Macrophage Cell-Cell Interactions Promoting HIV-1 Infection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
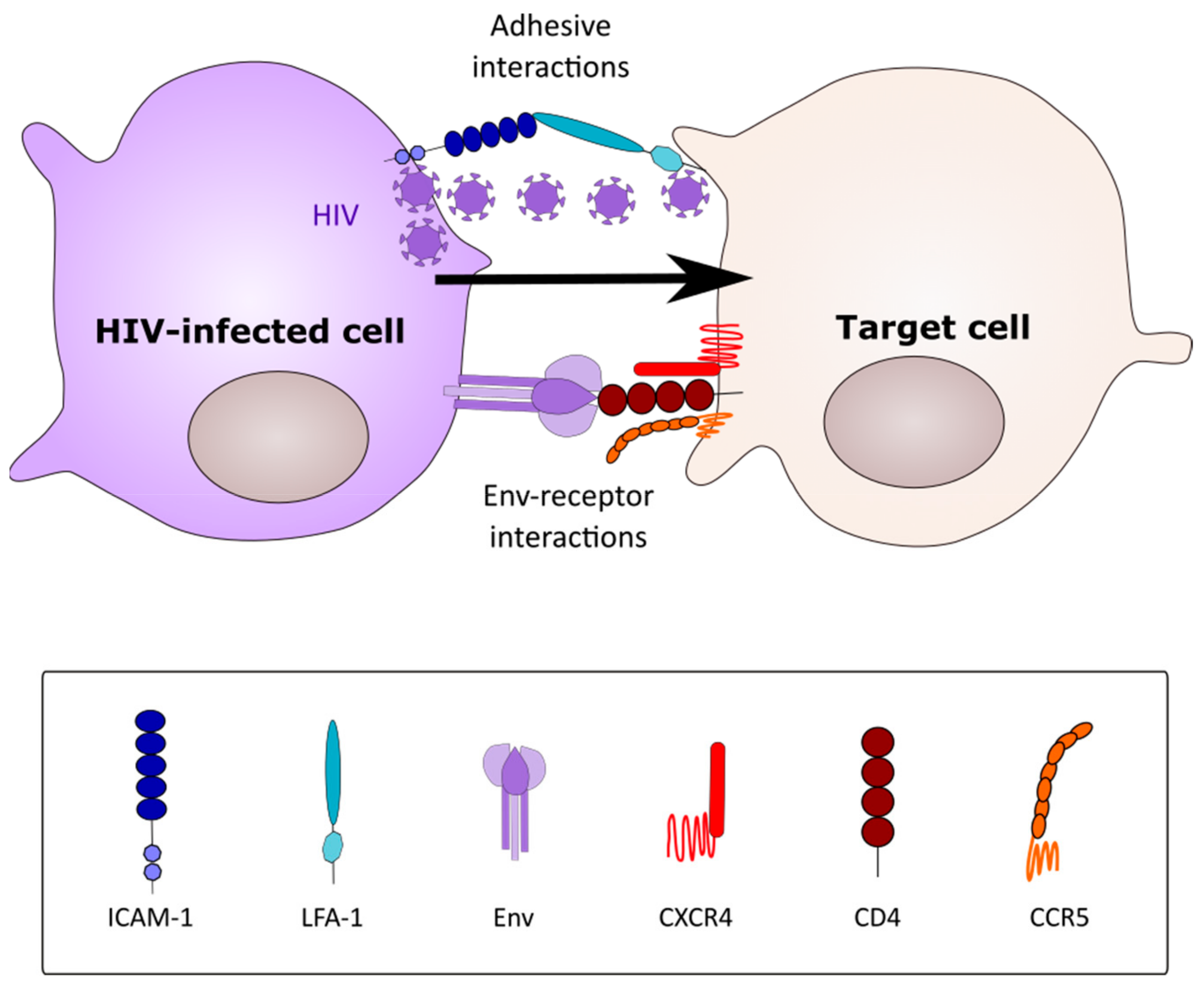
2. Modes of HIV-1 Cell-Cell Spread
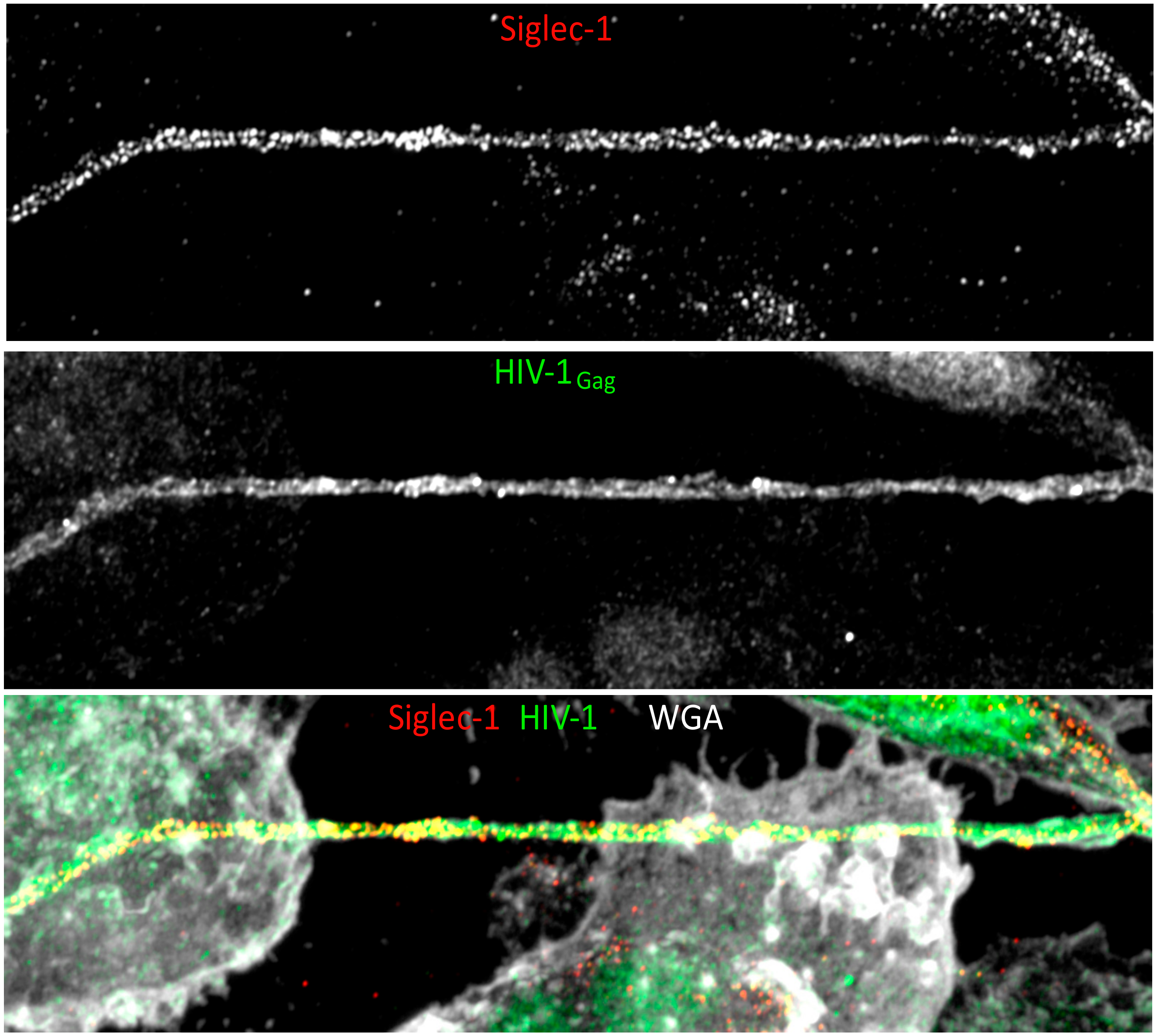
3. The Macrophage Virus-Containing Compartment
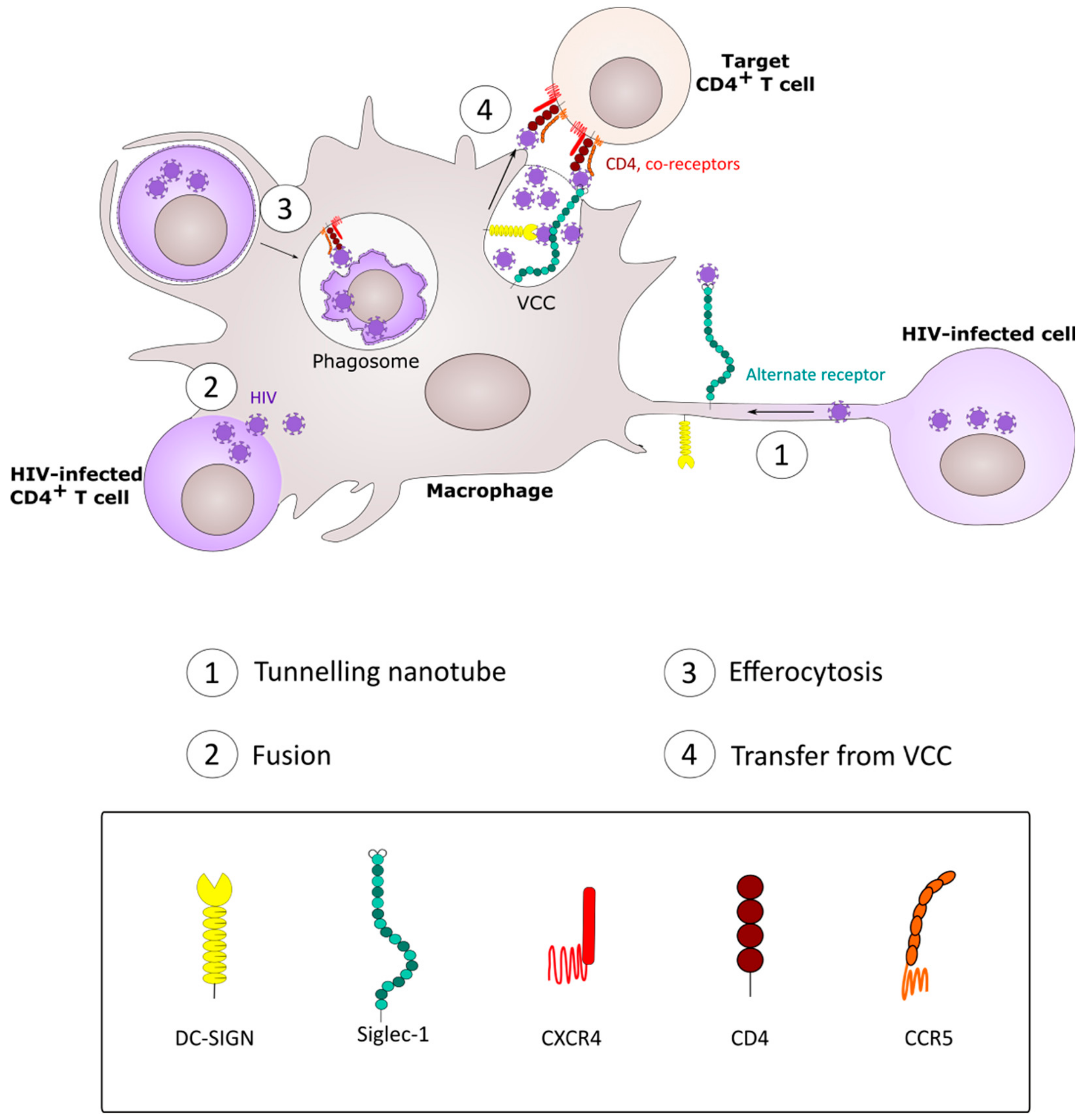
4. CD4+ T Cell Infection by Spread from Macrophages
5. Macrophage Infection by Phagocytic Uptake of HIV-1-Infected CD4+ T Cells.
6. Potential Consequences of Phagocytic Uptake of HIV-1 Infected Cells
7. Fusion between HIV-1-Infected CD4+ T Cells and Macrophages
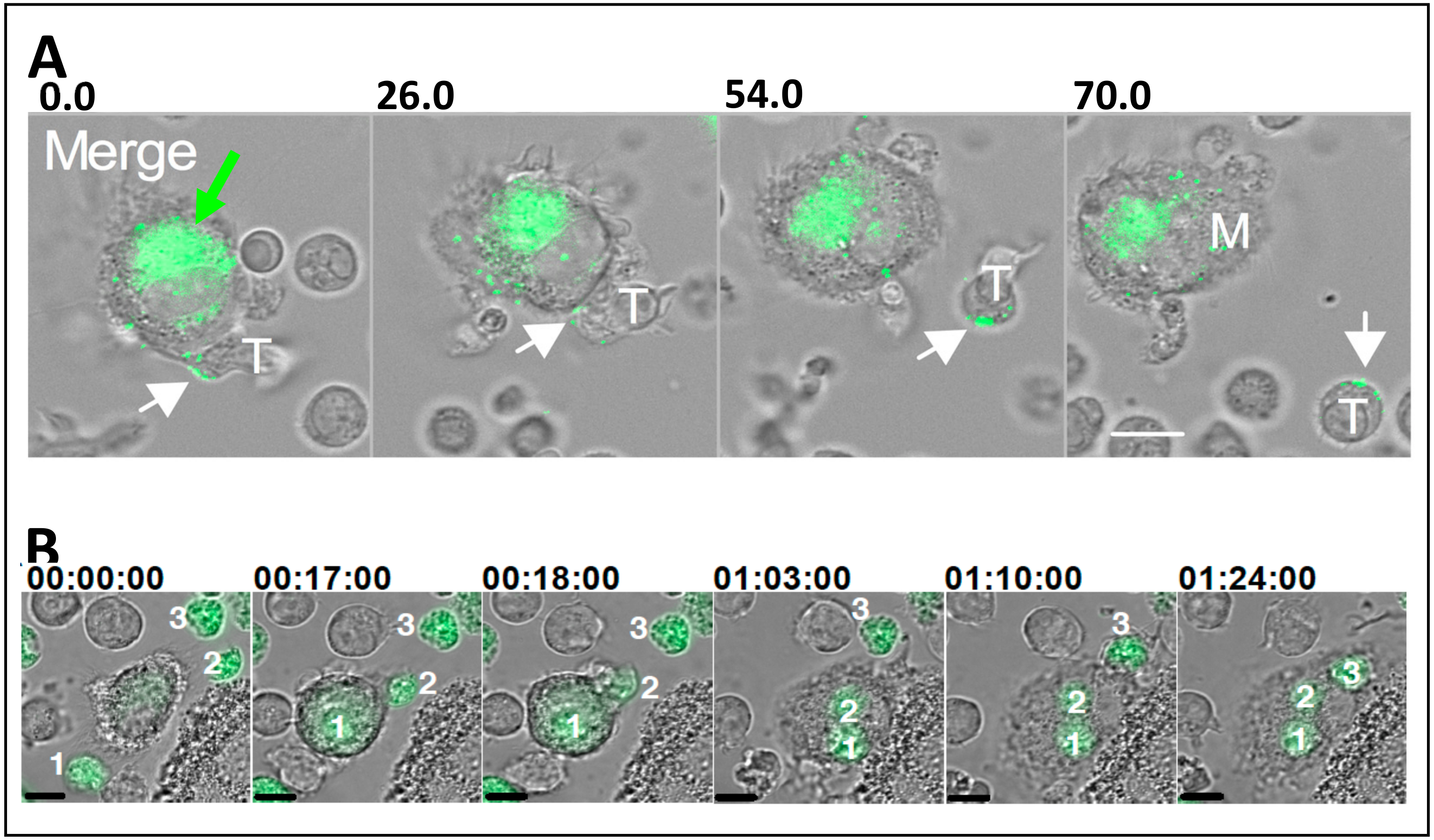
8. HIV-1 Spread between Macrophages: Implication of Tunneling Nanotubes.
9. Implication of Co-Infections in HIV-1 Intercellular Macrophage Interactions
10. In vivo Evidence for HIV-1 Cell-Cell Spread in Macrophage Infection and Dissemination
11. Discussion
Funding
Conflicts of Interest
References
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Sarantis, H.; Grinstein, S. Subversion of phagocytosis for pathogen survival. Cell Host Microbe 2012, 12, 419–431. [Google Scholar] [CrossRef]
- Weiss, G.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [CrossRef]
- Tan, J.; Sattentau, Q.J. The HIV-1-containing macrophage compartment: A perfect cellular niche? Trends Microbiol. 2013, 21, 405–412. [Google Scholar] [CrossRef]
- Mitchell, G.; Chen, C.; Portnoy, D.A. Strategies Used by Bacteria to Grow in Macrophages. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Chow, S.H.; Deo, P.; Naderer, T. Macrophage cell death in microbial infections. Cell. Microbiol. 2016, 18, 466–474. [Google Scholar] [CrossRef]
- Price, J.V.; Vance, R.E. The macrophage paradox. Immunity 2014, 41, 685–693. [Google Scholar] [CrossRef]
- Ren, Y.; Khan, F.A.; Pandupuspitasari, N.S.; Zhang, S. Immune Evasion Strategies of Pathogens in Macrophages: The Potential for Limiting Pathogen Transmission. Curr. Issues Mol. Biol. 2017, 21, 21–40. [Google Scholar]
- Chen, B. Molecular Mechanism of HIV-1 Entry. Trends Microbiol. 2019, 27, 878–891. [Google Scholar] [CrossRef]
- Li, G.H.; Henderson, L.; Nath, A. Astrocytes as an HIV Reservoir: Mechanism of HIV Infection. Curr. HIV Res. 2016, 14, 373–381. [Google Scholar] [CrossRef]
- Li, G.H.; Maric, D.; Major, E.O.; Nath, A. Productive HIV infection in astrocytes can be established via a non-classical mechanism. AIDS 2020. [Google Scholar] [CrossRef]
- Joseph, S.B.; Swanstrom, R. The evolution of HIV-1 entry phenotypes as a guide to changing target cells. J. Leukoc. Biol. 2018, 103, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.J.; Sattentau, Q.J. Viral determinants of HIV-1 macrophage tropism. Viruses 2011, 3, 2255–2279. [Google Scholar] [CrossRef]
- Bertram, K.M.; Tong, O.; Royle, C.; Turville, S.G.; Nasr, N.; Cunningham, A.L.; Harman, A.N. Manipulation of Mononuclear Phagocytes by HIV: Implications for Early Transmission Events. Front. Immunol. 2019, 10, 2263. [Google Scholar] [CrossRef]
- Salazar-Gonzalez, J.F.; Salazar, M.G.; Keele, B.F.; Learn, G.H.; Giorgi, E.E.; Li, H.; Decker, J.M.; Wang, S.; Baalwa, J.; Kraus, M.H.; et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 2009, 206, 1273–1289. [Google Scholar] [CrossRef]
- Ochsenbauer, C.; Edmonds, T.G.; Ding, H.; Keele, B.F.; Decker, J.; Salazar, M.G.; Salazar-Gonzalez, J.F.; Shattock, R.; Haynes, B.F.; Shaw, G.M.; et al. Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J. Virol. 2012, 86, 2715–2728. [Google Scholar] [CrossRef]
- Sattentau, Q. Avoiding the void: Cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 2008, 6, 815–826. [Google Scholar] [CrossRef]
- Martin, N.; Welsch, S.; Jolly, C.; Briggs, J.A.; Vaux, D.; Sattentau, Q.J. Virological synapse-mediated spread of human immunodeficiency virus type 1 between T cells is sensitive to entry inhibition. J. Virol. 2010, 84, 3516–3527. [Google Scholar] [CrossRef]
- Sourisseau, M.; Sol-Foulon, N.; Porrot, F.; Blanchet, F.; Schwartz, O. Inefficient human immunodeficiency virus replication in mobile lymphocytes. J. Virol. 2007, 81, 1000–1012. [Google Scholar] [CrossRef]
- Jolly, C.; Welsch, S.; Michor, S.; Sattentau, Q.J. The regulated secretory pathway in CD4(+) T cells contributes to human immunodeficiency virus type-1 cell-to-cell spread at the virological synapse. PLoS Pathog. 2011, 7, e1002226. [Google Scholar] [CrossRef]
- Jolly, C.; Kashefi, K.; Hollinshead, M.; Sattentau, Q.J. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 2004, 199, 283–293. [Google Scholar] [CrossRef]
- Jolly, C.; Booth, N.J.; Neil, S.J. Cell-cell spread of human immunodeficiency virus type 1 overcomes tetherin/BST-2-mediated restriction in T cells. J. Virol. 2010, 84, 12185–12199. [Google Scholar] [CrossRef]
- Zhong, P.; Agosto, L.M.; Ilinskaya, A.; Dorjbal, B.; Truong, R.; Derse, D.; Uchil, P.D.; Heidecker, G.; Mothes, W. Cell-to-cell transmission can overcome multiple donor and target cell barriers imposed on cell-free HIV. PLoS ONE 2013, 8, e53138. [Google Scholar] [CrossRef]
- Dufloo, J.; Bruel, T.; Schwartz, O. HIV-1 cell-to-cell transmission and broadly neutralizing antibodies. Retrovirology 2018, 15, 51. [Google Scholar] [CrossRef]
- Schiffner, T.; Sattentau, Q.J.; Duncan, C.J. Cell-to-cell spread of HIV-1 and evasion of neutralizing antibodies. Vaccine 2013, 31, 5789–5797. [Google Scholar] [CrossRef]
- Duncan, C.J.; Russell, R.A.; Sattentau, Q.J. High multiplicity HIV-1 cell-to-cell transmission from macrophages to CD4+ T cells limits antiretroviral efficacy. AIDS 2013, 27, 2201–2206. [Google Scholar] [CrossRef]
- Sigal, A.; Kim, J.T.; Balazs, A.B.; Dekel, E.; Mayo, A.; Milo, R.; Baltimore, D. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature 2011, 477, 95–98. [Google Scholar] [CrossRef]
- Agosto, L.M.; Zhong, P.; Munro, J.; Mothes, W. Highly active antiretroviral therapies are effective against HIV-1 cell-to-cell transmission. PLoS Pathog. 2014, 10, e1003982. [Google Scholar] [CrossRef]
- Jolly, C.; Sattentau, Q.J. Retroviral spread by induction of virological synapses. Traffic 2004, 5, 643–650. [Google Scholar] [CrossRef]
- Alvarez, R.A.; Barria, M.I.; Chen, B.K. Unique features of HIV-1 spread through T cell virological synapses. PLoS Pathog. 2014, 10, e1004513. [Google Scholar] [CrossRef]
- Sabatos, C.A.; Doh, J.; Chakravarti, S.; Friedman, R.S.; Pandurangi, P.G.; Tooley, A.J.; Krummel, M.F. A synaptic basis for paracrine interleukin-2 signaling during homotypic T cell interaction. Immunity 2008, 29, 238–248. [Google Scholar] [CrossRef]
- Pope, M.; Betjes, M.G.; Romani, N.; Hirmand, H.; Cameron, P.U.; Hoffman, L.; Gezelter, S.; Schuler, G.; Steinman, R.M. Conjugates of dendritic cells and memory T lymphocytes from skin facilitate productive infection with HIV-1. Cell 1994, 78, 389–398. [Google Scholar] [CrossRef]
- Izquierdo-Useros, N.; Lorizate, M.; McLaren, P.J.; Telenti, A.; Krausslich, H.G.; Martinez-Picado, J. HIV-1 capture and transmission by dendritic cells: The role of viral glycolipids and the cellular receptor Siglec-1. PLoS Pathog. 2014, 10, e1004146. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D. Dendritic Cells and HIV-1 Trans-Infection. Viruses 2010, 2, 1704–1717. [Google Scholar] [CrossRef] [PubMed]
- Sewald, X.; Ladinsky, M.S.; Uchil, P.D.; Beloor, J.; Pi, R.; Herrmann, C.; Motamedi, N.; Murooka, T.T.; Brehm, M.A.; Greiner, D.L.; et al. Retroviruses use CD169-mediated trans-infection of permissive lymphocytes to establish infection. Science 2015, 350, 563–567. [Google Scholar] [CrossRef]
- Yu, H.J.; Reuter, M.A.; McDonald, D. HIV traffics through a specialized, surface-accessible intracellular compartment during trans-infection of T cells by mature dendritic cells. PLoS Pathog. 2008, 4, e1000134. [Google Scholar] [CrossRef]
- Gendelman, H.E.; Baca, L.M.; Husayni, H.; Turpin, J.A.; Skillman, D.; Kalter, D.C.; Orenstein, J.M.; Hoover, D.L.; Meltzer, M.S. Macrophage-HIV interaction: Viral isolation and target cell tropism. AIDS 1990, 4, 221–228. [Google Scholar] [CrossRef]
- Orenstein, J.M.; Meltzer, M.S.; Phipps, T.; Gendelman, H.E. Cytoplasmic assembly and accumulation of human immunodeficiency virus types 1 and 2 in recombinant human colony-stimulating factor-1-treated human monocytes: An ultrastructural study. J. Virol. 1988, 62, 2578–2586. [Google Scholar] [CrossRef]
- Deneka, M.; Pelchen-Matthews, A.; Byland, R.; Ruiz-Mateos, E.; Marsh, M. In macrophages, HIV-1 assembles into an intracellular plasma membrane domain containing the tetraspanins CD81, CD9, and CD53. J. Cell. Biol. 2007, 177, 329–341. [Google Scholar] [CrossRef]
- Jouve, M.; Sol-Foulon, N.; Watson, S.; Schwartz, O.; Benaroch, P. HIV-1 buds and accumulates in “nonacidic” endosomes of macrophages. Cell Host Microbe 2007, 2, 85–95. [Google Scholar] [CrossRef]
- Welsch, S.; Keppler, O.T.; Habermann, A.; Allespach, I.; Krijnse-Locker, J.; Krausslich, H.G. HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog. 2007, 3, e36. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Groot, F.; Krausslich, H.G.; Keppler, O.T.; Sattentau, Q.J. Architecture and regulation of the HIV-1 assembly and holding compartment in macrophages. J. Virol. 2011, 85, 7922–7927. [Google Scholar] [CrossRef] [PubMed]
- Sharova, N.; Swingler, C.; Sharkey, M.; Stevenson, M. Macrophages archive HIV-1 virions for dissemination in trans. EMBO J. 2005, 24, 2481–2489. [Google Scholar] [CrossRef] [PubMed]
- Pelchen-Matthews, A.; Giese, S.; Mlcochova, P.; Turner, J.; Marsh, M. beta2 integrin adhesion complexes maintain the integrity of HIV-1 assembly compartments in primary macrophages. Traffic 2012, 13, 273–291. [Google Scholar] [CrossRef]
- Mlcochova, P.; Pelchen-Matthews, A.; Marsh, M. Organization and regulation of intracellular plasma membrane-connected HIV-1 assembly compartments in macrophages. BMC Biol. 2013, 11, 89. [Google Scholar] [CrossRef]
- Bennett, A.E.; Narayan, K.; Shi, D.; Hartnell, L.M.; Gousset, K.; He, H.; Lowekamp, B.C.; Yoo, T.S.; Bliss, D.; Freed, E.O.; et al. Ion-abrasion scanning electron microscopy reveals surface-connected tubular conduits in HIV-infected macrophages. PLoS Pathog. 2009, 5, e1000591. [Google Scholar] [CrossRef]
- Gaudin, R.; Berre, S.; Cunha de Alencar, B.; Decalf, J.; Schindler, M.; Gobert, F.X.; Jouve, M.; Benaroch, P. Dynamics of HIV-containing compartments in macrophages reveal sequestration of virions and transient surface connections. PLoS ONE 2013, 8, e69450. [Google Scholar] [CrossRef]
- Berre, S.; Gaudin, R.; Cunha de Alencar, B.; Desdouits, M.; Chabaud, M.; Naffakh, N.; Rabaza-Gairi, M.; Gobert, F.X.; Jouve, M.; Benaroch, P. CD36-specific antibodies block release of HIV-1 from infected primary macrophages and its transmission to T cells. J. Exp. Med. 2013, 210, 2523–2538. [Google Scholar] [CrossRef]
- Hammonds, J.E.; Beeman, N.; Ding, L.; Takushi, S.; Francis, A.C.; Wang, J.J.; Melikyan, G.B.; Spearman, P. Siglec-1 initiates formation of the virus-containing compartment and enhances macrophage-to-T cell transmission of HIV-1. PLoS Pathog. 2017, 13, e1006181. [Google Scholar] [CrossRef]
- Gousset, K.; Ablan, S.D.; Coren, L.V.; Ono, A.; Soheilian, F.; Nagashima, K.; Ott, D.E.; Freed, E.O. Real-time visualization of HIV-1 GAG trafficking in infected macrophages. PLoS Pathog. 2008, 4, e1000015. [Google Scholar] [CrossRef]
- Gaudin, R.; de Alencar, B.C.; Jouve, M.; Berre, S.; Le Bouder, E.; Schindler, M.; Varthaman, A.; Gobert, F.X.; Benaroch, P. Critical role for the kinesin KIF3A in the HIV life cycle in primary human macrophages. J. Cell. Biol. 2012, 199, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Ganor, Y.; Real, F.; Sennepin, A.; Dutertre, C.A.; Prevedel, L.; Xu, L.; Tudor, D.; Charmeteau, B.; Couedel-Courteille, A.; Marion, S.; et al. HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat. Microbiol. 2019, 4, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Carr, J.M.; Hocking, H.; Li, P.; Burrell, C.J. Rapid and efficient cell-to-cell transmission of human immunodeficiency virus infection from monocyte-derived macrophages to peripheral blood lymphocytes. Virology 1999, 265, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Swingler, S.; Brichacek, B.; Jacque, J.M.; Ulich, C.; Zhou, J.; Stevenson, M. HIV-1 Nef intersects the macrophage CD40L signalling pathway to promote resting-cell infection. Nature 2003, 424, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Groot, F.; Welsch, S.; Sattentau, Q.J. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 2008, 111, 4660–4663. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.J.; Williams, J.P.; Schiffner, T.; Gartner, K.; Ochsenbauer, C.; Kappes, J.; Russell, R.A.; Frater, J.; Sattentau, Q.J. High-multiplicity HIV-1 infection and neutralizing antibody evasion mediated by the macrophage-T cell virological synapse. J. Virol. 2014, 88, 2025–2034. [Google Scholar] [CrossRef]
- Herbein, G.; Gras, G.; Khan, K.A.; Abbas, W. Macrophage signaling in HIV-1 infection. Retrovirology 2010, 7, 34. [Google Scholar] [CrossRef]
- Kulpa, D.A.; Brehm, J.H.; Fromentin, R.; Cooper, A.; Cooper, C.; Ahlers, J.; Chomont, N.; Sekaly, R.P. The immunological synapse: The gateway to the HIV reservoir. Immunol. Rev. 2013, 254, 305–325. [Google Scholar] [CrossRef]
- Evans, V.A.; Kumar, N.; Filali, A.; Procopio, F.A.; Yegorov, O.; Goulet, J.P.; Saleh, S.; Haddad, E.K.; da Fonseca Pereira, C.; Ellenberg, P.C.; et al. Myeloid dendritic cells induce HIV-1 latency in non-proliferating CD4+ T cells. PLoS Pathog. 2013, 9, e1003799. [Google Scholar] [CrossRef]
- Kumar, N.A.; Cheong, K.; Powell, D.R.; da Fonseca Pereira, C.; Anderson, J.; Evans, V.A.; Lewin, S.R.; Cameron, P.U. The role of antigen presenting cells in the induction of HIV-1 latency in resting CD4(+) T-cells. Retrovirology 2015, 12, 76. [Google Scholar] [CrossRef]
- Kumar, N.A.; van der Sluis, R.M.; Mota, T.; Pascoe, R.; Evans, V.A.; Lewin, S.R.; Cameron, P.U. Myeloid Dendritic Cells Induce HIV Latency in Proliferating CD4(+) T Cells. J. Immunol. 2018, 201, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Cassol, E.; Cassetta, L.; Alfano, M.; Poli, G. Macrophage polarization and HIV-1 infection. J. Leukoc. Biol. 2010, 87, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Saidi, H.; Carbonneil, C.; Magri, G.; Eslahpazir, J.; Sekaly, R.P.; Belec, L. Differential modulation of CCR5-tropic human immunodeficiency virus-1 transfer from macrophages towards T cells under interleukin-4/interleukin-13 microenvironment. Hum. Immunol. 2010, 71, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cassol, E.; Cassetta, L.; Rizzi, C.; Gabuzda, D.; Alfano, M.; Poli, G. Dendritic cell-specific intercellular adhesion molecule-3 grabbing nonintegrin mediates HIV-1 infection of and transmission by M2a-polarized macrophages in vitro. AIDS 2013, 27, 707–716. [Google Scholar] [CrossRef]
- Baxter, A.E.; Russell, R.A.; Duncan, C.J.; Moore, M.D.; Willberg, C.B.; Pablos, J.L.; Finzi, A.; Kaufmann, D.E.; Ochsenbauer, C.; Kappes, J.C.; et al. Macrophage infection via selective capture of HIV-1-infected CD4+ T cells. Cell Host Microbe 2014, 16, 711–721. [Google Scholar] [CrossRef]
- Czuczman, M.A.; Fattouh, R.; van Rijn, J.M.; Canadien, V.; Osborne, S.; Muise, A.M.; Kuchroo, V.K.; Higgins, D.E.; Brumell, J.H. Listeria monocytogenes exploits efferocytosis to promote cell-to-cell spread. Nature 2014, 509, 230–234. [Google Scholar] [CrossRef]
- Davis, J.M.; Ramakrishnan, L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 2009, 136, 37–49. [Google Scholar] [CrossRef]
- Ritter, U.; Frischknecht, F.; van Zandbergen, G. Are neutrophils important host cells for Leishmania parasites? Trends Parasitol. 2009, 25, 505–510. [Google Scholar] [CrossRef]
- Karaji, N.; Sattentau, Q.J. Efferocytosis of Pathogen-Infected Cells. Front. Immunol. 2017, 8, 1863. [Google Scholar] [CrossRef]
- Russell, R.A.; Chojnacki, J.; Jones, D.M.; Johnson, E.; Do, T.; Eggeling, C.; Padilla-Parra, S.; Sattentau, Q.J. Astrocytes Resist HIV-1 Fusion but Engulf Infected Macrophage Material. Cell Rep. 2017, 18, 1473–1483. [Google Scholar] [CrossRef]
- Ma, M.; Geiger, J.D.; Nath, A. Characterization of a novel binding site for the human immunodeficiency virus type 1 envelope protein gp120 on human fetal astrocytes. J. Virol. 1994, 68, 6824–6828. [Google Scholar] [CrossRef] [PubMed]
- Sabri, F.; Tresoldi, E.; Di Stefano, M.; Polo, S.; Monaco, M.C.; Verani, A.; Fiore, J.R.; Lusso, P.; Major, E.; Chiodi, F.; et al. Nonproductive human immunodeficiency virus type 1 infection of human fetal astrocytes: Independence from CD4 and major chemokine receptors. Virology 1999, 264, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Willey, S.J.; Reeves, J.D.; Hudson, R.; Miyake, K.; Dejucq, N.; Schols, D.; De Clercq, E.; Bell, J.; McKnight, A.; Clapham, P.R. Identification of a subset of human immunodeficiency virus type 1 (HIV-1), HIV-2, and simian immunodeficiency virus strains able to exploit an alternative coreceptor on untransformed human brain and lymphoid cells. J. Virol. 2003, 77, 6138–6152. [Google Scholar] [CrossRef] [PubMed]
- Canki, M.; Thai, J.N.; Chao, W.; Ghorpade, A.; Potash, M.J.; Volsky, D.J. Highly productive infection with pseudotyped human immunodeficiency virus type 1 (HIV-1) indicates no intracellular restrictions to HIV-1 replication in primary human astrocytes. J. Virol. 2001, 75, 7925–7933. [Google Scholar] [CrossRef]
- Gray, L.R.; Tachedjian, G.; Ellett, A.M.; Roche, M.J.; Cheng, W.J.; Guillemin, G.J.; Brew, B.J.; Turville, S.G.; Wesselingh, S.L.; Gorry, P.R.; et al. The NRTIs lamivudine, stavudine and zidovudine have reduced HIV-1 inhibitory activity in astrocytes. PLoS ONE 2013, 8, e62196. [Google Scholar] [CrossRef]
- Chauhan, A. Enigma of HIV-1 latent infection in astrocytes: An in-vitro study using protein kinase C agonist as a latency reversing agent. Microbes Infect. 2015, 17, 651–659. [Google Scholar] [CrossRef]
- Chang, G.H.; Barbaro, N.M.; Pieper, R.O. Phosphatidylserine-dependent phagocytosis of apoptotic glioma cells by normal human microglia, astrocytes, and glioma cells. Neuro. Oncol. 2000, 2, 174–183. [Google Scholar] [CrossRef]
- Loov, C.; Hillered, L.; Ebendal, T.; Erlandsson, A. Engulfing astrocytes protect neurons from contact-induced apoptosis following injury. PLoS ONE 2012, 7, e33090. [Google Scholar] [CrossRef]
- Loov, C.; Mitchell, C.H.; Simonsson, M.; Erlandsson, A. Slow degradation in phagocytic astrocytes can be enhanced by lysosomal acidification. Glia 2015, 63, 1997–2009. [Google Scholar] [CrossRef]
- Gray, L.R.; Turville, S.G.; Hitchen, T.L.; Cheng, W.J.; Ellett, A.M.; Salimi, H.; Roche, M.J.; Wesselingh, S.L.; Gorry, P.R.; Churchill, M.J. HIV-1 entry and trans-infection of astrocytes involves CD81 vesicles. PLoS ONE 2014, 9, e90620. [Google Scholar] [CrossRef]
- Churchill, M.J.; Gorry, P.R.; Cowley, D.; Lal, L.; Sonza, S.; Purcell, D.F.; Thompson, K.A.; Gabuzda, D.; McArthur, J.C.; Pardo, C.A.; et al. Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J. Neurovirol. 2006, 12, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.J.; Wesselingh, S.L.; Cowley, D.; Pardo, C.A.; McArthur, J.C.; Brew, B.J.; Gorry, P.R. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann. Neurol. 2009, 66, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, L.; von Stockenstrom, S.; Faria, N.R.; Sinclair, E.; Bacchetti, P.; Killian, M.; Epling, L.; Tan, A.; Ho, T.; Lemey, P.; et al. The HIV-1 reservoir in eight patients on long-term suppressive antiretroviral therapy is stable with few genetic changes over time. Proc. Natl. Acad. Sci USA 2013, 110, E4987–E4996. [Google Scholar] [CrossRef] [PubMed]
- Sattentau, Q.J.; Stevenson, M. Macrophages and HIV-1: An Unhealthy Constellation. Cell Host Microbe 2016, 19, 304–310. [Google Scholar] [CrossRef]
- Ruggiero, E.; Bona, R.; Muratori, C.; Federico, M. Virological consequences of early events following cell-cell contact between human immunodeficiency virus type 1-infected and uninfected CD4+ cells. J. Virol. 2008, 82, 7773–7789. [Google Scholar] [CrossRef]
- Frankel, S.S.; Wenig, B.M.; Burke, A.P.; Mannan, P.; Thompson, L.D.; Abbondanzo, S.L.; Nelson, A.M.; Pope, M.; Steinman, R.M. Replication of HIV-1 in dendritic cell-derived syncytia at the mucosal surface of the adenoid. Science 1996, 272, 115–117. [Google Scholar] [CrossRef]
- Martinez-Mendez, D.; Rivera-Toledo, E.; Ortega, E.; Licona-Limon, I.; Huerta, L. Monocyte-lymphocyte fusion induced by the HIV-1 envelope generates functional heterokaryons with an activated monocyte-like phenotype. Exp. Cell. Res. 2017, 352, 9–19. [Google Scholar] [CrossRef]
- Bracq, L.; Xie, M.; Lambele, M.; Vu, L.T.; Matz, J.; Schmitt, A.; Delon, J.; Zhou, P.; Randriamampita, C.; Bouchet, J.; et al. T Cell-Macrophage Fusion Triggers Multinucleated Giant Cell Formation for HIV-1 Spreading. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Ono, A.; Freed, E.O. Cell-type-dependent targeting of human immunodeficiency virus type 1 assembly to the plasma membrane and the multivesicular body. J. Virol. 2004, 78, 1552–1563. [Google Scholar] [CrossRef]
- Xie, M.; Leroy, H.; Mascarau, R.; Woottum, M.; Dupont, M.; Ciccone, C.; Schmitt, A.; Raynaud-Messina, B.; Verollet, C.; Bouchet, J.; et al. Cell-to-Cell Spreading of HIV-1 in Myeloid Target Cells Escapes SAMHD1 Restriction. mBio 2019, 10. [Google Scholar] [CrossRef]
- Bell, J.E. The neuropathology of adult HIV infection. Rev. Neurol. (Paris) 1998, 154, 816–829. [Google Scholar] [PubMed]
- Mittal, R.; Karhu, E.; Wang, J.S.; Delgado, S.; Zukerman, R.; Mittal, J.; Jhaveri, V.M. Cell communication by tunneling nanotubes: Implications in disease and therapeutic applications. J. Cell. Physiol. 2019, 234, 1130–1146. [Google Scholar] [CrossRef] [PubMed]
- Dupont, M.; Souriant, S.; Lugo-Villarino, G.; Maridonneau-Parini, I.; Verollet, C. Tunneling Nanotubes: Intimate Communication between Myeloid Cells. Front. Immunol. 2018, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Sisakhtnezhad, S.; Khosravi, L. Emerging physiological and pathological implications of tunneling nanotubes formation between cells. Eur. J. Cell. Biol. 2015, 94, 429–443. [Google Scholar] [CrossRef] [PubMed]
- McCoy-Simandle, K.; Hanna, S.J.; Cox, D. Exosomes and nanotubes: Control of immune cell communication. Int. J. Biochem. Cell. Biol. 2016, 71, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Sowinski, S.; Jolly, C.; Berninghausen, O.; Purbhoo, M.A.; Chauveau, A.; Kohler, K.; Oddos, S.; Eissmann, P.; Brodsky, F.M.; Hopkins, C.; et al. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell. Biol. 2008, 10, 211–219. [Google Scholar] [CrossRef]
- Eugenin, E.A.; Gaskill, P.J.; Berman, J.W. Tunneling nanotubes (TNT) are induced by HIV-infection of macrophages: A potential mechanism for intercellular HIV trafficking. Cell. Immunol. 2009, 254, 142–148. [Google Scholar] [CrossRef]
- Kadiu, I.; Gendelman, H.E. Human immunodeficiency virus type 1 endocytic trafficking through macrophage bridging conduits facilitates spread of infection. J. Neuroimmune. Pharmacol. 2011, 6, 658–675. [Google Scholar] [CrossRef]
- Kadiu, I.; Gendelman, H.E. Macrophage bridging conduit trafficking of HIV-1 through the endoplasmic reticulum and Golgi network. J. Proteome Res. 2011, 10, 3225–3238. [Google Scholar] [CrossRef]
- Okafo, G.; Prevedel, L.; Eugenin, E. Tunneling nanotubes (TNT) mediate long-range gap junctional communication: Implications for HIV cell to cell spread. Sci. Rep. 2017, 7, 16660. [Google Scholar] [CrossRef]
- Hashimoto, M.; Bhuyan, F.; Hiyoshi, M.; Noyori, O.; Nasser, H.; Miyazaki, M.; Saito, T.; Kondoh, Y.; Osada, H.; Kimura, S.; et al. Potential Role of the Formation of Tunneling Nanotubes in HIV-1 Spread in Macrophages. J. Immunol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Uhl, J.; Gujarathi, S.; Waheed, A.A.; Gordon, A.; Freed, E.O.; Gousset, K. Myosin-X is essential to the intercellular spread of HIV-1 Nef through tunneling nanotubes. J. Cell. Commun. Signal. 2019, 13, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Hase, K.; Kimura, S.; Takatsu, H.; Ohmae, M.; Kawano, S.; Kitamura, H.; Ito, M.; Watarai, H.; Hazelett, C.C.; Yeaman, C.; et al. M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat. Cell. Biol. 2009, 11, 1427–1432. [Google Scholar] [CrossRef] [PubMed]
- Imle, A.; Abraham, L.; Tsopoulidis, N.; Hoflack, B.; Saksela, K.; Fackler, O.T. Association with PAK2 Enables Functional Interactions of Lentiviral Nef Proteins with the Exocyst Complex. mBio 2015, 6, e01309–e01315. [Google Scholar] [CrossRef]
- Mukerji, J.; Olivieri, K.C.; Misra, V.; Agopian, K.A.; Gabuzda, D. Proteomic analysis of HIV-1 Nef cellular binding partners reveals a role for exocyst complex proteins in mediating enhancement of intercellular nanotube formation. Retrovirology 2012, 9, 33. [Google Scholar] [CrossRef]
- Xu, W.; Santini, P.A.; Sullivan, J.S.; He, B.; Shan, M.; Ball, S.C.; Dyer, W.B.; Ketas, T.J.; Chadburn, A.; Cohen-Gould, L.; et al. HIV-1 evades virus-specific IgG2 and IgA responses by targeting systemic and intestinal B cells via long-range intercellular conduits. Nat. Immunol. 2009, 10, 1008–1017. [Google Scholar] [CrossRef]
- Diou, J.; Tardif, M.R.; Barat, C.; Tremblay, M.J. Malaria hemozoin modulates susceptibility of immature monocyte-derived dendritic cells to HIV-1 infection by inducing a mature-like phenotype. Cell. Microbiol. 2010, 12, 615–625. [Google Scholar] [CrossRef]
- Diou, J.; Tardif, M.R.; Barat, C.; Tremblay, M.J. Dendritic cells derived from hemozoin-loaded monocytes display a partial maturation phenotype that promotes HIV-1 trans-infection of CD4+ T cells and virus replication. J. Immunol. 2010, 184, 2899–2907. [Google Scholar] [CrossRef]
- Nakata, K.; Rom, W.N.; Honda, Y.; Condos, R.; Kanegasaki, S.; Cao, Y.; Weiden, M. Mycobacterium tuberculosis enhances human immunodeficiency virus-1 replication in the lung. Am. J. Respir. Crit. Care Med. 1997, 155, 996–1003. [Google Scholar] [CrossRef]
- Goletti, D.; Weissman, D.; Jackson, R.W.; Graham, N.M.; Vlahov, D.; Klein, R.S.; Munsiff, S.S.; Ortona, L.; Cauda, R.; Fauci, A.S. Effect of Mycobacterium tuberculosis on HIV replication. Role of immune activation. J. Immunol. 1996, 157, 1271–1278. [Google Scholar]
- Toossi, Z.; Johnson, J.L.; Kanost, R.A.; Wu, M.; Luzze, H.; Peters, P.; Okwera, A.; Joloba, M.; Mugyenyi, P.; Mugerwa, R.D.; et al. Increased replication of HIV-1 at sites of Mycobacterium tuberculosis infection: Potential mechanisms of viral activation. J. Acquir. Immune. Defic. Syndr. 2001, 28, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lawn, S.D.; Pisell, T.L.; Hirsch, C.S.; Wu, M.; Butera, S.T.; Toossi, Z. Anatomically compartmentalized human immunodeficiency virus replication in HLA-DR+ cells and CD14+ macrophages at the site of pleural tuberculosis coinfection. J. Infect. Dis. 2001, 184, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Duh, E.J.; Maury, W.J.; Folks, T.M.; Fauci, A.S.; Rabson, A.B. Tumor necrosis factor alpha activates human immunodeficiency virus type 1 through induction of nuclear factor binding to the NF-kappa B sites in the long terminal repeat. Proc. Natl. Acad. Sci USA 1989, 86, 5974–5978. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, Y.; Hoshino, S.; Gold, J.A.; Raju, B.; Prabhakar, S.; Pine, R.; Rom, W.N.; Nakata, K.; Weiden, M. Mechanisms of polymorphonuclear neutrophil-mediated induction of HIV-1 replication in macrophages during pulmonary tuberculosis. J. Infect. Dis. 2007, 195, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Souriant, S.; Balboa, L.; Dupont, M.; Pingris, K.; Kviatcovsky, D.; Cougoule, C.; Lastrucci, C.; Bah, A.; Gasser, R.; Poincloux, R.; et al. Tuberculosis Exacerbates HIV-1 Infection through IL-10/STAT3-Dependent Tunneling Nanotube Formation in Macrophages. Cell Rep. 2019, 26, 3586–3599.e7. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Picado, J.; McLaren, P.J.; Telenti, A.; Izquierdo-Useros, N. Retroviruses As Myeloid Cell Riders: What Natural Human Siglec-1 “Knockouts” Tell Us About Pathogenesis. Front. Immunol. 2017, 8, 1593. [Google Scholar] [CrossRef] [PubMed]
- Dupont, M.; Souriant, S.; Balboa, L.; Vu Manh, T.P.; Pingris, K.; Rousset, S.; Cougoule, C.; Rombouts, Y.; Poincloux, R.; Ben Neji, M.; et al. Tuberculosis-associated IFN-I induces Siglec-1 on tunneling nanotubes and favors HIV-1 spread in macrophages. Elife 2020, 9. [Google Scholar] [CrossRef]
- Ugolini, S.; Mondor, I.; Sattentau, Q.J. HIV-1 attachment: Another look. Trends Microbiol. 1999, 7, 144–149. [Google Scholar] [CrossRef]
- Lastrucci, C.; Benard, A.; Balboa, L.; Pingris, K.; Souriant, S.; Poincloux, R.; Al Saati, T.; Rasolofo, V.; Gonzalez-Montaner, P.; Inwentarz, S.; et al. Tuberculosis is associated with expansion of a motile, permissive and immunomodulatory CD16(+) monocyte population via the IL-10/STAT3 axis. Cell Res. 2015, 25, 1333–1351. [Google Scholar] [CrossRef]
- Lai, J.; Bernhard, O.K.; Turville, S.G.; Harman, A.N.; Wilkinson, J.; Cunningham, A.L. Oligomerization of the macrophage mannose receptor enhances gp120-mediated binding of HIV-1. J. Biol. Chem. 2009, 284, 11027–11038. [Google Scholar] [CrossRef]
- Rosas-Taraco, A.G.; Arce-Mendoza, A.Y.; Caballero-Olin, G.; Salinas-Carmona, M.C. Mycobacterium tuberculosis upregulates coreceptors CCR5 and CXCR4 while HIV modulates CD14 favoring concurrent infection. AIDS Res. Hum. Retrovir. 2006, 22, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Murooka, T.T.; Deruaz, M.; Marangoni, F.; Vrbanac, V.D.; Seung, E.; von Andrian, U.H.; Tager, A.M.; Luster, A.D.; Mempel, T.R. HIV-infected T cells are migratory vehicles for viral dissemination. Nature 2012, 490, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Calantone, N.; Wu, F.; Klase, Z.; Deleage, C.; Perkins, M.; Matsuda, K.; Thompson, E.A.; Ortiz, A.M.; Vinton, C.L.; Ourmanov, I.; et al. Tissue myeloid cells in SIV-infected primates acquire viral DNA through phagocytosis of infected T cells. Immunity 2014, 41, 493–502. [Google Scholar] [CrossRef] [PubMed]
- DiNapoli, S.R.; Ortiz, A.M.; Wu, F.; Matsuda, K.; Twigg, H.L., 3rd; Hirsch, V.M.; Knox, K.; Brenchley, J.M. Tissue-resident macrophages can contain replication-competent virus in antiretroviral-naive, SIV-infected Asian macaques. JCI Insight 2017, 2, e91214. [Google Scholar] [CrossRef]
- Orenstein, J.M. In vivo cytolysis and fusion of human immunodeficiency virus type 1-infected lymphocytes in lymphoid tissue. J. Infect. Dis. 2000, 182, 338–342. [Google Scholar] [CrossRef]
- Haldar, M.; Murphy, K.M. Origin, development, and homeostasis of tissue-resident macrophages. Immunol. Rev. 2014, 262, 25–35. [Google Scholar] [CrossRef]
- Amit, I.; Winter, D.R.; Jung, S. The role of the local environment and epigenetics in shaping macrophage identity and their effect on tissue homeostasis. Nat. Immunol. 2016, 17, 18–25. [Google Scholar] [CrossRef]
- Lavin, Y.; Mortha, A.; Rahman, A.; Merad, M. Regulation of macrophage development and function in peripheral tissues. Nat. Rev. Immunol. 2015, 15, 731–744. [Google Scholar] [CrossRef]
- Lee, C.Z.W.; Kozaki, T.; Ginhoux, F. Studying tissue macrophages in vitro: Are iPSC-derived cells the answer? Nat. Rev. Immunol. 2018, 18, 716–725. [Google Scholar] [CrossRef]
- Verollet, C.; Souriant, S.; Bonnaud, E.; Jolicoeur, P.; Raynaud-Messina, B.; Kinnaer, C.; Fourquaux, I.; Imle, A.; Benichou, S.; Fackler, O.T.; et al. HIV-1 reprograms the migration of macrophages. Blood 2015, 125, 1611–1622. [Google Scholar] [CrossRef]
- Van Goethem, E.; Poincloux, R.; Gauffre, F.; Maridonneau-Parini, I.; Le Cabec, V. Matrix architecture dictates three-dimensional migration modes of human macrophages: Differential involvement of proteases and podosome-like structures. J. Immunol. 2010, 184, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, T.M. Discovering Macrophage Functions Using In Vivo Optical Imaging Techniques. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dupont, M.; Sattentau, Q.J. Macrophage Cell-Cell Interactions Promoting HIV-1 Infection. Viruses 2020, 12, 492. https://doi.org/10.3390/v12050492
Dupont M, Sattentau QJ. Macrophage Cell-Cell Interactions Promoting HIV-1 Infection. Viruses. 2020; 12(5):492. https://doi.org/10.3390/v12050492
Chicago/Turabian StyleDupont, Maeva, and Quentin James Sattentau. 2020. "Macrophage Cell-Cell Interactions Promoting HIV-1 Infection" Viruses 12, no. 5: 492. https://doi.org/10.3390/v12050492
APA StyleDupont, M., & Sattentau, Q. J. (2020). Macrophage Cell-Cell Interactions Promoting HIV-1 Infection. Viruses, 12(5), 492. https://doi.org/10.3390/v12050492