Pantoea agglomerans-Infecting Bacteriophage vB_PagS_AAS21: A Cold-Adapted Virus Representing a Novel Genus within the Family Siphoviridae

 , , ,
, , ,  and
and 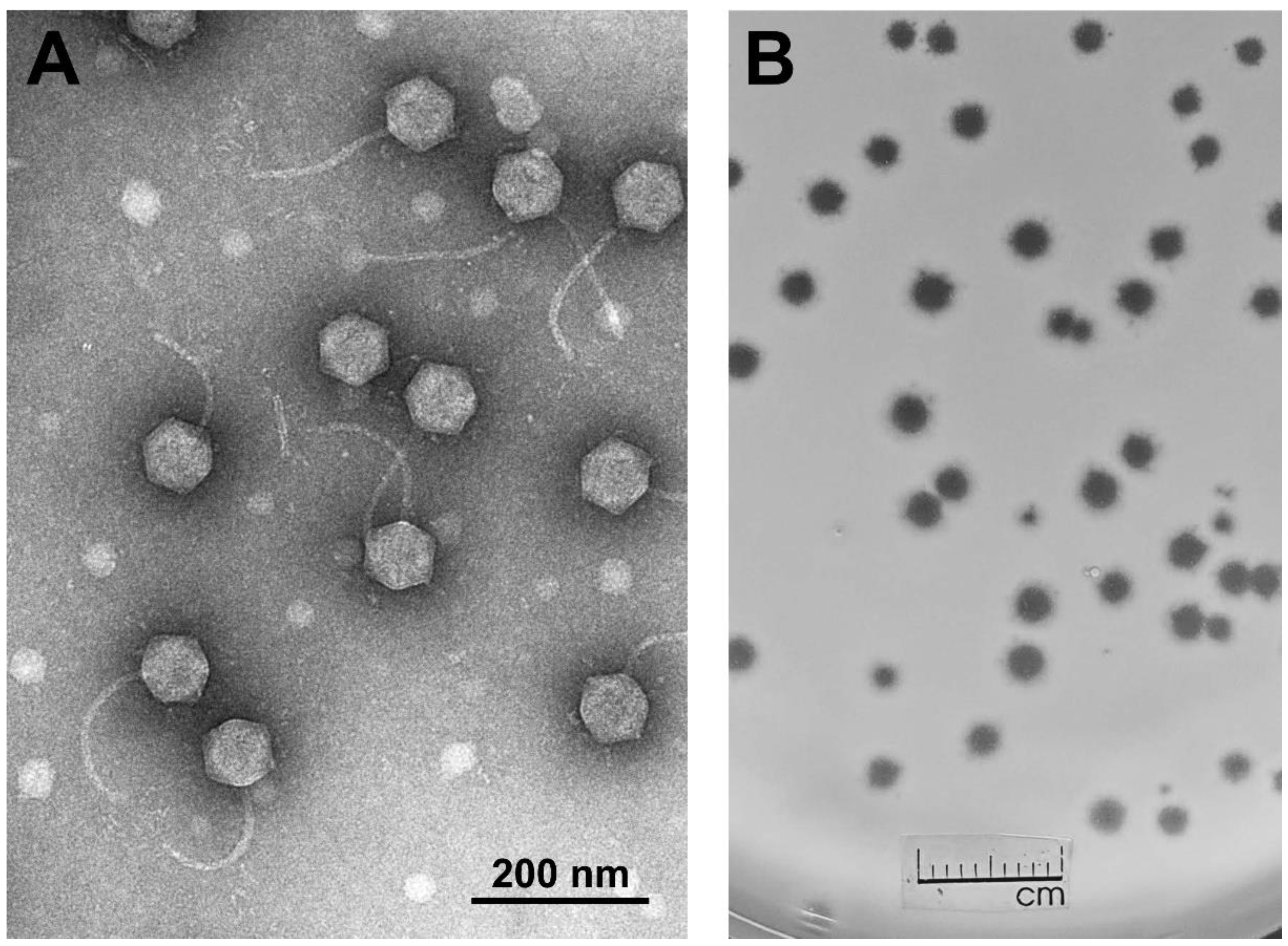{kind=link}
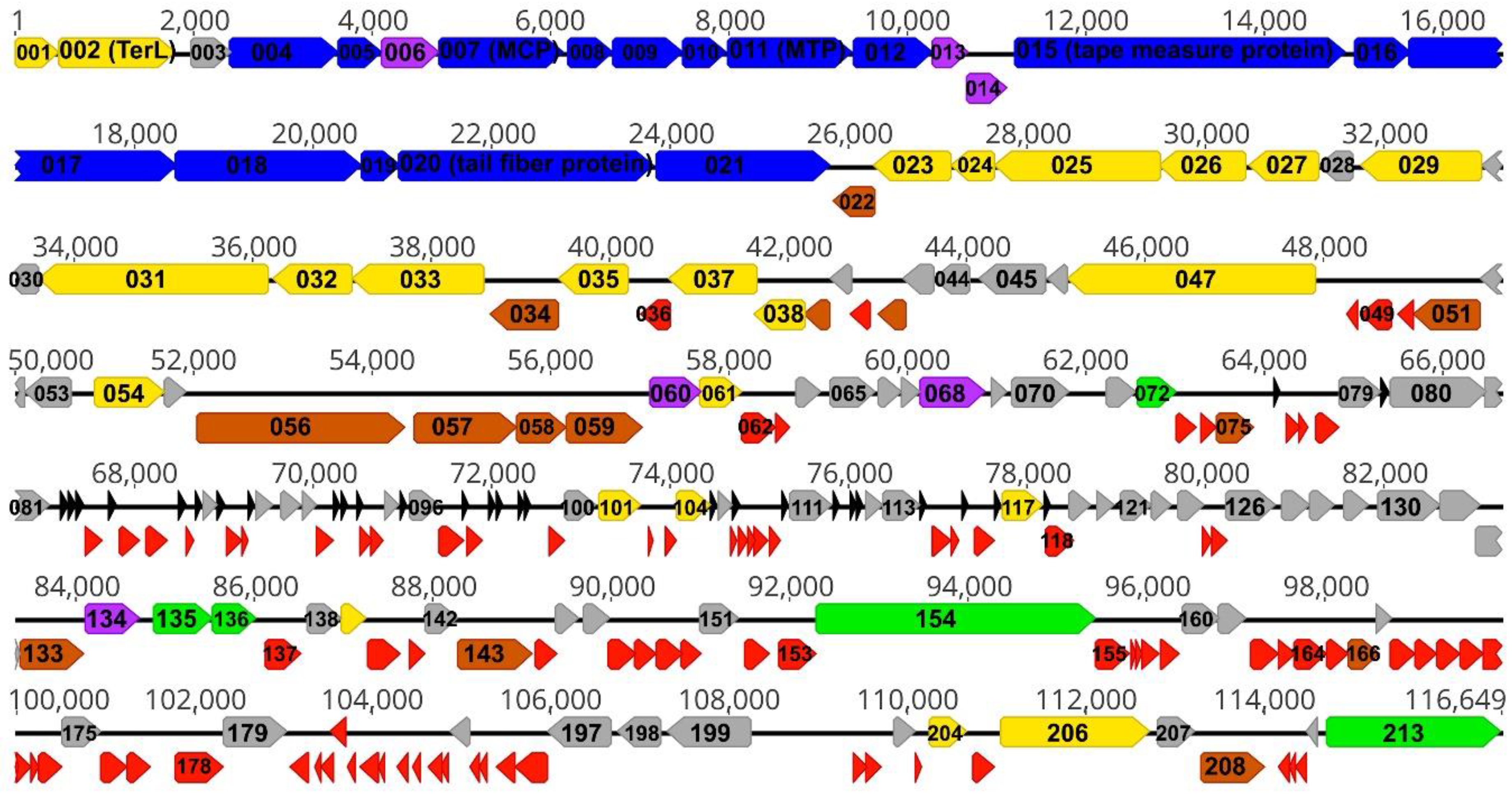{kind=link}
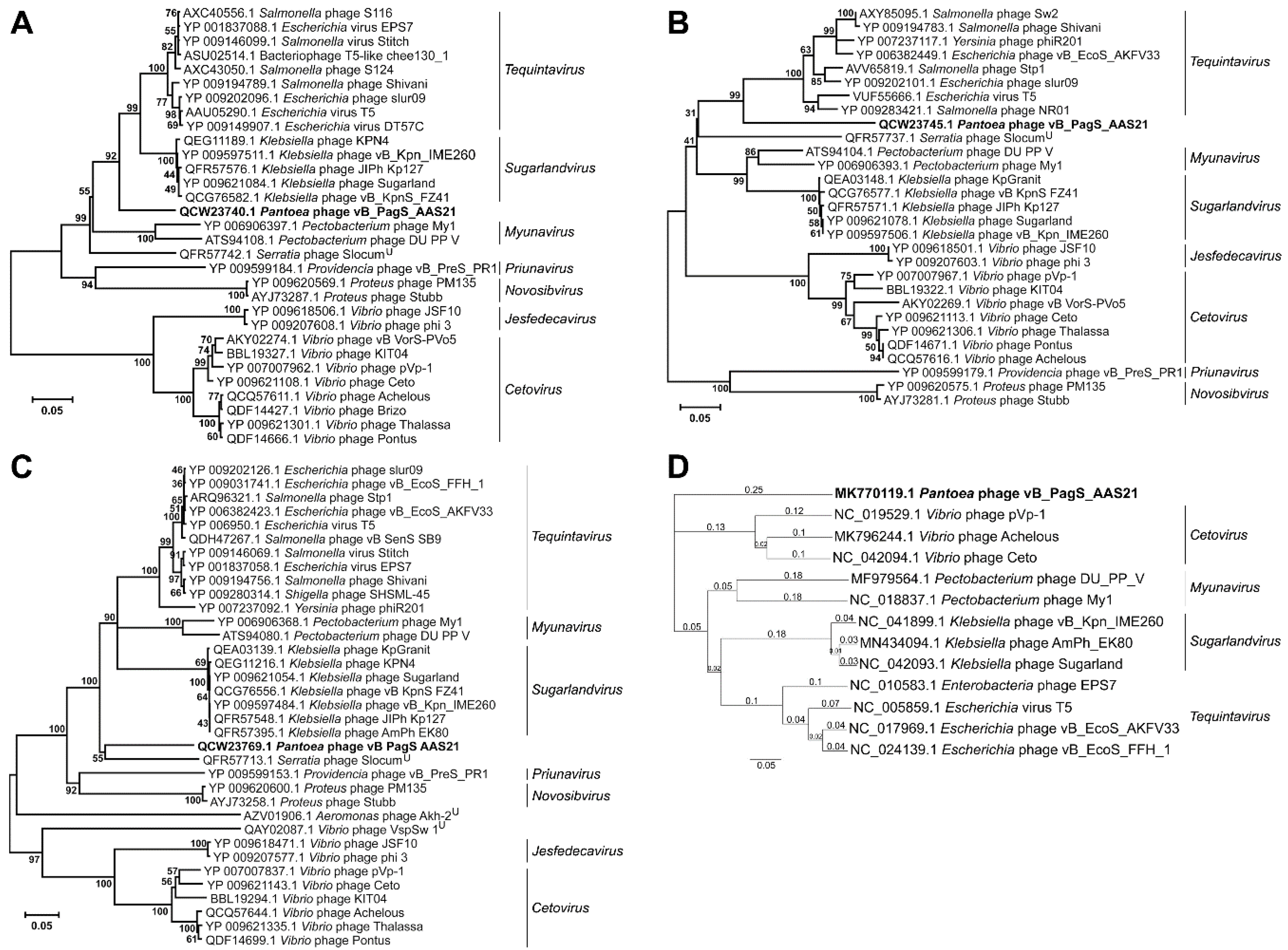{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Walterson, A.M.; Stavrinides, J. Pantoea: Insights into a highly versatile and diverse genus within the Enterobacteriaceae. FEMS Microbiol. Rev. 2015, 39, 968–984. [Google Scholar] [CrossRef] [PubMed]
- Dutkiewicz, J.; Mackiewicz, B.; Lemieszek, M.; Golec, M.; Milanowski, J. Pantoea agglomerans—A mysterious bacterium of evil and good. Part I. Deleterious effects: Dust-borne endotoxins and allergens—Focus on cotton dust. Ann. Agric. Environ. Med. 2015, 22, 576–588. [Google Scholar] [CrossRef] [PubMed]
- Meczker, K.; Domotor, D.; Vass, J.; Rakhely, G.; Schneider, G.; Kovacs, T. The genome of the Erwinia amylovora phage PhiEaH1 reveals greater diversity and broadens the applicability of phages for the treatment of fire blight. FEMS Microbiol. Lett. 2014, 350, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Moye, Z.; Woolston, J.; Sulakvelidze, A. Bacteriophage applications for food production and processing. Viruses 2018, 10, 205. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Ceyssens, P.J.; Dunon, V.; Ackermann, H.W.; Van Vaerenbergh, J.; Maes, M.; De Proft, M.; Lavigne, R. Bacteriophages LIME-light and LIMEzero of Pantoea agglomerans, belonging to the “phiKMV-like viruses”. Appl. Environ. Microbiol. 2011, 77, 3443–3450. [Google Scholar] [CrossRef]
- Šimoliūnas, E.; Šimoliūnienė, M.; Kaliniene, L.; Zajančkauskaitė, A.; Skapas, M.; Meškys, R.; Kaupinis, A.; Valius, M.; Truncaitė, L. Pantoea bacteriophage vB_PagS_Vid5: A low-temperature siphovirus that harbors a cluster of genes involved in the biosynthesis of archaeosin. Viruses 2018, 10, 583. [Google Scholar] [CrossRef]
- Thompson, D.W.; Casjens, S.R.; Sharma, R.; Grose, J.H. Genomic comparison of 60 completely sequenced bacteriophages that infect Erwinia and/or Pantoea bacteria. Virology 2019, 535, 59–73. [Google Scholar] [CrossRef]
- McDougall, D.L.; Soutar, C.D.; Perry, B.J.; Brown, C.; Alexander, D.; Yost, C.K.; Stavrinides, J. Isolation and characterization of vB_PagP-SK1, a T7-like phage infecting Pantoea agglomerans. J. PHAGE 2020, 1, 45–56. [Google Scholar] [CrossRef]
- Kropinski, A.M. Measurement of the rate of attachment of bacteriophage to cells. In Bacteriophages: Methods and protocols; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: New York, NY, USA, 2009; pp. 151–155. [Google Scholar]
- Šimoliūnas, E.; Kaliniene, L.; Truncaitė, L.; Zajančkauskaitė, A.; Staniulis, J.; Kaupinis, J.; Ger, M.; Valius, M.; Meškys, R. Klebsiella phage vB_KleM-RaK2—A giant singleton virus of the family Myoviridae. PLoS ONE 2013, 8, e60717. [Google Scholar] [CrossRef]
- Carlson, K.; Miller, E. Experiments in T4 genetics. In Bacteriophage T4; Karam, J.D., Ed.; ASM Press: Washington, DC, USA, 1994; pp. 419–483. [Google Scholar]
- Šimoliūnas, E.; Kaliniene, L.; Stasilo, M.; Truncaitė, L.; Zajančkauskaitė, A.; Staniulis, J.; Nainys, J.; Kaupinis, A.; Valius, M.; Meškys, R. Isolation and characterization of vB_ArS-ArV2—First Arthrobacter sp. infecting bacteriophage with completely sequenced genome. PLoS ONE 2014, 9, e111230. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Alva, V.; Nam, S.Z.; Söding, J.; Lupas, A.N. The MPI bioinformatics toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res. 2016, 44, W410–W415. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kubler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A completely reimplemented mpi bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Bao, Y.; Chetvernin, V.; Tatusova, T. Improvements to pairwise sequence comparison (PASC): A genome-based web tool for virus classification. Arch. Virol. 2014, 159, 3293–3304. [Google Scholar] [CrossRef]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Seeley, N.D.; Primrose, S.B. The effect of temperature on the ecology of aquatic bacteriophages. J. Gen. Virol. 1980, 46, 87–95. [Google Scholar] [CrossRef]
- Kaliniene, L.; Truncaite, L.; Šimoliūnas, E.; Zajančkauskaitė, A.; Vilkaitytė, M.; Kaupinis, A.; Skapas, M.; Meškys, R. Molecular analysis of the low-temperature Escherichia coli phage vB_EcoS_NBD2. Arch. Virol. 2017, 163, 105–114. [Google Scholar] [CrossRef]
- Kaliniene, L.; Klausa, V.; Truncaite, L. Low-temperature T4-like coliphages vB_EcoM-VR5, vB_EcoM-VR7 and vB_EcoM-VR20. Arch. Virol. 2010, 155, 871–880. [Google Scholar] [CrossRef]
- Jurczak-Kurek, A.; Gasior, T.; Nejman-Falenczyk, B.; Bloch, S.; Dydecka, A.; Topka, G.; Necel, A.; Jakubowska-Deredas, M.; Narajczyk, M.; Richert, M.; et al. Biodiversity of bacteriophages: Morphological and biological properties of a large group of phages isolated from urban sewage. Sci. Rep. 2016, 6, 34338. [Google Scholar] [CrossRef]
- Morgado, S.; Vicente, A.C. Global in-silico scenario of tRNA genes and their organization in virus genomes. Viruses 2019, 11, 180. [Google Scholar] [CrossRef]
- Pope, W.H.; Anders, K.R.; Baird, M.; Bowman, C.A.; Boyle, M.M.; Broussard, G.W.; Chow, T.; Clase, K.L.; Cooper, S.; Cornely, K.A.; et al. Cluster M mycobacteriophages Bongo, PegLeg, and Rey with unusually large repertoires of tRNA isotypes. J. Virol. 2014, 88, 2461–2480. [Google Scholar] [CrossRef][Green Version]
- Bailly-Bechet, M.; Vergassola, M.; Rocha, E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007, 17, 1486–1495. [Google Scholar] [CrossRef]
- Delesalle, V.A.; Tanke, N.T.; Vill, A.C.; Krukonis, G.P. Testing hypotheses for the presence of tRNA genes in mycobacteriophage genomes. Bacteriophage 2016, 6, e1219441. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. GtRNAdb 2.0: An expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016, 44, D184–D189. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Cowan, D.A. Using signature genes as tools to assess environmental viral ecology and diversity. Appl. Environ. Microbiol. 2014, 80, 4470–4480. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Brister, J.R. How to name and classify your phage: An informal guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šimoliūnienė, M.; Truncaitė, L.; Petrauskaitė, E.; Zajančkauskaitė, A.; Meškys, R.; Skapas, M.; Kaupinis, A.; Valius, M.; Šimoliūnas, E. Pantoea agglomerans-Infecting Bacteriophage vB_PagS_AAS21: A Cold-Adapted Virus Representing a Novel Genus within the Family Siphoviridae. Viruses 2020, 12, 479. https://doi.org/10.3390/v12040479
Šimoliūnienė M, Truncaitė L, Petrauskaitė E, Zajančkauskaitė A, Meškys R, Skapas M, Kaupinis A, Valius M, Šimoliūnas E. Pantoea agglomerans-Infecting Bacteriophage vB_PagS_AAS21: A Cold-Adapted Virus Representing a Novel Genus within the Family Siphoviridae. Viruses. 2020; 12(4):479. https://doi.org/10.3390/v12040479
Chicago/Turabian StyleŠimoliūnienė, Monika, Lidija Truncaitė, Emilija Petrauskaitė, Aurelija Zajančkauskaitė, Rolandas Meškys, Martynas Skapas, Algirdas Kaupinis, Mindaugas Valius, and Eugenijus Šimoliūnas. 2020. "Pantoea agglomerans-Infecting Bacteriophage vB_PagS_AAS21: A Cold-Adapted Virus Representing a Novel Genus within the Family Siphoviridae" Viruses 12, no. 4: 479. https://doi.org/10.3390/v12040479
APA StyleŠimoliūnienė, M., Truncaitė, L., Petrauskaitė, E., Zajančkauskaitė, A., Meškys, R., Skapas, M., Kaupinis, A., Valius, M., & Šimoliūnas, E. (2020). Pantoea agglomerans-Infecting Bacteriophage vB_PagS_AAS21: A Cold-Adapted Virus Representing a Novel Genus within the Family Siphoviridae. Viruses, 12(4), 479. https://doi.org/10.3390/v12040479