External Quality Assessment for Next-Generation Sequencing-Based HIV Drug Resistance Testing: Unique Requirements and Challenges



Abstract
1. Introduction
2. Ongoing EQA is Critical for SS-Based HIVDR Genotyping
3. The Development of EQA for NGS-Based HIVDR is Essential
4. EQA for NGS-Based HIVDR Assays: Unique Requirements and Challenges
4.1. PT Panel Design for NGS-Based HIVDR Testing
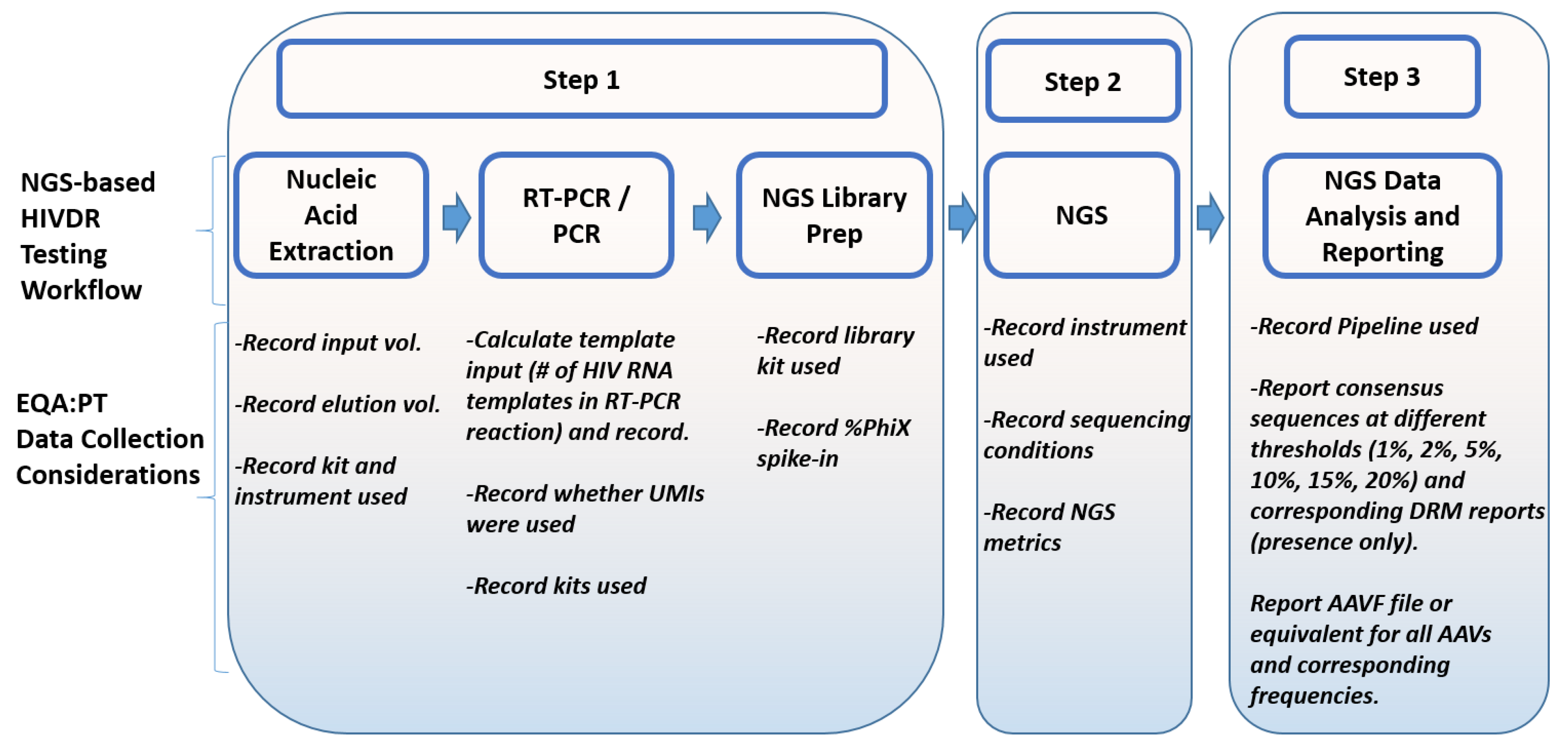
4.2. Data Collection for NGS-Based HIVDR Testing
4.3. EQA: Data Assessment and Scoring Strategies for NGS-Based HIVDR Testing
4.3.1. Inconsistencies in Detecting DRMs
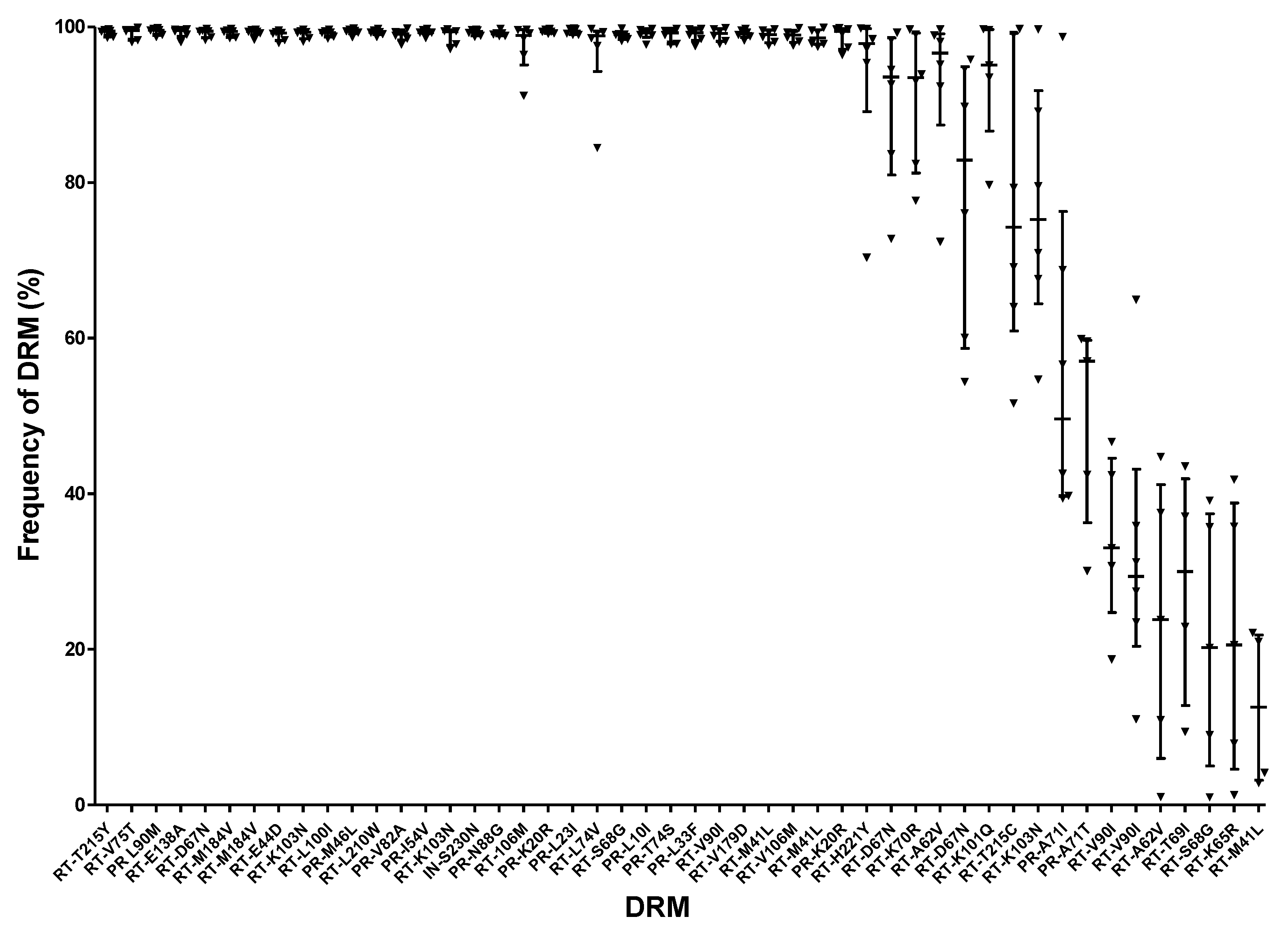
4.3.2. Large Variations in DRM Frequencies
4.3.3. Variations in Wet-Lab Methods, NGS Platforms and Bioinformatics Pipelines
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Voelkerding, K.V.; Dames, S.A.; Durtschi, J.D. Next-generation sequencing:from basic research to diagnostics. Clin. Chem. 2009, 55, 641–658. [Google Scholar] [CrossRef] [PubMed]
- Maljkovic Berry, I.; Melendrez, M.C.; Bishop-Lilly, K.A.; Rutvisuttinunt, W.; Pollett, S.; Talundzic, E.; Morton, L.; Jarman, R.G. Next Generation Sequencing and Bioinformatics Methodologies for Infectious Disease Research and Public Health: Approaches, Applications, and Considerations for Development of Laboratory Capacity. J. Infect. Dis. 2020, 221, S292–S307. [Google Scholar] [CrossRef] [PubMed]
- Brumme, C.J.; Poon, A.F.Y. Promises and pitfalls of Illumina sequencing for HIV resistance genotyping. Virus Res. 2017, 239, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Casadellà, M.; Paredes, R. Deep sequencing for HIV-1 clinical management. Virus Res. 2017, 239, 69–81. [Google Scholar] [CrossRef]
- Hamers, R.L.; Schuurman, R.; Sigaloff, K.C.E.; Wallis, C.L.; Kityo, C.; Siwale, M.; Mandaliya, K.; Ive, P.; Botes, M.E.; Wellington, M.; et al. Effect of pretreatment HIV-1 drug resistance on immunological, virological, and drug-resistance outcomes of first-line antiretroviral treatment in sub-Saharan Africa: A multicentre cohort study. Lancet Infect. Dis. 2012, 12, 307–317. [Google Scholar] [CrossRef]
- Boender, T.S.; Hoenderboom, B.M.; Sigaloff, K.C.E.; Hamers, R.L.; Wellington, M.; Shamu, T.; Siwale, M.; Labib Maksimos, E.E.F.; Nankya, I.; Kityo, C.M.; et al. Pretreatment HIV drug resistance increases regimen switches in sub-saharan Africa. Clin. Infect. Dis. 2015, 61, 1749–1758. [Google Scholar] [CrossRef]
- Ávila-Ríos, S.; García-Morales, C.; Matías-Florentino, M.; Romero-Mora, K.A.; Tapia-Trejo, D.; Quiroz-Morales, V.S.; Reyes-Gopar, H.; Ji, H.; Sandstrom, P.; Casillas-Rodríguez, J.; et al. Pretreatment HIV-drug resistance in Mexico and its impact on the effectiveness of first-line antiretroviral therapy: A nationally representative 2015 WHO survey. Lancet HIV 2016, 3, e579–e591. [Google Scholar] [CrossRef]
- Vandenhende, M.A.; Bellecave, P.; Recordon-Pinson, P.; Reigadas, S.; Bidet, Y.; Bruyand, M.; Bonnet, F.; Lazaro, E.; Neau, D.; Fleury, H.; et al. Prevalence and evolution of low frequency HIV drug resistance mutations detected by ultra deep sequencing in patients experiencing first line antiretroviral therapy failure. PLoS ONE 2014, 9, e86771. [Google Scholar] [CrossRef]
- Kantor, R.; Smeaton, L.; Vardhanabhuti, S.; Hudelson, S.E.; Wallis, C.L.; Tripathy, S.; Morgado, M.G.; Saravanan, S.; Balakrishnan, P.; Reitsma, M.; et al. Pretreatment HIV Drug Resistance and HIV-1 Subtype C Are Independently Associated with Virologic Failure: Results from the Multinational PEARLS (ACTG A5175) Clinical Trial. Clin. Infect. Dis. 2015, 60, 1541–1549. [Google Scholar] [CrossRef]
- Cozzi-Lepri, A.; Noguera-Julian, M.; Di Giallonardo, F.; Schuurman, R.; Däumer, M.; Aitken, S.; Ceccherini-Silberstein, F.; Monforte, A.D.A.; Geretti, A.M.; Booth, C.L.; et al. Low-frequency drug-resistant HIV-1 and risk of virological failure to first-line NNRTI-based ART: A multicohort European case-control study using centralized ultrasensitive 454 pyrosequencing. J. Antimicrob. Chemother. 2015, 70, 930–940. [Google Scholar] [CrossRef]
- Inzaule, S.C.; Hamers, R.L.; Noguera-Julian, M.; Casadellà, M.; Parera, M.; Kityo, C.; Steegen, K.; Naniche, D.; Clotet, B.; Rinke de Wit, T.F.; et al. Clinically relevant thresholds for ultrasensitive HIV drug resistance testing: A multi-country nested case-control study. Lancet HIV 2018, 5, e638–e646. [Google Scholar] [CrossRef]
- Endrullat, C.; Glökler, J.; Franke, P.; Frohme, M. Standardization and quality management in next-generation sequencing. Appl. Transl. Genom. 2016, 10, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Gargis, A.S.; Kalman, L.; Lubin, I.M. Assuring the quality of next-generation sequencing in clinical microbiology and public health laboratories. J. Clin. Microbiol. 2016, 54, 2857–2865. [Google Scholar] [CrossRef] [PubMed]
- Gargis, A.S.; Kalman, L.; Berry, M.W.; Bick, D.P.; Dimmock, D.P.; Hambuch, T.; Lu, F.; Lyon, E.; Voelkerding, K.V.; Zehnbauer, B.A.; et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat. Biotechnol. 2012, 30, 1033–1036. [Google Scholar] [CrossRef]
- Hutchins, R.J.; Phan, K.L.; Saboor, A.; Miller, J.D.; Muehlenbachs, A. Practical Guidance to Implementing Quality Management Systems in Public Health Laboratories Performing Next-Generation Sequencing: Personnel, Equipment, and Process Management (Phase 1). J. Clin. Microbiol. 2019, 5, e00261-19. [Google Scholar] [CrossRef]
- WHO: Content Sheet 10-1: Overview of External Quality Assessment (EQA). Available online: https://www.who.int/ihr/training/laboratory_quality/10_b_eqa_contents.pdf (accessed on 10 February 2020).
- Wayne, P.; CSLI. Quality System Regulation for Laboratory-Developed Tests: A Practical Guide for the Laboratory; Clinical Laboratory Standards Institute: Stanford, CA, USA, 2015. [Google Scholar]
- Genzen, J.R. Regulation of Laboratory-Developed Tests. Am. J. Clin. Pathol. 2019, 152, 122–131. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services; Rockville, M.; CMS. Clinical Laboratory Improvement Ammendments of 1988 (part 493); Centers for Medicare and Medicaid Services: Baltimore, MD, USA, 1988. [Google Scholar]
- (CLSI 2014) Nucleic Acid Sequencing Methods in Diagnostic Laboratory Medicine; Approved Guideline; MM09-A2 2014; CLSI: Stanford, CA, USA, 2014; p. 34.
- Kalman, L.V.; Lubin, I.M.; Barker, S.; Du Sart, D.; Elles, R.; Grody, W.W.; Zehnbauer, B. Current Landscape and New Paradigms of Proficiency Testing and External Quality Assessment for Molecular Genetics. Arch. Pathol. Lab. Med. 2013, 137, 983–988. [Google Scholar] [CrossRef]
- Parkin, N.; Bremer, J.; Bertagnolio, S. Genotyping external quality assurance in the world health organization HIV drug resistance laboratory network during 2007–2010. Clin. Infect. Dis. 2012, 54, S266–S272. [Google Scholar] [CrossRef]
- Pandit, A.; Mackay, W.G.; Steel, C.; van Loon, A.M.; Schuurman, R. HIV-1 drug resistance genotyping quality assessment: Results of the ENVA7 Genotyping Proficiency Programme. J. Clin. Virol. 2008, 43, 401–406. [Google Scholar] [CrossRef]
- Land, S.; Cunningham, P.; Zhou, J.; Frost, K.; Katzenstein, D.; Kantor, R.; Chen, Y.-M.A.; Oka, S.; DeLong, A.; Sayer, D.; et al. TREAT Asia Quality Assessment Scheme (TAQAS) to standardize the outcome of HIV genotypic resistance testing in a group of Asian laboratories. J. Virol. Methods 2009, 159, 185–193. [Google Scholar] [CrossRef]
- Yoshida, S.; Hattori, J.; Matsuda, M.; Okada, K.; Kazuyama, Y.; Hashimoto, O.; Ibe, S.; Fujisawa, S.I.; Chiba, H.; Tatsumi, M.; et al. Japanese external quality assessment program to standardize HIV-1 drug-resistance testing (JEQS2010 program) using in vitro transcribed RNA as reference material. AIDS Res. Hum. Retrovir. 2015, 31, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, P.R.; Trevino, C.; Hall, T.A.; Chui, C.K.S.; Wynhoven, B.; Woods, C.K.; Liu, T.F.; Chu, A.L.; Shafer, R.W.; Brumme, C.J. Automating HIV Drug Resistance Genotyping with RECall, a Freely Accessible Sequence Analysis Tool. J. Clin. Microbiol. 2012, 50, 1936–1942. [Google Scholar]
- Liu, T.F.; Shafer, R.W. Web Resources for HIV Type 1 Genotypic-Resistance Test Interpretation. Clin. Infect. Dis. 2006, 42, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Wensing, A.M.; Calvez, V.; Ceccherini-Silberstein, F.; Charpentier, C.; Günthard, H.F.; Paredes, R.; Shafer, R.W.; Richman, D.D. 2019 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 2019, 27, 111–121. [Google Scholar] [PubMed]
- WHO. Who/Hivresnet HIV Drug Resistance; WHO: Geneva, Switzerland, 2017; ISBN 9789241512879. [Google Scholar]
- Wang, C.; Mitsuya, Y.; Gharizadeh, B.; Ronaghi, M.; Shafer, R.W. Characterization of mutation spectra with ultra-deep pyrosequencing: Application to HIV-1 drug resistance. Genome Res. 2007, 17, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Minkah, N.; Leipzig, J.; Wang, G.; Arens, M.Q.; Tebas, P.; Bushman, F.D. DNA bar coding and pyrosequencing to identify rare HIV drug resistance mutations. Nucleic Acids Res. 2007, 35, e91. [Google Scholar] [CrossRef]
- Taylor, T.; Lee, E.R.; Nykoluk, M.; Enns, E.; Liang, B.; Capina, R.; Gauthier, M.K.; Van Domselaar, G.; Sandstrom, P.; Brooks, J.; et al. A MiSeq-HyDRA platform for enhanced HIV drug resistance genotyping and surveillance. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Noguera-Julian, M.; Edgil, D.; Harrigan, P.R.; Sandstrom, P.; Godfrey, C.; Paredes, R. Next-Generation Human Immunodeficiency Virus Sequencing for Patient Management and Drug Resistance Surveillance. J. Infect. Dis. 2017, 216, S829–S833. [Google Scholar] [CrossRef]
- Chui, C.K.S.; Dong, W.W.Y.; Joy, J.B.; Poon, A.F.Y.; Dong, W.Y.; Mo, T.; Woods, C.K.; Beatty, C.; Hew, H.; Harrigan, P.R.; et al. Development and validation of two screening assays for the hepatitis C virus NS3 Q80K polymorphism associated with reduced response to combination treatment regimens containing simeprevir. J. Clin. Microbiol. 2015, 53, 2942–2950. [Google Scholar] [CrossRef][Green Version]
- Howison, M.; Coetzer, M.; Kantor, R. Measurement error and variant-calling in deep Illumina sequencing of HIV. Bioinformatics 2019, 35, 2029–2035. [Google Scholar] [CrossRef]
- Aziz, N.; Zhao, Q.; Bry, L.; Driscoll, D.K.; Funke, B.; Gibson, J.S.; Grody, W.W.; Hegde, M.R.; Hoeltge, G.A.; Leonard, D.G.B.; et al. College of American pathologists’ laboratory standards for next-generation sequencing clinical tests. Arch. Pathol. Lab. Med. 2015, 139, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Lefterova, M.I.; Suarez, C.J.; Banaei, N.; Pinsky, B.A. Next-Generation Sequencing for Infectious Disease Diagnosis and Management: A Report of the Association for Molecular Pathology. J. Mol. Diagn. 2015, 17, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Parkin, N.; Zaccaro, D.; Avila-Rios, S.; Brumme, C.; Hunt, G.; Ji, H.; Kantor, R.; Mbisa, J.; Paredes, R.; Rivera-Amill, V.; et al. Multi-Laboratory Comparison of Next-Generation to Sanger-Based Sequencing for HIV-1 Drug Resistance Genotyping. In Proceedings of the XXVII International HIV Drug Resistance and Treatment Strategies Workshop, Johannesburg, South Africa, 22–23 October 2018. [Google Scholar]
- WHO. World Health Organization Global Strategy For The Surveillance And Monitoring Of HIV Drug Resistance. HIV/AIDS Programme. 2012. Available online: http:/www.who.int/hiv/pub/drugresistance/drug_resistance_strategy/en/ (accessed on 10 February 2020).
- Palmer, S.; Kearney, M.; Maldarelli, F.; Halvas, E.K.; Bixby, C.J.; Bazmi, H.; Rock, D.; Falloon, J.; Davey, R.T.; Dewar, R.L.; et al. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J. Clin. Microbiol. 2005, 43, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Cai, F.; Chen, H.; Hicks, C.B.; Bartlett, J.A.; Zhu, J.; Gao, F. Detection of minor drug-resistant populations by parallel allele-specific sequencing. Nat. Methods 2007, 4, 123–125. [Google Scholar] [CrossRef]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef]
- Song, H.; Pavlicek, J.W.; Cai, F.; Bhattacharya, T.; Li, H.; Iyer, S.S.; Bar, K.J.; Decker, J.M.; Goonetilleke, N.; Liu, M.K.P.; et al. Impact of immune escape mutations on HIV-1 fitness in the context of the cognate transmitted/founder genome. Retrovirology 2012, 9, 1. [Google Scholar] [CrossRef]
- Kireev, D.E.; Lopatukhin, A.E.; Murzakova, A.V.; Pimkina, E.V.; Speranskaya, A.S.; Neverov, A.D.; Fedonin, G.G.; Fantin, Y.S.; Shipulin, G.A. Evaluating the accuracy and sensitivity of detecting minority HIV-1 populations by Illumina next-generation sequencing. J. Virol. Methods 2018, 261, 40–45. [Google Scholar] [CrossRef]
- Orton, R.J.; Wright, C.F.; Morelli, M.J.; King, D.J.; Paton, D.J.; King, D.P.; Haydon, D.T. Distinguishing low frequency mutations from RT-PCR and sequence errors in viral deep sequencing data. BMC Genom. 2015, 16, 229. [Google Scholar] [CrossRef]
- Kugelman, J.R.; Wiley, M.R.; Nagle, E.R.; Reyes, D.; Pfeffer, B.P.; Kuhn, J.H.; Sanchez-Lockhart, M.; Palacios, G.F. Error baseline rates of five sample preparation methods used to characterize RNA virus populations. PLoS ONE 2017, 12, e0171333. [Google Scholar] [CrossRef]
- Santiago, G.A.; Vergne, E.; Quiles, Y.; Cosme, J.; Vazquez, J.; Medina, J.F.; Medina, F.; Colón, C.; Margolis, H.; Muñoz-Jordán, J.L. Analytical and Clinical Performance of the CDC Real Time RT-PCR Assay for Detection and Typing of Dengue Virus. PLoS Negl. Trop. Dis. 2013, 7, e2311. [Google Scholar] [CrossRef]
- Long, C.; Chao, E.; Da, L.T.; Yu, J. A Viral T7 RNA Polymerase Ratcheting Along DNA With Fidelity Control. Comput. Struct. Biotechnol. J. 2019, 17, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Brieba, L.G.; Sousa, R. Misincorporation by wild-type and mutant T7 RNA polymerases: Identification of interactions that reduce misincorporation rates by stabilizing the catalytically incompetent open conformation. Biochemistry 2000, 39, 11571–11580. [Google Scholar] [CrossRef] [PubMed]
- Mcelroy, K.; Thomas, T.; Luciani, F. Deep sequencing of evolving pathogen populations : Applications, errors, and bioinformatic solutions. Microb. Inform. Exp. 2014, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Andino, R.; Domingo, E. Viral Quasi Species. Virology 2015, 344, 1173–1178. [Google Scholar]
- Boltz, V.F.; Rausch, J.; Shao, W.; Hattori, J.; Luke, B.; Maldarelli, F.; Mellors, J.W.; Kearney, M.F.; Coffin, J.M. Ultrasensitive single-genome sequencing: Accurate, targeted, next generation sequencing of hiv-1 rna. Retrovirology 2016, 13, 87. [Google Scholar] [CrossRef]
- Zhou, S.; Jones, C.; Mieczkowski, P.; Swanstrom, R. Primer ID Validates Template Sampling Depth and Greatly Reduces the Error Rate of Next-Generation Sequencing of HIV-1 Genomic RNA Populations. J. Virol. 2015, 89, 8540–8555. [Google Scholar] [CrossRef]
- Kou, R.; Lam, H.; Duan, H.; Ye, L.; Jongkam, N.; Chen, W.; Zhang, S.; Li, S. Benefits and Challenges with Applying Unique Molecular Identifiers in Next Generation Sequencing to Detect Low Frequency Mutations. PLoS ONE 2016, 11, e0146638. [Google Scholar] [CrossRef]
- Lee, E.R.; Enns, E.; Parkin, N.; Brumme, C.J.; Casadella, M.; Howison, M.; Avila Rios, S.; Jennings, R.; Capina, R.; Marinier, E.; et al. Characterization and data assessment of next generation sequencing-based genotyping using existing HIV-1 drug resistance proficiency panels. In Proceedings of the XXVII International HIV Drug Resistance and Treatment Strategies Workshop, Johannesburg, South Africa, 22–23 October 2018. [Google Scholar]
- Ji, H.; Parkin, N.; Gao, F.; Denny, T.; Jennings, C.; Bremer, J.; Kantor, R. External quality assessment program for next generation sequencing-based HIV drug resistance testing: Logistical considerations. Viruses 2020. (accepted on 14 May 2020).. [Google Scholar]
- Gibson, R.M.; Meyer, A.M.; Winner, D.; Archer, J.; Feyertag, F.; Ruiz-Mateos, E.; Leal, M.; Robertson, D.L.; Schmotzer, C.L.; Quiñones-Mateu, M.E. Sensitive Deep-Sequencing-Based HIV-1 Genotyping Assay To Simultaneously Determine Susceptibility to Protease, Reverse Transcriptase, Integrase, and Maturation Inhibitors, as Well as HIV-1 Coreceptor Tropism. Antimicrob. Agents Chemother. 2014, 58, 2167–2185. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Yang, X.; Charlebois, P.; Macalalad, A.; Henn, M.R.; Zody, M.C. V-Phaser 2: Variant inference for viral populations. BMC Genom. 2013, 14, 674. [Google Scholar] [CrossRef] [PubMed]
- Zagordi, O.; Bhattacharya, A.; Eriksson, N.; Beerenwinkel, N. ShoRAH: Estimating the genetic diversity of a mixed sample from next-generation sequencing data. BMC Bioinform. 2011, 12, 119. [Google Scholar] [CrossRef] [PubMed]
- Verbist, B.M.P.; Thys, K.; Reumers, J.; Wetzels, Y.; Van Der Borght, K.; Talloen, W.; Aerssens, J.; Clement, L.; Thas, O. VirVarSeq: A low-frequency virus variant detection pipeline for Illumina sequencing using adaptive base-calling accuracy filtering. Bioinformatics 2015, 31, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Cassarino, T.G.; Frampton, D.; Sugar, R.; Charles, E.; Zisis Kozlakidid, P.K. High-throughput pipeline for de-novo assembly and drug resistance mutations identifcation from next-generation sequencing viral data of residual diagnostic samples. bioRxiv 2016. [Google Scholar] [CrossRef]
- Huber, M.; Metzner, K.J.; Geissberger, F.D.; Shah, C.; Leemann, C.; Klimkait, T.; Böni, J.; Trkola, A.; Zagordi, O. MinVar: A rapid and versatile tool for HIV-1 drug resistance genotyping by deep sequencing. J. Virol. Methods 2017, 240, 7–13. [Google Scholar] [CrossRef]
- Lee, E.R.; Parkin, N.; Jennings, C.; Brumme, C.J.; Enns, E.; Casadellà, M.; Howison, M.; Coetzer, M.; Avila-Rios, S.; Capina, R.; et al. Performance comparison of next generation sequencing analysis pipelines for HIV-1 drug resistance testing. Sci. Rep. 2020, 10, 1634. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Item | Sanger Sequencing | NGS |
|---|---|---|
| Extraction | Required | Required |
| RT-PCR | Required | Required |
| PCR | Required | Required |
| Specific sequencing primers | Multiple specific primers | Not required |
| Library preparation | Not required | Required |
| Sequencing reaction | Single reaction | Massive parallel clonal sequencing |
| Data output | One sequence per sample | Thousands of sequences per sample |
| DRM frequency detection threshold | ~20% | ~1% |
| Qualitative DRM detection | Enabled | Enabled |
| Quantitative DRM detection | Not applicable | Enabled |
| Sample Type | Advantages | Disadvantages |
|---|---|---|
| Donor Specimens (plasma, serum, DBS) | Real specimens | Unpredictable DRMs |
| Quasispecies population* | Unknown DRM frequency | |
| Limited supply | ||
| Complicated and expensive to acquire | ||
| Clinical Viral Isolates | Quasispecies population | Unpredictable DRMs |
| Known DRMs | Unknown DRM frequency | |
| Unlimited amount | Viral culture required | |
| Reusable | Expensive and complicated to prepare | |
| Minor DRMs may arise during viral culture | ||
| Infectious Molecular Clones | Culture of clone-derived isolates | Homogenous population with defined DRMs |
| Clone mixtures can be produced | Viral culture required | |
| Abundant Supply | Minor DRMs may arise during viral culture | |
| Known DRMs | ||
| Any DRMs in any genes | ||
| Any DRM frequency | ||
| Plasmids, Plasmid Mixtures, Synthetic RNA | Known sequences | Homogenous population |
| Known DRMs | Plasmids are DNA-based and are not suitable | |
| Any DRMs in any genes | for RNA related protocol validations | |
| Any DRM frequency | Plasmids underestimate PCR bias | |
| Ideal for low-frequency DRMs | ||
| Ideal for NGS standard | ||
| Ideal for monitoring systematic error | ||
| Economical | ||
| Unlimited amount | ||
| Stable for storage and transportation |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, E.R.; Gao, F.; Sandstrom, P.; Ji, H. External Quality Assessment for Next-Generation Sequencing-Based HIV Drug Resistance Testing: Unique Requirements and Challenges. Viruses 2020, 12, 550. https://doi.org/10.3390/v12050550
Lee ER, Gao F, Sandstrom P, Ji H. External Quality Assessment for Next-Generation Sequencing-Based HIV Drug Resistance Testing: Unique Requirements and Challenges. Viruses. 2020; 12(5):550. https://doi.org/10.3390/v12050550
Chicago/Turabian StyleLee, Emma R., Feng Gao, Paul Sandstrom, and Hezhao Ji. 2020. "External Quality Assessment for Next-Generation Sequencing-Based HIV Drug Resistance Testing: Unique Requirements and Challenges" Viruses 12, no. 5: 550. https://doi.org/10.3390/v12050550
APA StyleLee, E. R., Gao, F., Sandstrom, P., & Ji, H. (2020). External Quality Assessment for Next-Generation Sequencing-Based HIV Drug Resistance Testing: Unique Requirements and Challenges. Viruses, 12(5), 550. https://doi.org/10.3390/v12050550