
- Ford, J.; Khoo, S.H.; Back, D.J. The intracellular pharmacology of antiretroviral protease inhibitors. J. Antimicrob. Chemother. 2004, 54, 982–990. [Google Scholar] [CrossRef]
- Gong, Y.; Chowdhury, P.; Nagesh, P.K.B.; Rahman, M.A.; Zhi, K.; Yallapu, M.; Kumar, S. Novel elvitegravir nanoformulation for drug delivery across the blood-brain barrier to achieve HIV-1 suppression in the CNS macrophages. Sci. Rep. 2020, 10, 3835. [Google Scholar] [CrossRef]
- Gong, Y.; Chowdhury, P.; Midde, N.M.; Rahman, M.A.; Yallapu, M.M.; Kumar, S. Novel elvitegravir nanoformulation approach to suppress the viral load in HIV-infected macrophages. Biochem. Biophys. Rep. 2017, 12, 214–219. [Google Scholar] [CrossRef]
- Joyce, A.P.; Wang, M.; Lawrence-Henderson, R.; Filliettaz, C.; Leung, S.; Xu, X.; O’Hara, D.M. One Mouse, One Pharmacokinetic Profile: Quantitative Whole Blood Serial Sampling for Biotherapeutics. Pharm. Res. 2014, 31, 1823–1833. [Google Scholar] [CrossRef]
- Midde, N.M.; Rahman, M.A.; Rathi, C.; Li, J.; Meibohm, B.; Li, W.; Kumar, S. Effect of Ethanol on the Metabolic Characteristics of HIV-1 Integrase Inhibitor Elvitegravir and Elvitegravir/Cobicistat with CYP3A: An Analysis Using a Newly Developed LC-MS/MS Method. PLoS ONE 2016, 11, e0149225. [Google Scholar] [CrossRef]
- Poluektova, L.Y.; Gorantla, S.; Faraci, J.; Birusingh, K.; Dou, H.; Gendelman, H.E. Neuroregulatory events follow adaptive immune-mediated elimination of HIV-1-infected macrophages: Studies in a murine model of viral encephalitis. J. Immunol. 2004, 172, 7610–7617. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Laurent, S.; Tawil, N.; Mahmoudi, M.; Yahia, L. Nanoparticle and Protein Corona. In Electrophysiology of Unconventional Channels and Pores; Springer Science and Business Media LLC: Berlin, Germany, 2013; Volume 15, pp. 21–44. [Google Scholar]
- Yallapu, M.M.; Khan, S.; Maher, D.M.; Ebeling, M.C.; Sundram, V.; Chauhan, N.; Ganju, A.; Balakrishna, S.; Gupta, B.K.; Zafar, N.; et al. Anti-cancer activity of curcumin loaded nanoparticles in prostate cancer. Biomaterials 2014, 35, 8635–8648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, X.; Lv, Y.; Xin, X.; Qin, C.; Han, X.; Yang, L.; He, W.; Yin, L. Cytosolic co-delivery of miRNA-34a and docetaxel with core-shell nanocarriers via caveolae-mediated pathway for the treatment of metastatic breast cancer. Sci. Rep. 2017, 7, 46186. [Google Scholar] [CrossRef] [PubMed]
- Castellano, P.; Prevedel, L.; Eugenin, E. HIV-infected macrophages and microglia that survive acute infection become viral reservoirs by a mechanism involving Bim. Sci. Rep. 2017, 7, 12866. [Google Scholar] [CrossRef]
- Etemad, S.; Zamin, R.M.; Ruitenberg, M.J.; Filgueira, L. A novel in vitro human microglia model: Characterization of human monocyte-derived microglia. J. Neurosci. Methods 2012, 209, 79–89. [Google Scholar] [CrossRef]
- Nair, M.; Jayant, R.D.; Kaushik, A.; Sagar, V. Getting into the brain: Potential of nanotechnology in the management of NeuroAIDS. Adv. Drug Deliv. Rev. 2016, 103, 202–217. [Google Scholar] [CrossRef]
- Aalinkeel, R.; Mangum, C.S.; Abou-Jaoude, E.; Reynolds, J.L.; Liu, M.; Sundquist, K.; Parikh, N.U.; Chaves, L.D.; Mammen, M.J.; Schwartz, S.A.; et al. Galectin-1 Reduces Neuroinflammation via Modulation of Nitric Oxide-Arginase Signaling in HIV-1 Transfected Microglia: A Gold Nanoparticle-Galectin-1 “Nanoplex” a Possible Neurotherapeutic? J. Neuroimmune Pharmacol. 2016, 12, 133–151. [Google Scholar] [CrossRef]
- Vader, P.; Mol, E.A.; Pasterkamp, G.; Schiffelers, R.M. Extracellular vesicles for drug delivery. Adv. Drug Deliv. Rev. 2016, 106, 148–156. [Google Scholar] [CrossRef]
- Rodriguez, M.; Kaushik, A.; Lapierre, J.; Dever, S.M.; El-Hage, N.; Nair, M. Electro-Magnetic Nano-Particle Bound Beclin1 siRNA Crosses the Blood–Brain Barrier to Attenuate the Inflammatory Effects of HIV-1 Infection in Vitro. J. Neuroimmune Pharmacol. 2016, 12, 120–132. [Google Scholar] [CrossRef]
- Pommier, Y.; Johnson, A.A.; Marchand, C. Integrase inhibitors to treat HIV/Aids. Nat. Rev. Drug Discov. 2005, 4, 236–248. [Google Scholar] [CrossRef]
- Calcagno, A.; Simiele, M.; Motta, I.; Pinna, S.M.; Bertucci, R.; D’Avolio, A.; Di Perri, G.; Bonora, S. Elvitegravir/Cobicistat/Tenofovir/Emtricitabine Penetration in the Cerebrospinal Fluid of Three HIV-Positive Patients. AIDS Res. Hum. Retrovir. 2016, 32, 409–411. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Wang, L.; Deng, G.; Liu, J.; Chen, Q.; Chen, Z. Systemic delivery to central nervous system by engineered PLGA nanoparticles. Am. J. Transl. Res. 2016, 8, 749–764. [Google Scholar] [PubMed]
- DeMarino, C.; Schwab, A.; Pleet, M.; Mathiesen, A.; Friedman, J.; El-Hage, N.; Kashanchi, F. Biodegradable Nanoparticles for Delivery of Therapeutics in CNS Infection. J. Neuroimmune Pharmacol. 2016, 12, 31–50. [Google Scholar] [CrossRef]
- Patel, B.K.; Parikh, R.H.; Patel, N. Targeted delivery of mannosylated-PLGA nanoparticles of antiretroviral drug to brain. Int. J. Nanomed. 2018, 13, 97–100. [Google Scholar] [CrossRef]
- FDA. Inactive Ingredient Search for Approved Drug Products. Available online: https://www.accessdata.fda.gov/scripts/cder/iig/index.cfm (accessed on 21 April 2020).
- Plough-Center. Plough Center for Sterile Drug Delivery Solutions; Plough-Center: Memphis, TN, USA, 2020; Available online: https://www.uthsc.edu/plough-center/ (accessed on 21 April 2020).
- Breakthrough-Therapy. FDA Breakthrough Therapy; Breakthrough-Therapy: Silver Spring, MD, USA, 2020. Available online: https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/breakthrough-therapy (accessed on 21 April 2020).
- Liberti, L.; Breckenridge, A.; Hoekman, J.; Leufkens, H.; Lumpkin, M.; McAuslane, N.; Stolk, P.; Zhi, K.; Rägo, L. Practical aspects of developing, implementing and using facilitated regulatory pathways in the emerging markets. In Proceedings of the Poster Drug Information Association Annual Meeting, Philadelphia, PA, USA. [CrossRef]
- Liberti, L.; Breckenridge, A.; Hoekman, J.; Leufkens, H.; Lumpkin, M.; McAuslane, N.; Stolk, P.; Zhi, K.; Rägo, L.; Liberti, A.B.L. Accelerating access to new medicines: Current status of facilitated regulatory pathways used by emerging regulatory authorities. J. Public Health Policy 2016, 37, 315–333. [Google Scholar] [CrossRef]
- Iversen, T.-G.; Skotland, T.; Sandvig, K. Endocytosis and intracellular transport of nanoparticles: Present knowledge and need for future studies. Nano Today 2011, 6, 176–185. [Google Scholar] [CrossRef]
- Papa, S.; Ferrari, R.; De Paola, M.; Rossi, F.; Mariani, A.; Caron, I.; Sammali, E.; Peviani, M.; Dell’Oro, V.; Colombo, C.; et al. Polymeric nanoparticle system to target activated microglia/macrophages in spinal cord injury. J. Control. Release 2014, 174, 15–26. [Google Scholar] [CrossRef]
- Papa, S.; Rossi, F.; Ferrari, R.; Mariani, A.; De Paola, M.; Caron, I.; Fiordaliso, F.; Bisighini, C.; Sammali, E.; Colombo, C.; et al. Selective Nanovector Mediated Treatment of Activated Proinflammatory Microglia/Macrophages in Spinal Cord Injury. ACS Nano 2013, 7, 9881–9895. [Google Scholar] [CrossRef]
- Zhang, F.; Lin, Y.-A.; Kannan, S.; Kannan, R.M. Targeting specific cells in the brain with nanomedicines for CNS therapies. J. Control. Release 2015, 240, 212–226. [Google Scholar] [CrossRef]
- Selby, L.I.; Cortez-Jugo, C.; Such, G.K.; Johnston, A.P.R. Nanoescapology: Progress toward understanding the endosomal escape of polymeric nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1452. [Google Scholar] [CrossRef] [PubMed]
- Mathews, S.; Branch-Woods, A.; Katano, I.; Makarov, E.; Thomas, M.B.; Gendelman, H.E.; Poluektova, L.Y.; Ito, M.; Gorantla, S. Human Interleukin-34 facilitates microglia-like cell differentiation and persistent HIV-1 infection in humanized mice. Mol. Neurodegener. 2019, 14, 12. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, Y.; Zhi, K.; Nagesh, P.K.B.; Sinha, N.; Chowdhury, P.; Chen, H.; Gorantla, S.; Yallapu, M.M.; Kumar, S. An Elvitegravir Nanoformulation Crosses the Blood–Brain Barrier and Suppresses HIV-1 Replication in Microglia. Viruses 2020, 12, 564. https://doi.org/10.3390/v12050564
Gong Y, Zhi K, Nagesh PKB, Sinha N, Chowdhury P, Chen H, Gorantla S, Yallapu MM, Kumar S. An Elvitegravir Nanoformulation Crosses the Blood–Brain Barrier and Suppresses HIV-1 Replication in Microglia. Viruses. 2020; 12(5):564. https://doi.org/10.3390/v12050564
Chicago/Turabian StyleGong, Yuqing, Kaining Zhi, Prashanth K. B. Nagesh, Namita Sinha, Pallabita Chowdhury, Hao Chen, Santhi Gorantla, Murali M. Yallapu, and Santosh Kumar. 2020. "An Elvitegravir Nanoformulation Crosses the Blood–Brain Barrier and Suppresses HIV-1 Replication in Microglia" Viruses 12, no. 5: 564. https://doi.org/10.3390/v12050564
APA StyleGong, Y., Zhi, K., Nagesh, P. K. B., Sinha, N., Chowdhury, P., Chen, H., Gorantla, S., Yallapu, M. M., & Kumar, S. (2020). An Elvitegravir Nanoformulation Crosses the Blood–Brain Barrier and Suppresses HIV-1 Replication in Microglia. Viruses, 12(5), 564. https://doi.org/10.3390/v12050564